1,3,5-Triaryl-1,3,5-Triazinane-2,4,6-Trithiones: Synthesis, Electronic Structure and Linear Optical Properties


 , , and
, , and 
Abstract
:
1. Introduction
2. Results and Discussion
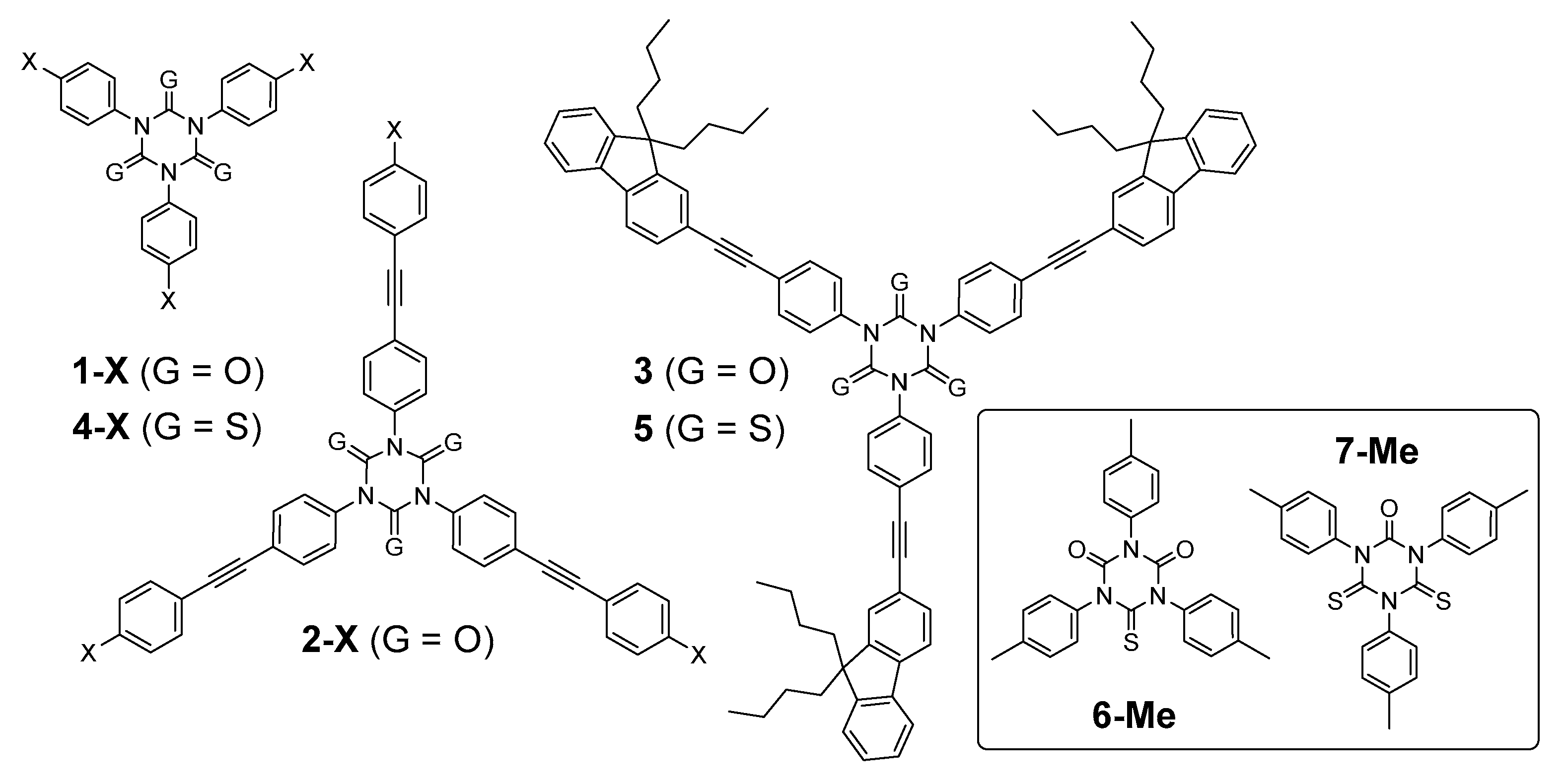
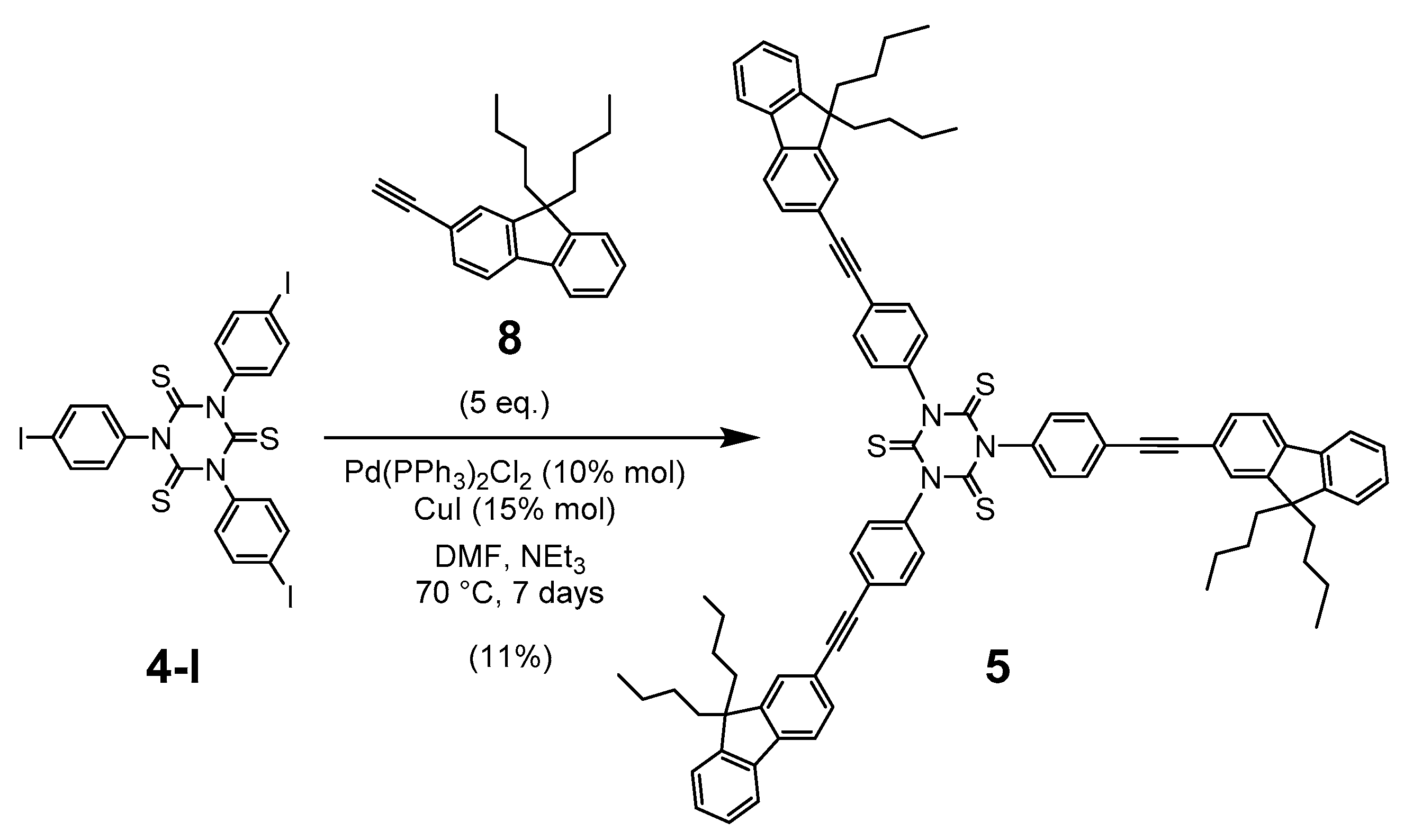
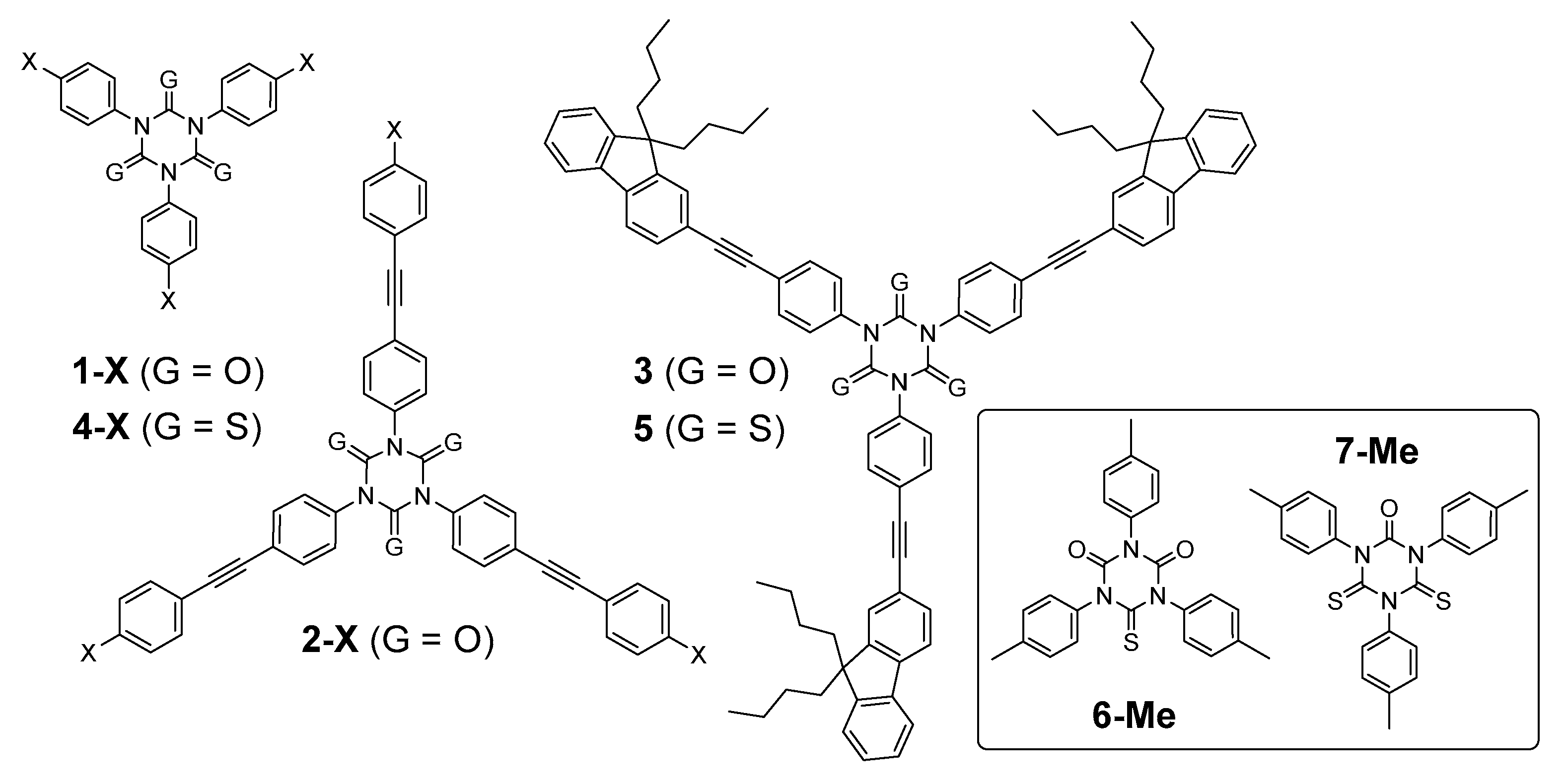
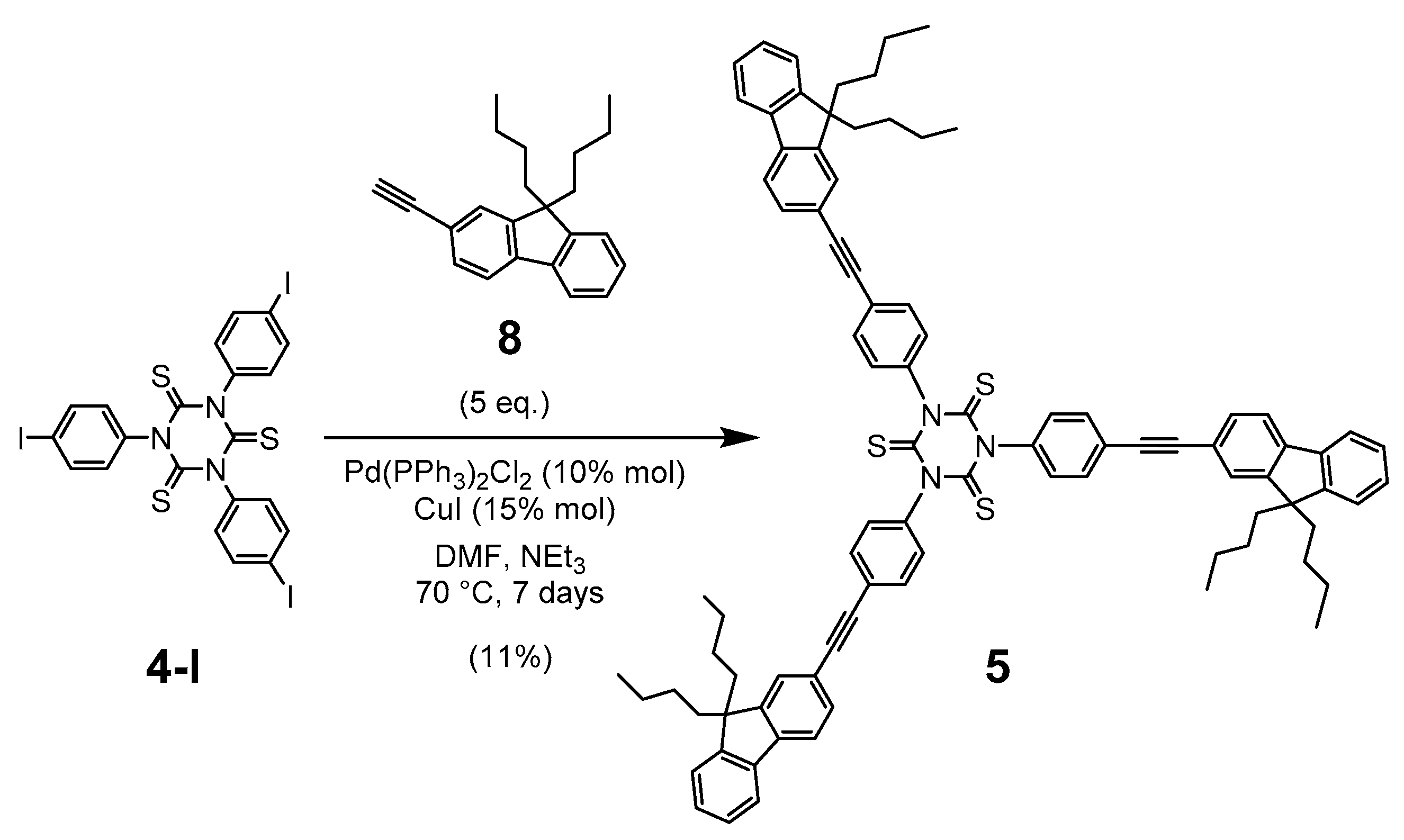
2.1. Synthesis
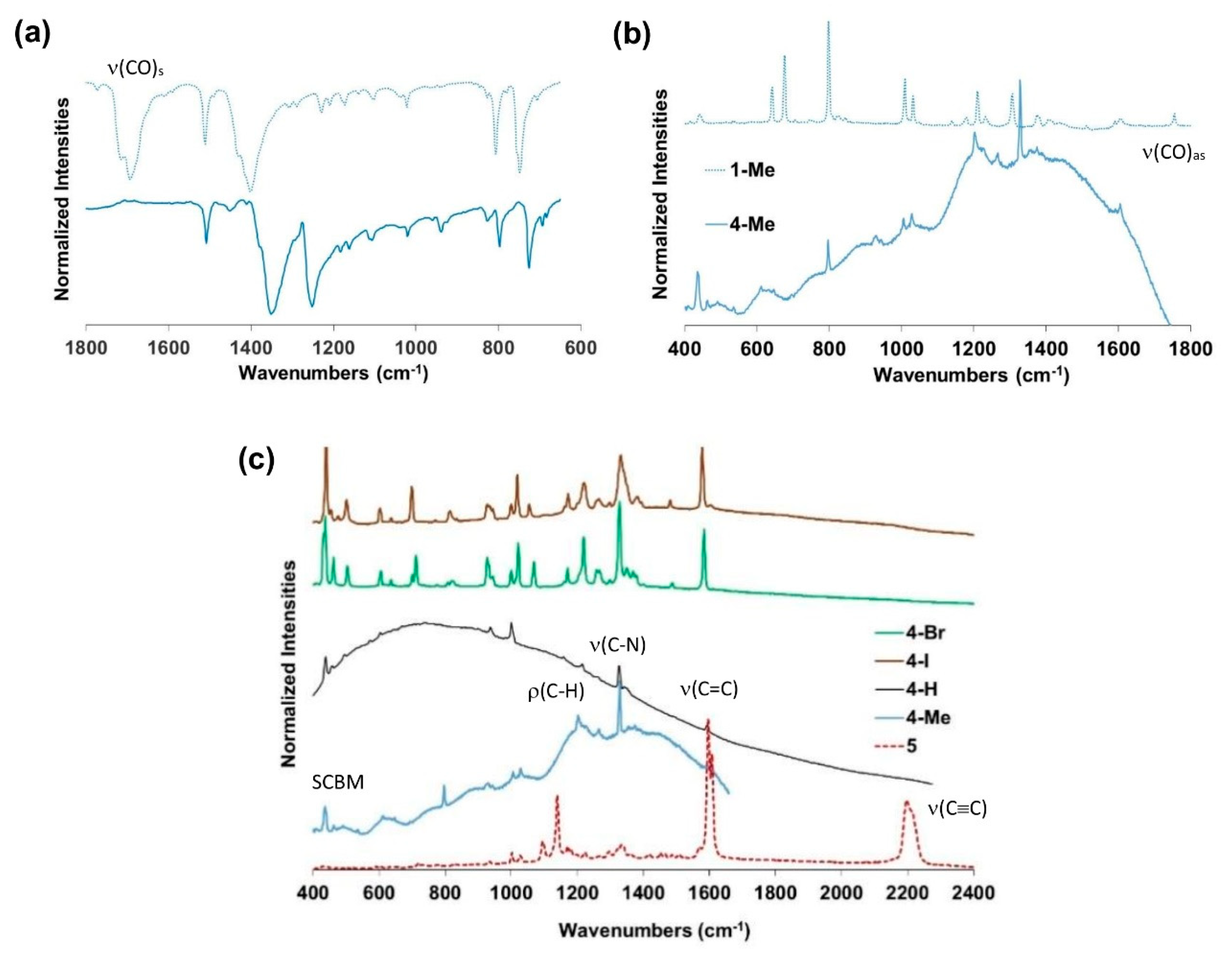
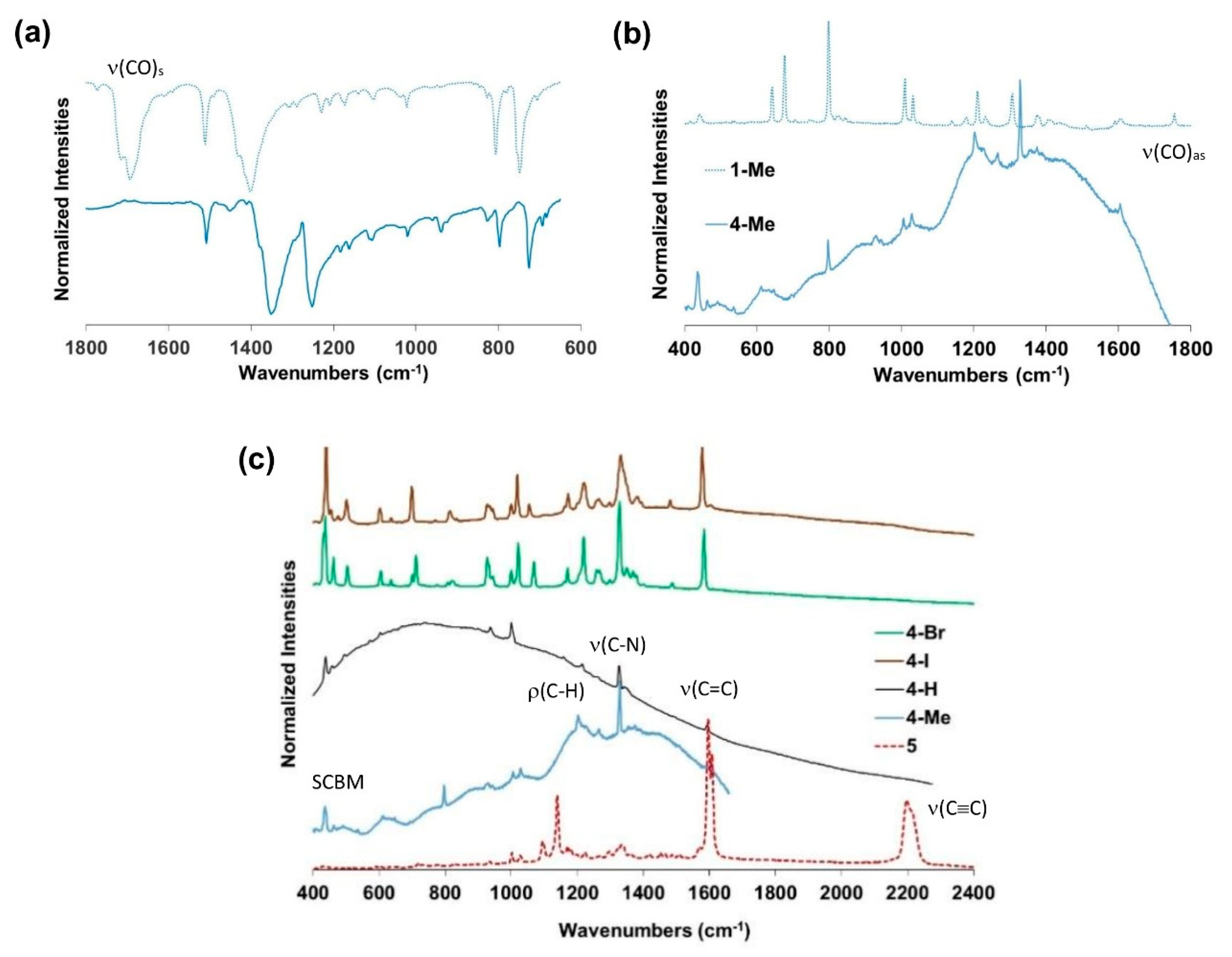
2.2. Vibrational Properties
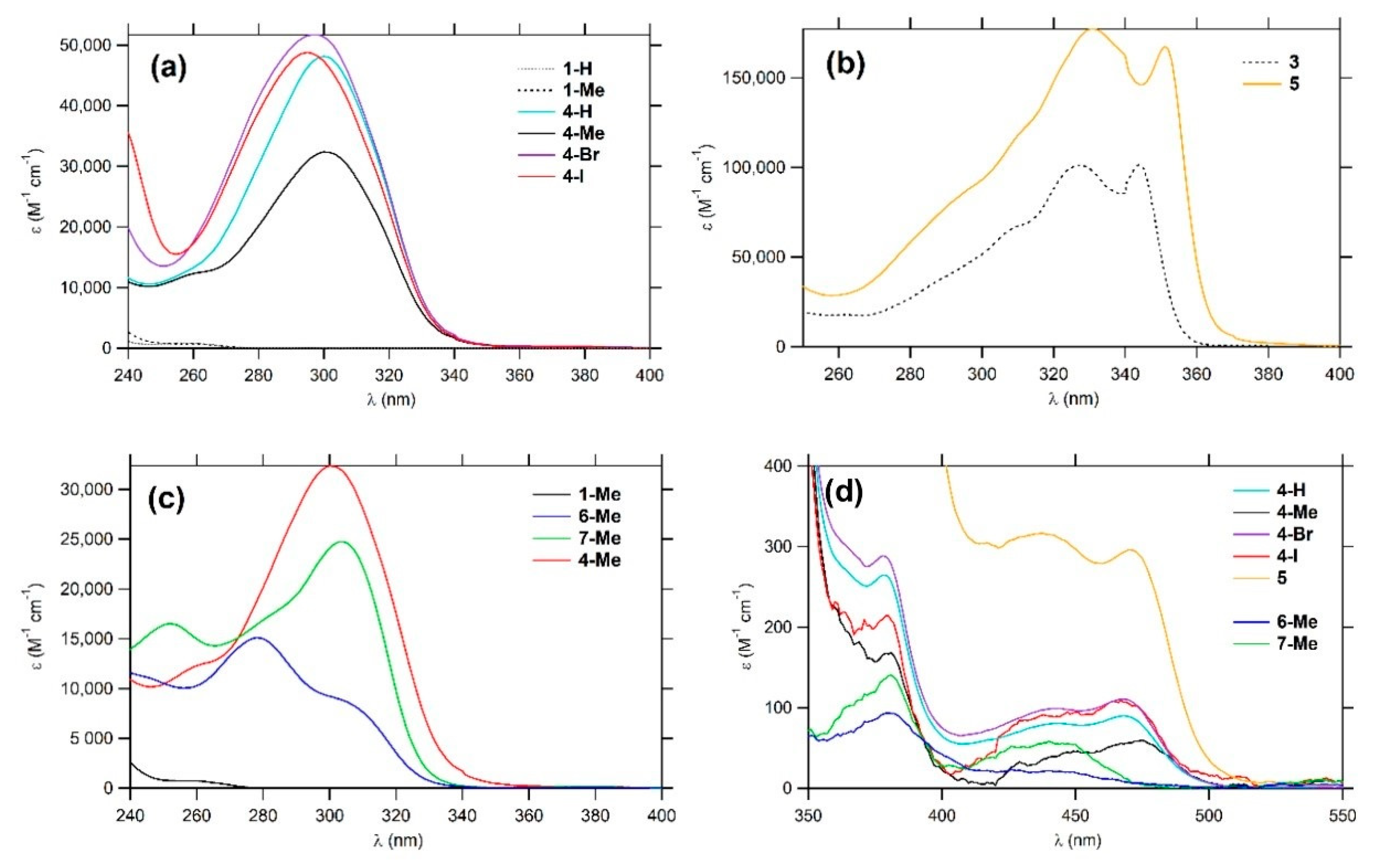
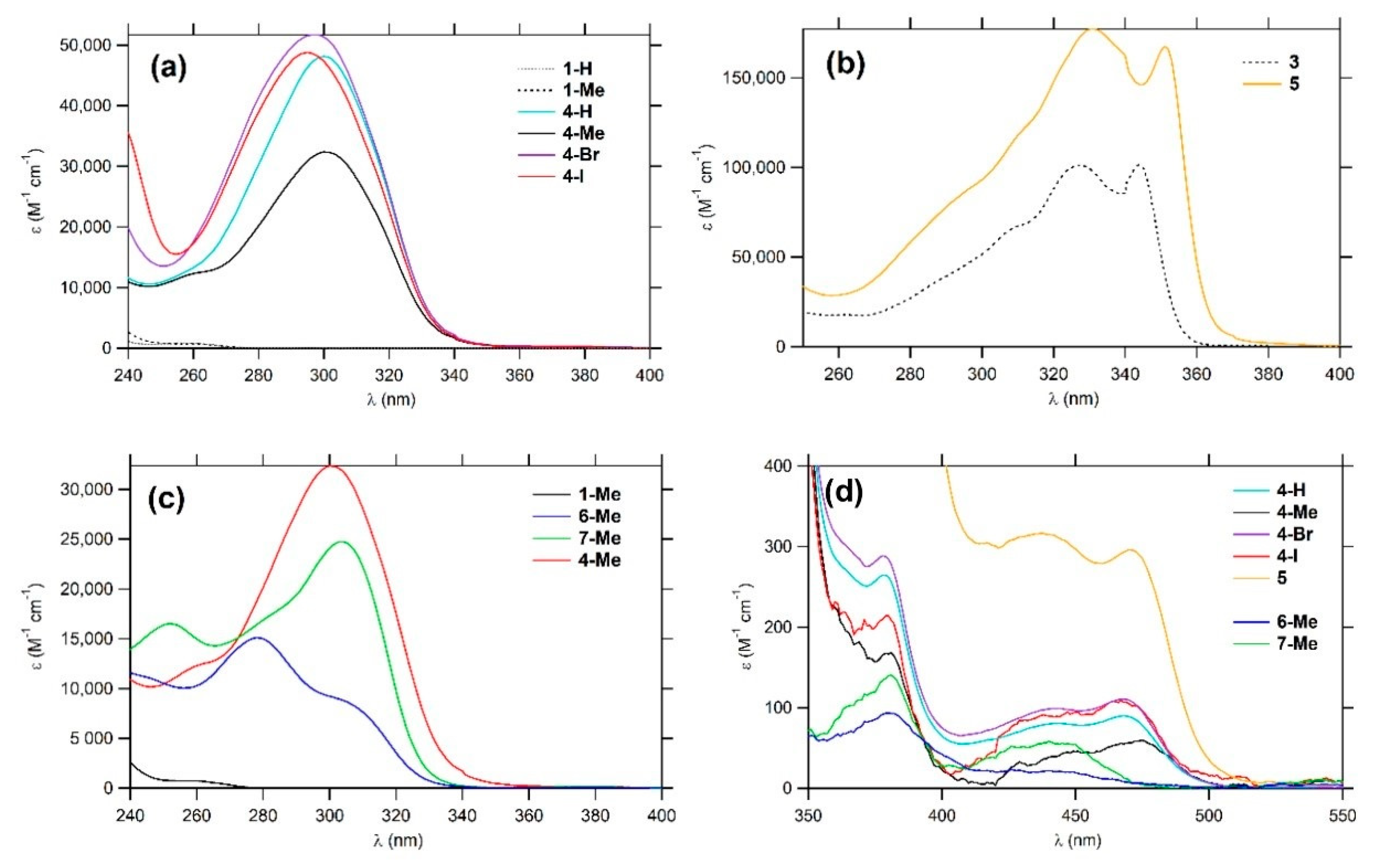
2.3. Linear Photophysical Properties
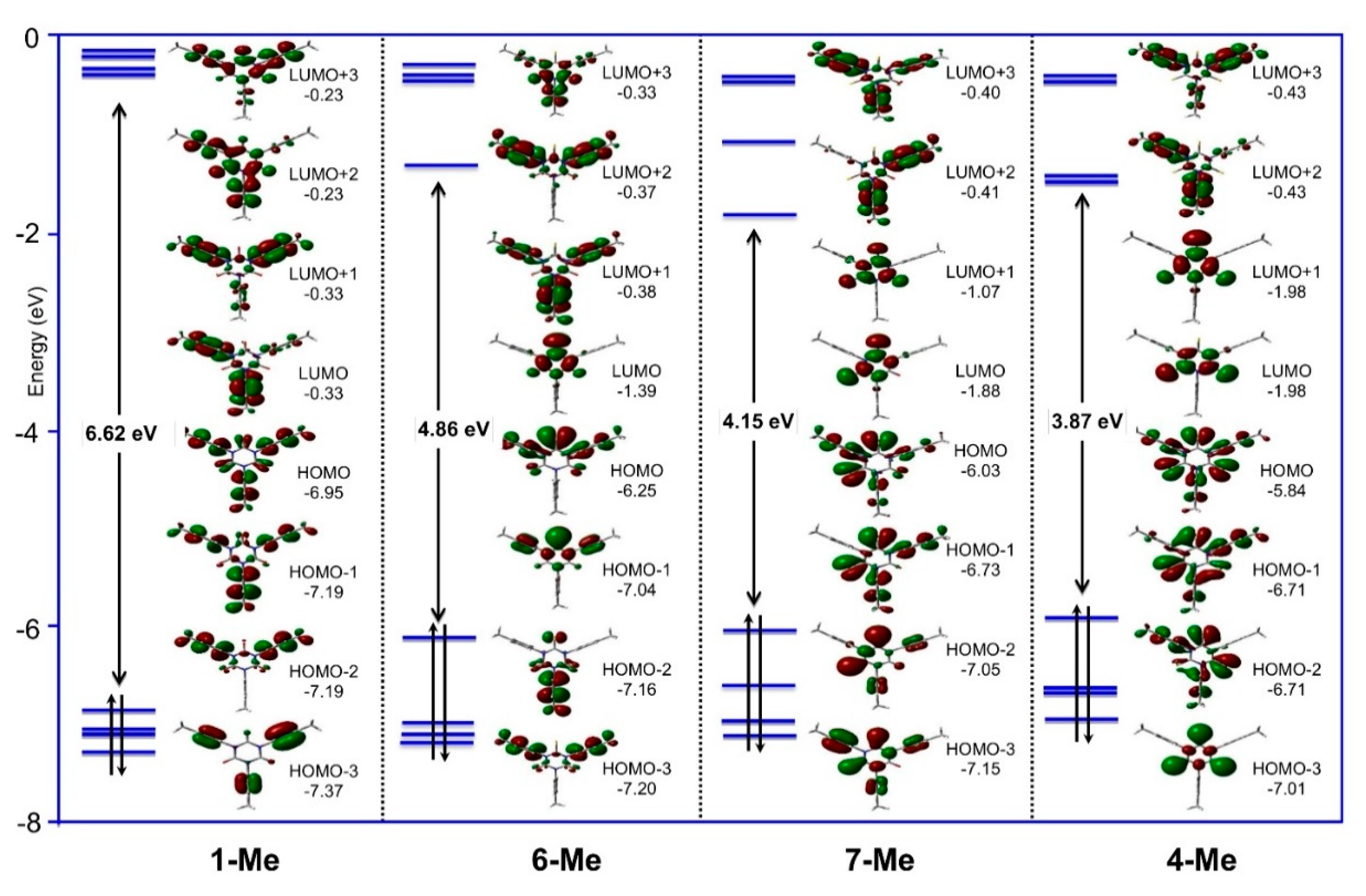
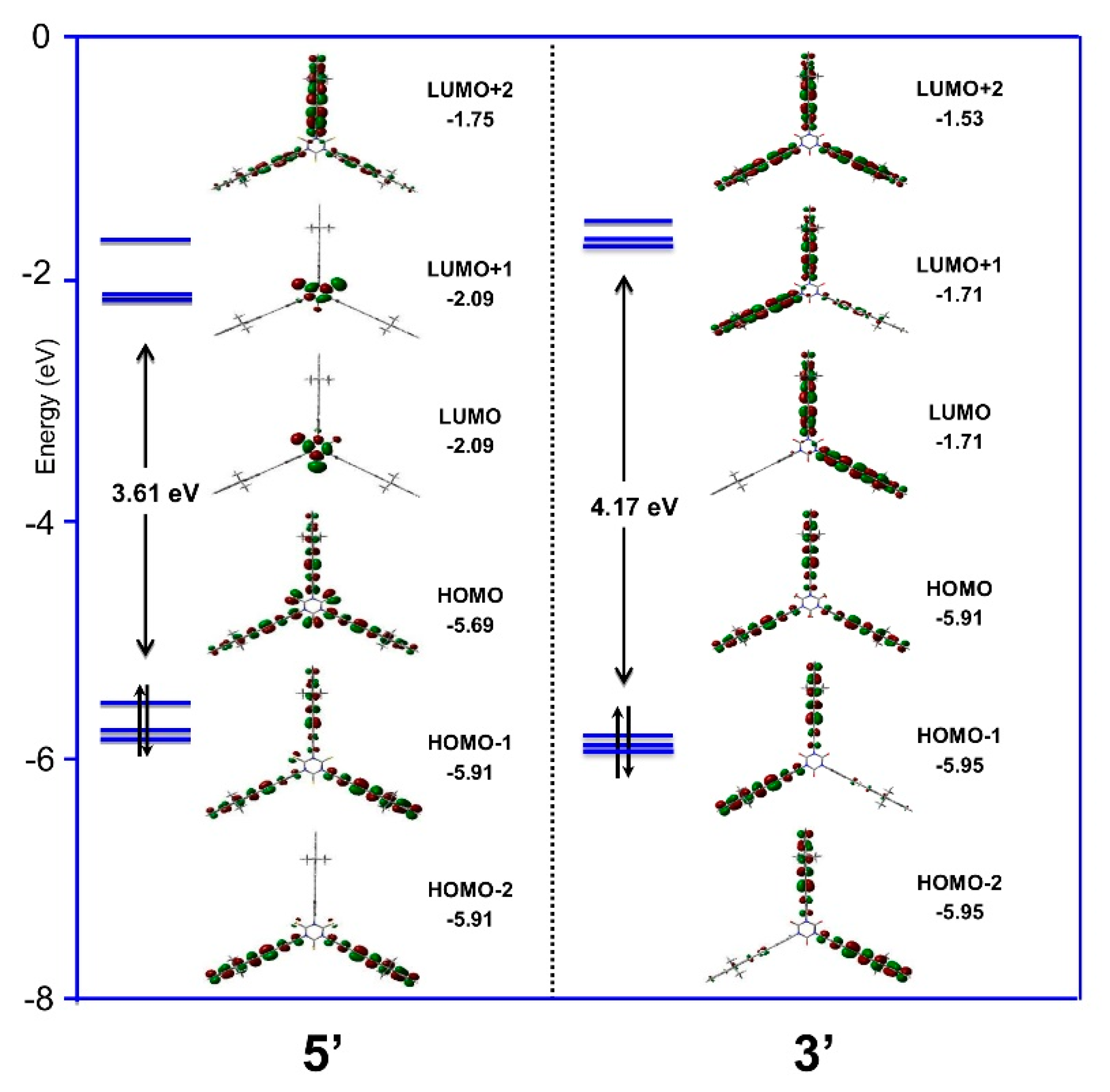
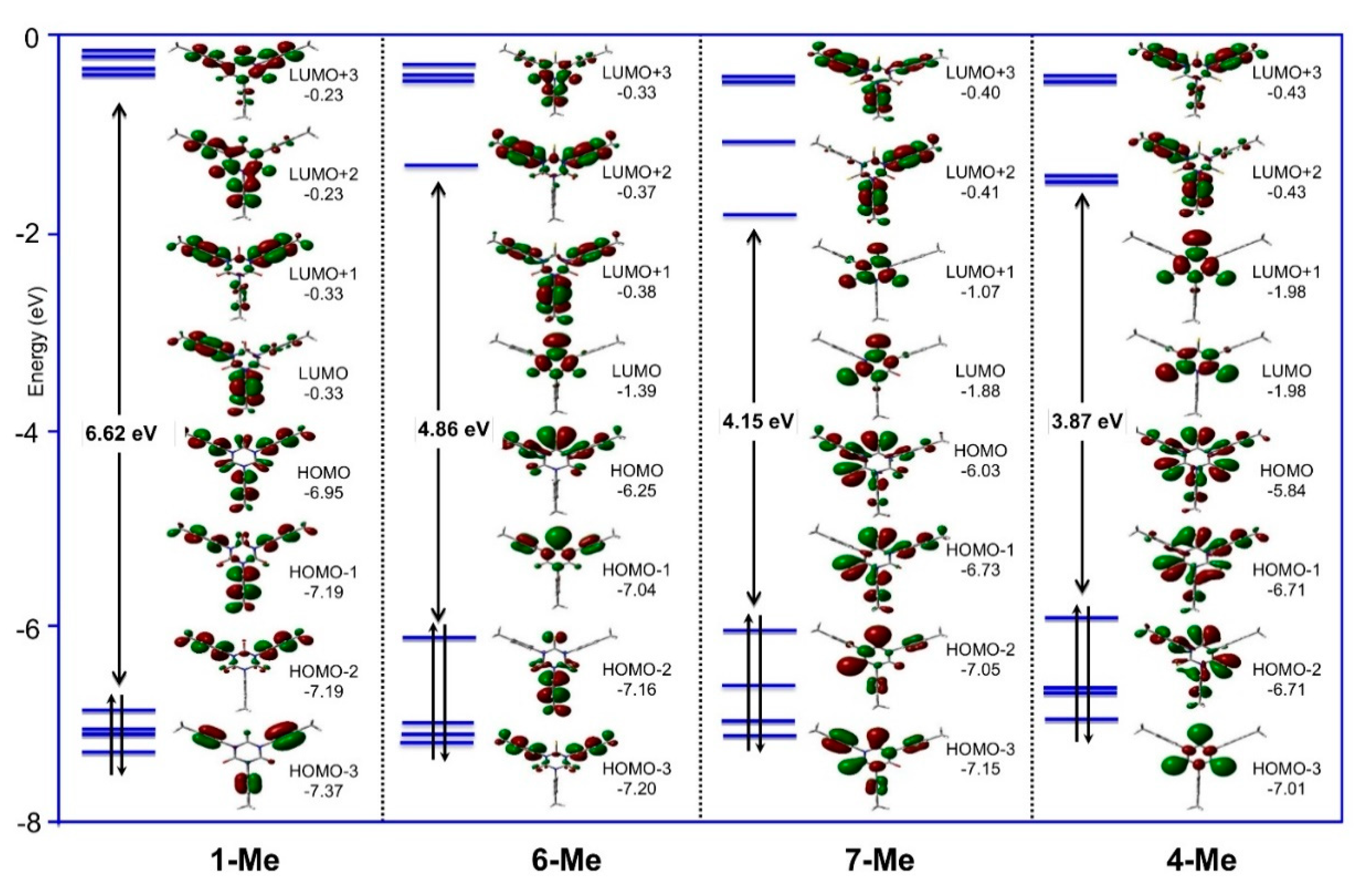
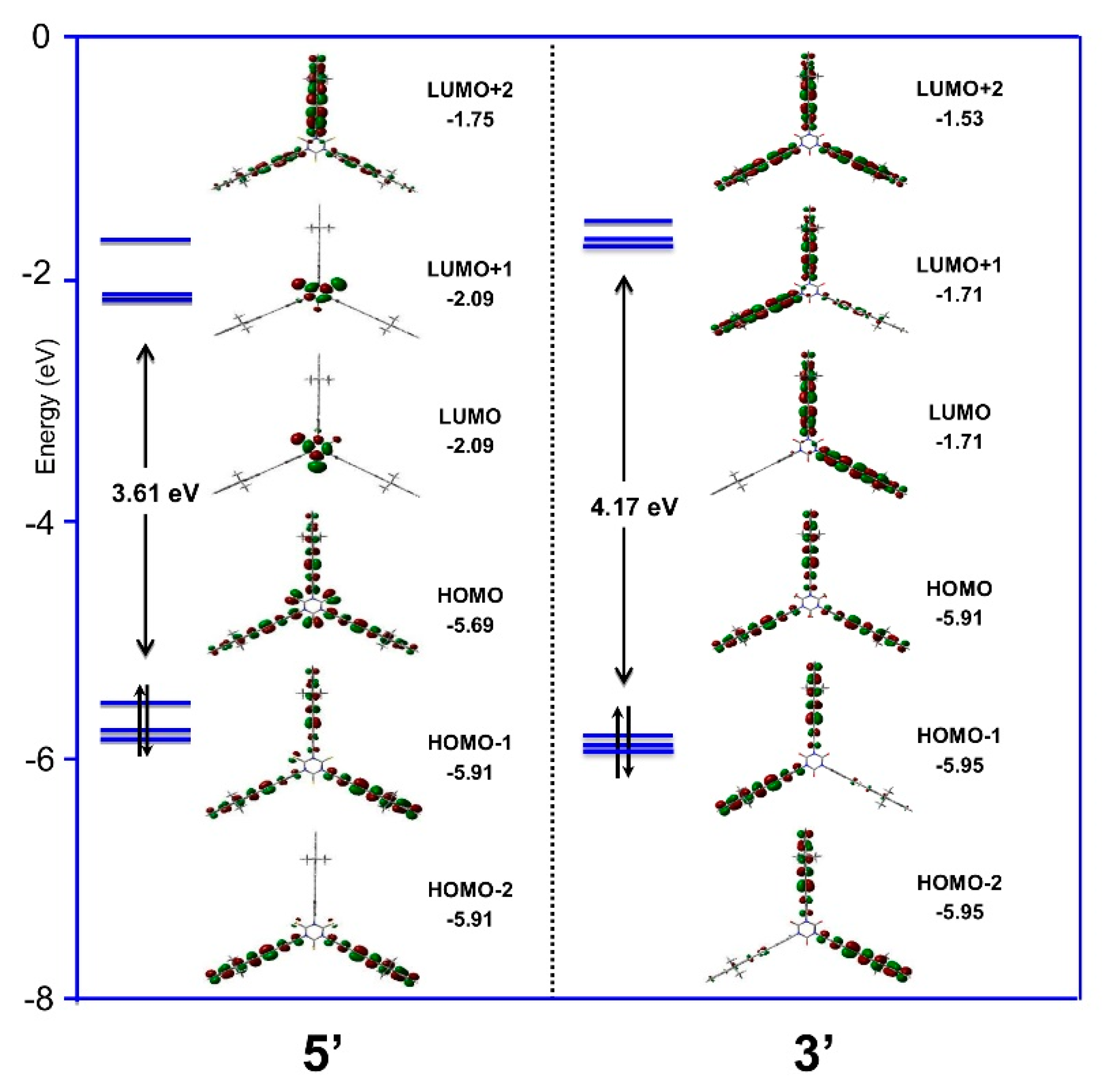
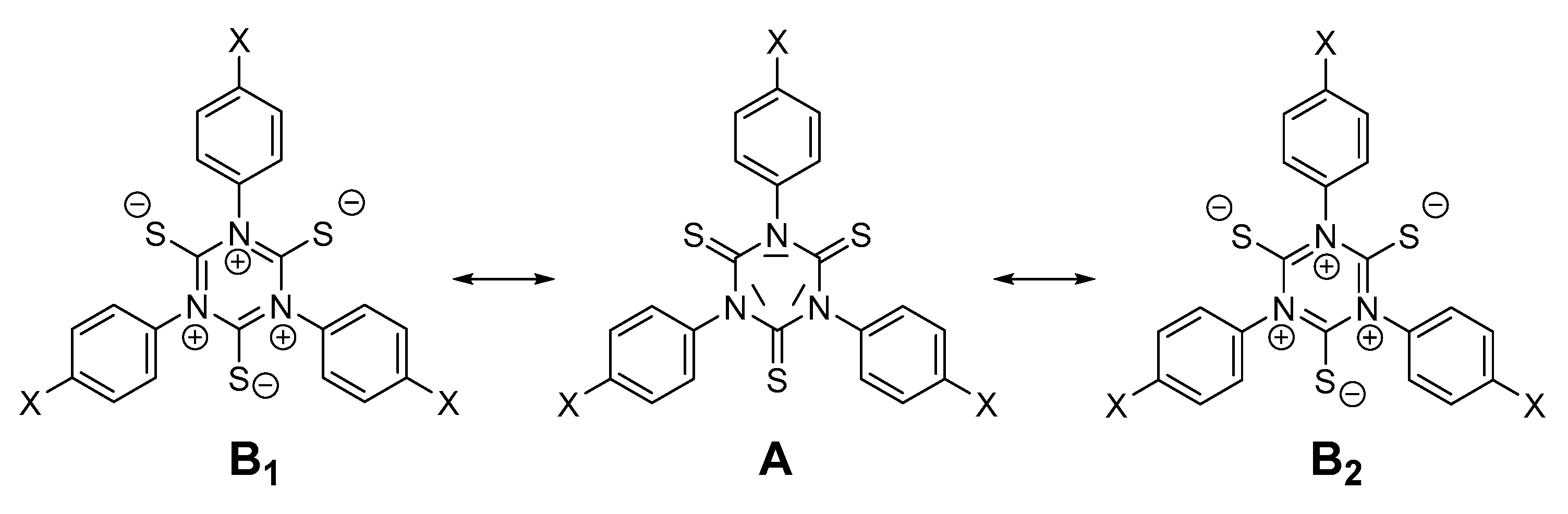
2.4. Density Functional Theory (DFT) Calculations
3. Materials and Methods
3.1. General
3.2. Synthesis of Thioisocyanurates from Isocyanurate Precursors; General Procedure
3.3. Isolation of the Mono-Thioisocyanurate Derivative 6-Me and of the Di-thioisocyanurate Derivative 7-Me
3.4. Synthesis of 1,3,5-Tris(4-((9,9-dibutyl-9H-fluoren-2-yl)ethynyl)-phenyl)-1,3,5-triazinane-2,4,6-trithione (5)
3.5. Spectroscopic Measurements
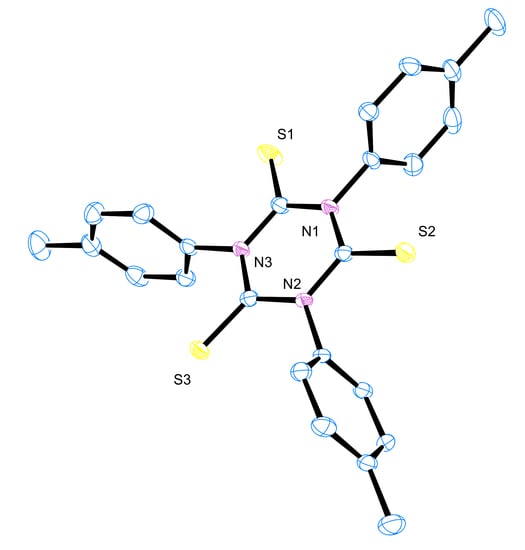
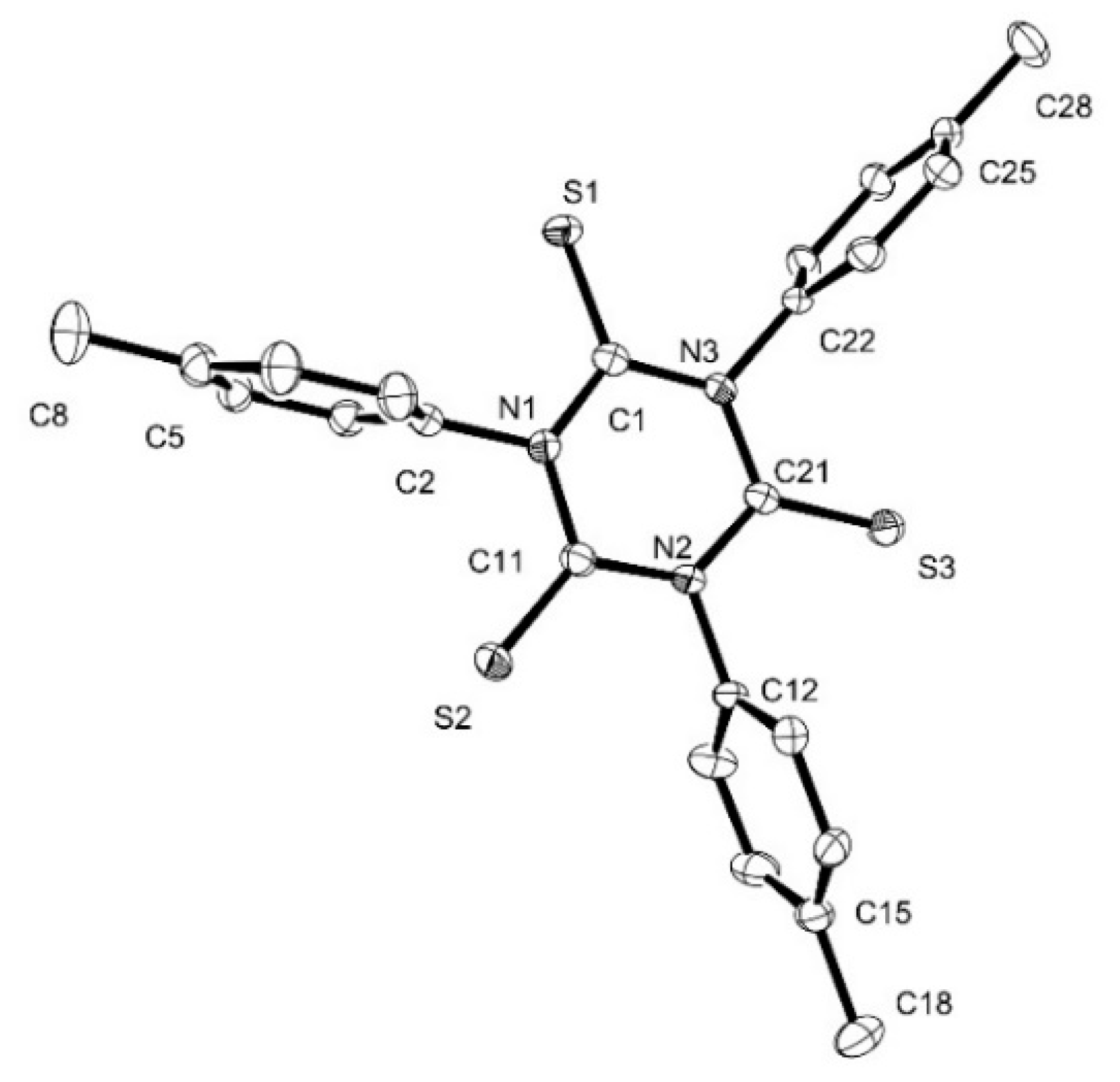
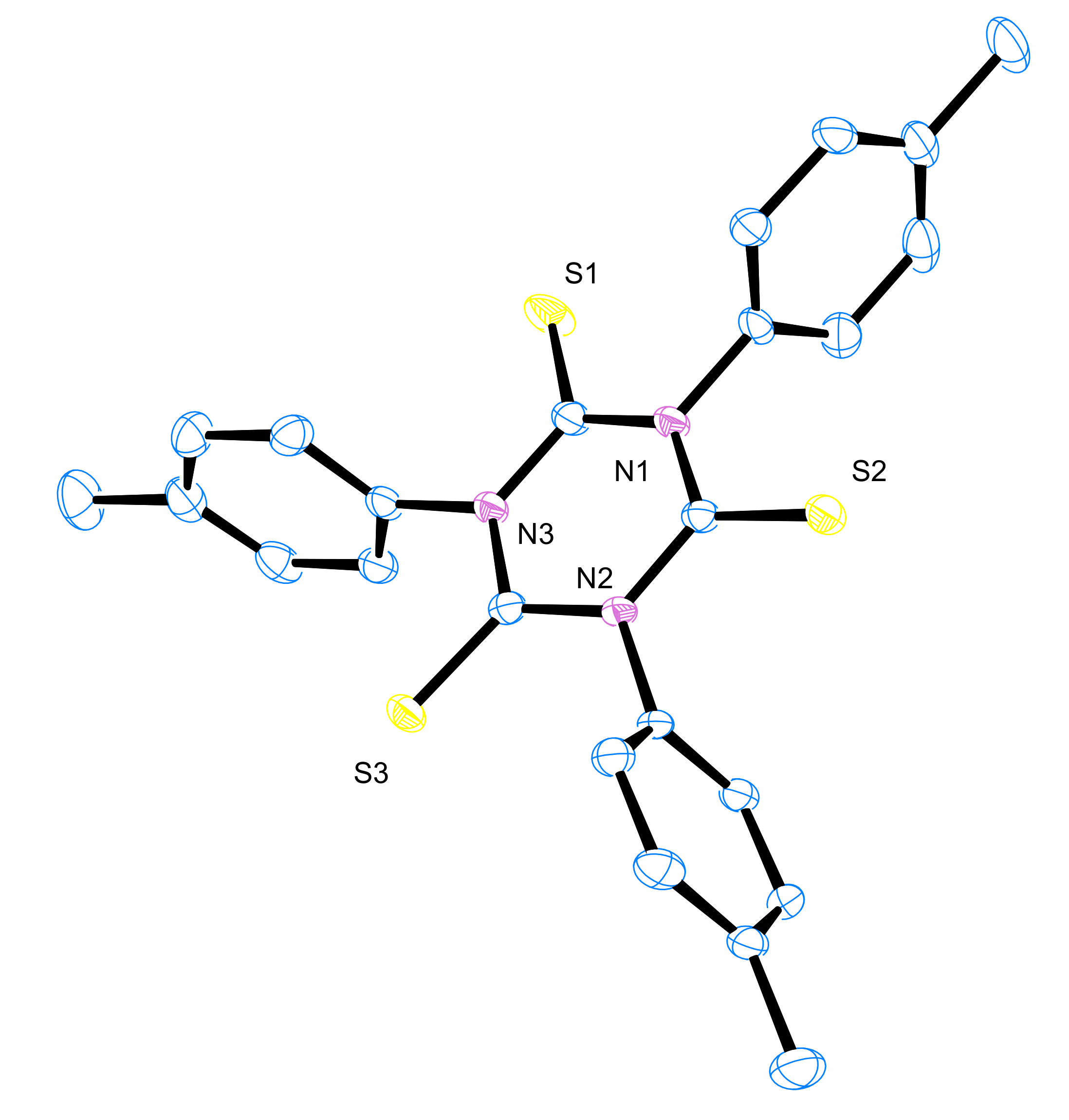
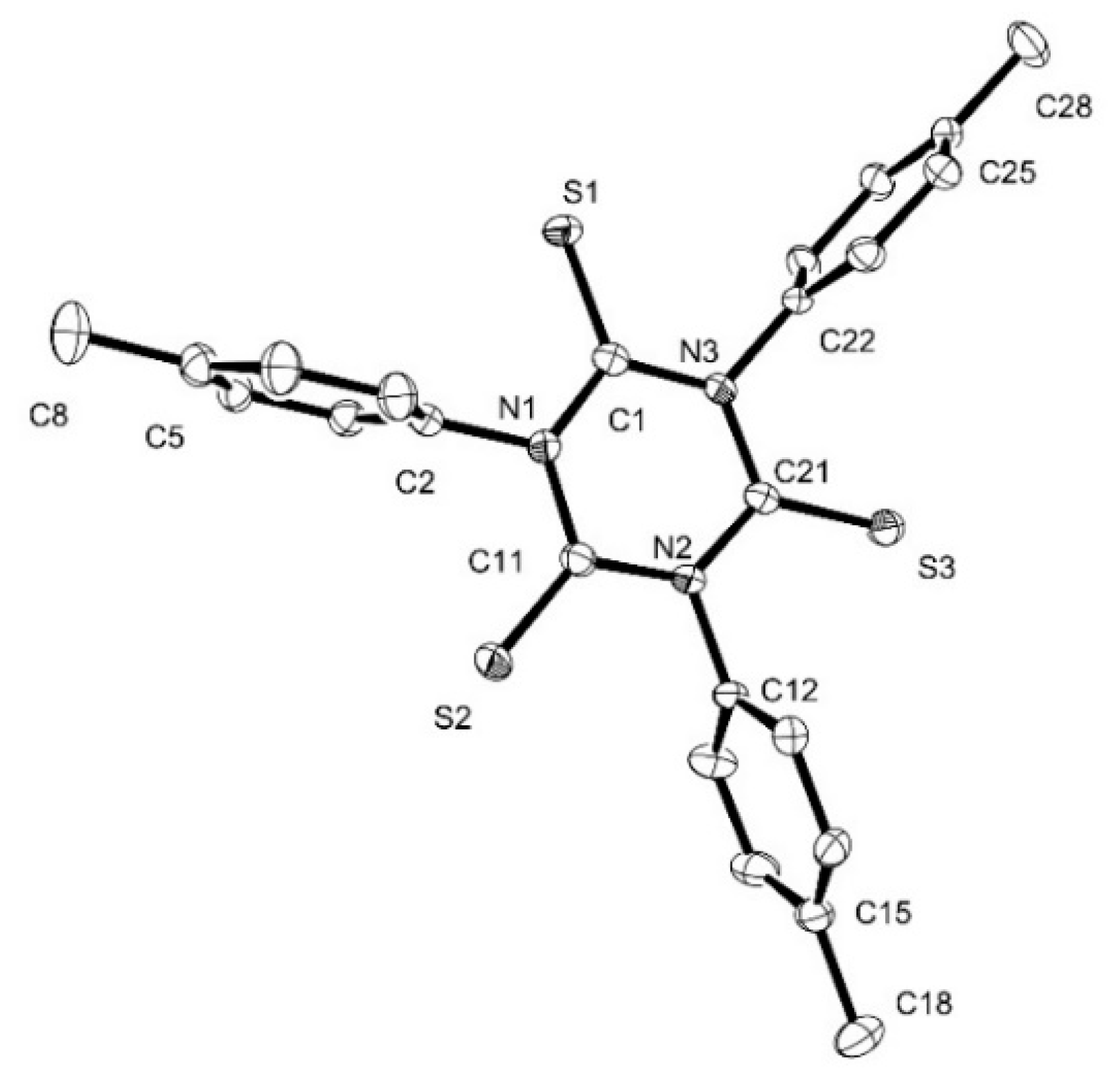
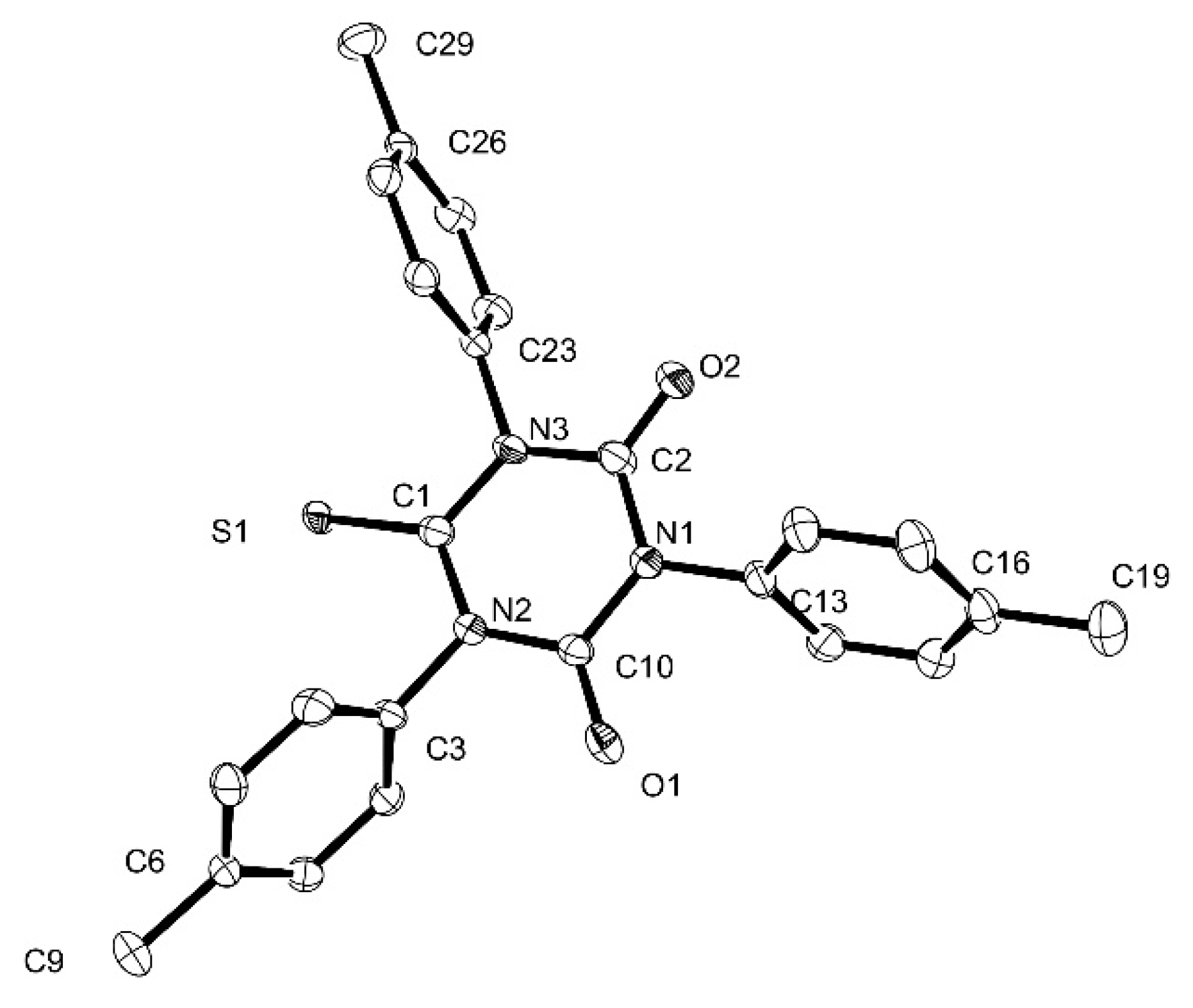
3.6. X-ray Crystallography
3.7. DFT Computations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Hofmann, A.W. Beobachtungen vermischten Inhalt (Aus dem Berl. Universitätslaboratorium LXI). Chem. Ber. 1870, 3, 761–772. [Google Scholar]
- Zyss, J.; Ledoux, I. Nonlinear Optics in Multipolar Media: Theory and Experiment. Chem. Rev. 1994, 94, 77–105. [Google Scholar]
- Argouarch, G.; Veillard, R.; Roisnel, T.; Amar, A.; Meghezzi, H.; Boucekkine, A.; Hugues, V.; Mongin, O.; Blanchard-Desce, M.; Paul, F. Triaryl-1,3,5-Triazinane-2,4,6-Triones (Isocyanurates) Peripherally Functionalized by Donor Groups: Synthesis and Study of their Linear and Nonlinear Optical Properties. Chem. Eur. J. 2012, 18, 11811–11826. [Google Scholar] [PubMed]
- Gautier, Y.; Argouarch, G.; Malvolti, F.; Blondeau, B.; Richy, N.; Amar, A.; Boucekkine, A.; Nawara, K.; Chlebowicz, K.; Orzanowska, G.; et al. Triarylisocyanurate-Based Fluorescent Two-Photon Absorbers. ChemPlusChem 2020, 85, 411–425. [Google Scholar] [PubMed]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–3266. [Google Scholar]
- Nalwa, H.S. Organic Materials for Third-Order Nonlinear Optics. Adv. Mater. 1993, 5, 341–358. [Google Scholar]
- Breitzer, J.G.; Dlott, D.D.; Iwaki, L.K.; Kirkpatrick, S.M.; Rauchfuss, T.B. Third-Order Nonlinear Optical Properties of Sulfur-Rich Compounds. J. Phys. Chem. A 1999, 103, 6930–6937. [Google Scholar]
- Mishira, A.; Ma, C.Q.; Bäuerle, P. Functional Oligothiophenes: Molecular Design for Multidimensional Nanoarchitectures and Their Applications. Chem. Rev. 2009, 109, 1141–1272. [Google Scholar]
- Tugolukova, L.F.; Ignat’eva, E.K.; Kuznetsov, E.V. Reactions of isocyanates in the presence of substituted orthotitanates. Sbornik Nauchnykh Trudov, Kuzbasskii Politekhnicheskii Institut 1974, 69, 197–201. [Google Scholar]
- Tripolt, R.; Nachbaur, E. The Oxidation of Trithiocyanuric Acid and of its N- and S-esters. Phosphorus Sulfur Silicon 1992, 65, 173–176. [Google Scholar]
- Shunichiro, Y.; Kazuaki, S. Charge-Controlling Agent and Toner for Developing Electrostatic Image. Patent No. JP10010787, 24 June 1996. [Google Scholar]
- Lang, H.; Herres, M.; Köhler, K.; Blau, S.; Weinmann, S.; Weinmann, M.; Reinwald, G.; Imhof, W. Monomere Alkin-stabilisierte Kupfer(I)-Halogenid- und Kupfer(I)-Pseudohalogenid-Verbindungen; Kristallstruktur von [(5-C5H4SiMe3)2Ti(CCPh)2]CuCl. J. Organomet. Chem. 1995, 505, 85–94. [Google Scholar]
- Chang, C.-W.; Lin, Y.-C.; Lee, G.-H.; Huang, S.-L.; Wang, Y. Reactions of Ruthenium Acetylide Complexes with Isothiocyanate. Organometallics 1998, 17, 2534–2542. [Google Scholar]
- Akira, K.; Shunichiro, Y. Preparation of triazinetrithione derivatives. Patent No. JP05255283, 9 March 1992. [Google Scholar]
- Lunazzi, L.; Mancinelli, M.; Mazzanti, A. Atropisomers of Hindered Triarylisocyanurates: Structure, Conformation, Stereodynamics, and Absolute Configuration. J. Org. Chem. 2012, 77, 3373–3380. [Google Scholar] [PubMed]
- The weak solubility of 4-I in the usual solvents precluded recording its 13C-NMR spectrum.
- Raper, E.S.; Creighton, J.R.; Bell, N.A.; Clegg, W.; Cucurull-Sanchez, L. Complexes of heterocyclic thiones and group twelve metals Part 1. Preparation and Characterization of 1:1 Complexes of Mercury(II) Halides with 1-Methylimidazoline-2(3H)-thione: The Crystal Structure of [(μ2-dibromo)bis(trans{(bromo)(1-methyl-imidazoline-2(3H)-thione)}mercury(II))] at 160 K. Inorg. Chim. Acta 1998, 277, 14–20. [Google Scholar]
- Sudha, L.V.; Sathyanarayana, D.N. 1H and 13C Dynamic NMR Studies of N,N’-Diethyl N’-Aryl Thioureas. J. Mol. Struct. 1985, 127, 313–317. [Google Scholar]
- Sudha, L.V.; Sathyanarayana, D.N. 13C-NMR Study of 1,3-Pyridylmethyl Ureas and Thioureas. J. Mol. Struct. 1985, 131, 259. [Google Scholar]
- Martin, M.; Frilleux-Blanchard, M.L.; Martin, G.J.; Webb, G.A. Application of 15N Spectroscopy and Dynamic NMR to the Study of Ureas, Thioureas and their Lewis Acid Adducts. Org. Mag. Res. 1980, 13, 396–402. [Google Scholar]
- Crystal Data for C24H21N3S3 (M = 447.62 g), Yellow plates, monoclinic, space group P21/n (no. 14), a = 14.3342(12), b = 19.3756(13), c = 16.7906(12) Å, β = 107.008(3)°, V = 4459.4(6) Å3. Z = 8, Dcalc = 1.333 g.cm3 and μ(Mo-K) = 0.349 mm−1.
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths determined by X-ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar]
- Arjunan, P.; Ramamurthy, V.; Venkatesan, K. 4-Biphenylyl Phenyl Thioketone (I), Cl9H14S, and l-Naphthyl Phenyl Thioketone (II), C17H12S. Acta Cryst. Sect. C 1984, 40, 556–558. [Google Scholar]
- This C-N bond length is comparable with those observed in isocyanurates (1.39 Å).
- Crystal Data for C24H21N3O2S (M = 415.50 g), Colourless sticks, orthorhombic, space group Pbcn (no. 60), a = 19.4944(10), b = 9.5037(5), c = 22.3666(11) Å, V = 4143.8(4) Å3, Z = 8, Dcalc = 1.332 g.cm3 and μ(Mo-K) = 0.182 mm−1.
- Bahili, M.A.; Stokes, E.C.; Amesbury, R.C.; Ould, D.M.C.; Christo, B.; Horne, R.J.; Kariuki, B.M.; Stewart, J.A.; Taylor, R.L.; Williams, P.A.; et al. Aluminum-Catalyzed Isocyanate Trimerization, Enhanced by exploiting a Dynamic Coordination Sphere. Chem. Commun. 2019, 55, 7679–7682. [Google Scholar]
- Hernan-Gomez, A.; Bradle, T.D.; Kennedy, A.R.; Livingstone, Z.; Robertson, S.D.; Hevia, E. Developing Catalytic Applications of Cooperative Bimetallics: Competitive Hydroamination/Trimerization Reactions of Isocyanates catalysed by Sodium Magnesiates. Chem. Commun. 2013, 49, 8659–8661. [Google Scholar]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar]
- Malvolti, F.; Rouxel, C.; Triadon, A.; Grelaud, G.; Richy, N.; Mongin, O.; Blanchard-Desce, M.; Toupet, L.; Abdul Razak, F.I.; Stranger, R.; et al. 2,7-Fluorenediyl-bridged Complexes Containing Electroactive “Fe(η5-C5Me5)(η2-dppe)C≡C-” Endgroups: Molecular Wires and Remarkable Nonlinear Electrochromes. Organometallics 2015, 34, 5418–5437. [Google Scholar]
- Zhou, A.; Cao, L.; Li, H.; Liu, Z.; Cho, H.; Henry, W.P.; Pittman, C.U., Jr. “Push-pull” and Spirobicyclic Structures by reacting N-Methyl Cyclic Ketene-N,X (X=S,O)-Acetals with Isocyanates and Isothiocyanates. Tetrahedron Lett. 2006, 62, 4188–4200. [Google Scholar]
- Desseyn, H.O.; Van der Veken, B.J.; Herman, M.A. The Characteristic Pattern of Thioamides in Infared and Raman. Appl. Spectrosc. 1978, 31, 101–105. [Google Scholar]
- Panicker, C.Y.; Varghese, H.T.; Georged, A.; Thomas, P.K.V. FT-IR, FT-Raman and ab-initio Studies of 1,3-Diphenyl Thiourea. Eur. J. Chem. 2010, 1, 173–178. [Google Scholar]
- Estevez-Hernandez, O.; Otazo-Sanchez, E.; Hidalgo-Hidalgo de Cisneros, J.L.; Naranjo-Rodrıguez, I.; Reguera, E. A Raman and Infrared Study of 1-Furoyl-3-monosubstituted and 3,3-Disubstituted thioureas. Spectrochim. Acta A 2005, 62, 964–971, and refs therein. [Google Scholar]
- Mido, Y.; Kimura, S.; Sugano, Y.; Machida, K. Infrared and Raman Spectra of N-n-alkylthioureas. Spectrochem. Acta A 1988, 44, 661–668. [Google Scholar]
- Silverstein, R.M.; Morrill, T.C.; Bassler, G.C. Spectrometric Identification of Organic Compounds; John Wiley & Sons: New York, NY, USA, 1991. [Google Scholar]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules; Methuen & Co Ltd.: London, UK, 1955. [Google Scholar]
- Chary, C.A.I.; Ramiah, K.V. Infrared and Raman Spectra of Normal Vibrations of N,N-Dimethylthioformamide and N,N-Dimethylthioacetamide. Indian Acad. Sci. 1969, 69, 18–35. [Google Scholar]
- Brouwer, A.M. Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar]
- Ndimba, A.N.; Barnes, A.; Richy, N.; Mongin, O.; Dudek, M.; Matczyszyn, K.; Cifuentes, M.P.; Humphrey, M.G.; Paul, F. Work in progress.
- They often have a significant π*C=X←nX character (X = S, O).
- Demas, N.; Crosby, G.A. Measurement of Photoluminescence Quantum Yields. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar]
- Eaton, G.R. Reference Materials for Fluorescence Measurement. Pure Appl. Chem. 1988, 60, 1107–1114. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar]
- These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336033; email: Deposit@ccdc.cam.ac.uk).
- Gaussian 09; Revision A.2; Gaussian, Inc.: Pittsburgh, PA, USA, 2009.
- Adamo, C.; Barone, V. Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar]
- SWizard Program; Revision 4.5; University of Ottawa: Ottawa, ON, Canada, 2013.
- GaussView; Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009.
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
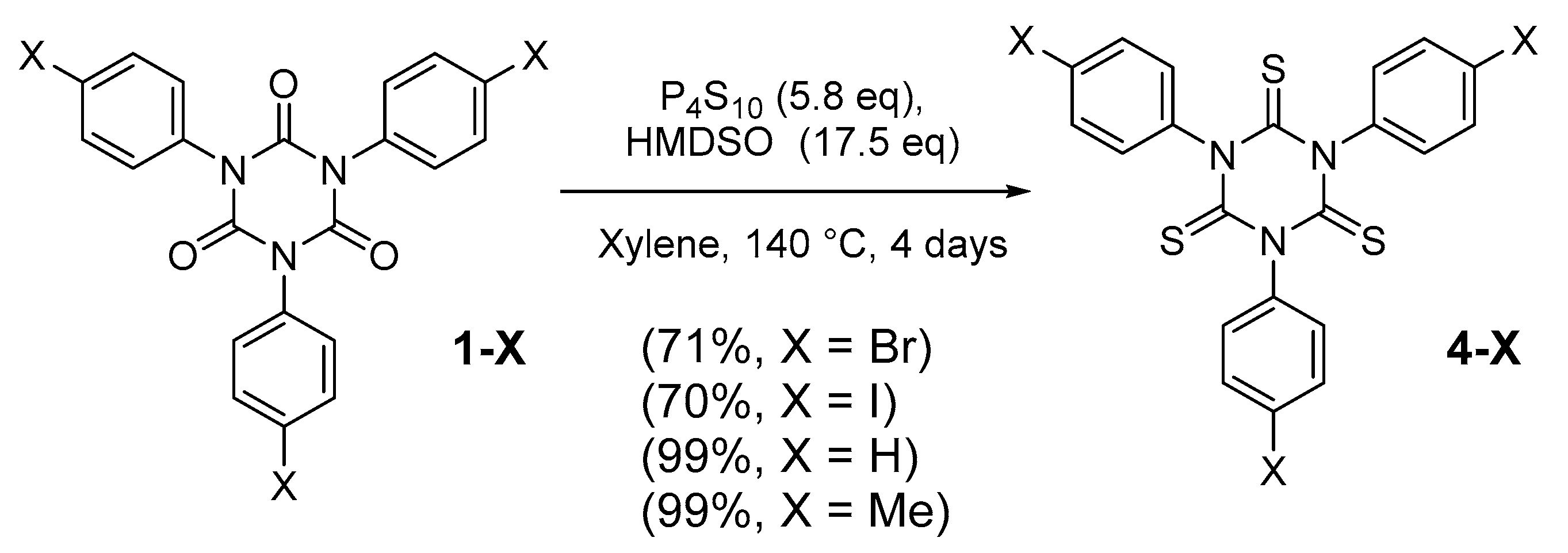{kind=link}
{kind=link}
{kind=link}
| Cmpd. | υ(C≡C) (cm−1) | υ (C=X) (X = O, S) (cm−1) | δ(C≡C) a (ppm) | δ(C=X) a (ppm) |
|---|---|---|---|---|
| 1-Me b | / | 1772 (s) c, 1694 (vs) d,e,f | / | 148.8 |
| 3 | 2203 c,d | 1777 (s) c, 1720 (vs) d | 92.8, 88.9 | 148.4 |
| 4-Br | / | 438 (vs) d | / | / |
| 4-I | / | 440 (vs) d | / | / |
| 4-H | / | 439 (vs) d,g | / | 172.8 h |
| 4-Me | / | 436 (s) d,g | / | 172.8 |
| 5 | 2198 c,d | 428 (vw) d | 93.0, 89.1 | 172.8 |
| 6-Me | / | 1755 (s) c,d,g, 1709 (s) c | / | 178.8, 147.5 |
| 7-Me | / | 1728 (s) c,d,g | / | 176.5, 145.1 |
| Cmpd. | λmaxabs4 | εmax4 | λmaxabs3 | εmax3 | λmaxabs2 | εmax2 | λmaxabs1 | εmax1 | λmaxem | ΦFa | Stokes Shift b | DFT c,d | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (λmax in nm and εmax in 1 × 103 cm−1 M−1) | (nm) | (cm−1) | λmax (nm) [f ] | ||||||||||
| 1-Br | / | / | / | / | <240 | / | 262 (sh) | 1.5 | 306 | 0.0006 | 5200 | nc | This work and [3] |
| 1-I | / | / | / | / | <240 | / | 266 (sh) | 6.3 | / | 0.0 | / | nc | [39] |
| 1-H | / | / | / | / | 251 | 74.5 | 256 (sh) | 0.8 | 283 | 0.005 | 3730 | nc | [3] |
| 1-Me | / | / | / | / | <240 | / | 261 (sh) | 1.0 | 290 | 0.004 | 3830 | 207 [0.30] 207 [0.30] | This work and [3] |
| 3 | / | / | / | / | 327 | 101 | 344 | 101 | 355 | 0.60 | 900 | 345 [2.83] e 345 [2.81] e | This work and [4] |
| 4-Br | <240 | / | 297 | 51.7 | 378 (sh) | 0.3 | 468 | 0.1 | / | 0.0 | / | nc | This work |
| 4-I | <240 | / | 295 | 48.8 | 380 (sh) | 0.2 | 467 | 0.1 | / | 0.0 | / | nc | This work |
| 4-H | <240 | / | 300 | 48.1 | 378 (sh) | 0.3 | 468 | 0.1 | / | 0.0 | / | nc | This work |
| 4-Me | 260 (sh) | 12.3 | 300 | 32.4 | 380 (sh) | 0.2 | 475 | 0.05 | / | 0.0 | / | 279 [0.40] 279 [0.40] | This work |
| 5 | 331 | 177 | 351 | 167 | 438 | 0.3 | 470 | 0.3 | / | 0.0 | / | 367 [2.36] 367 [2.37] | This work |
| 6-Me | 278 | 15.1 | 304 (sh) | 8.8 | 380 | 0.1 | 440 (sh) | 0.02 | / | 0.0 | / | 252 [0.16] | This work |
| 7-Me | 252 | 16.5 | 304 | 24.7 | 381 | 0.1 | 440 | 0.06 | / | 0.0 | / | 279 [0.35] | This work |
| Cmpds. | μ (D) | HOMO-LUMO Gap (eV) | Ref. |
|---|---|---|---|
| 1-Me | 0.10 | 6.62 | This work |
| 3′ a | 0.77 | 4.17 | This work |
| 4-Me | 0.09 | 3.87 | This work |
| 5′ | 0.73 | 3.61 | This work |
| 6-Me | 1.62 | 4.86 | This work |
| 7-Me | 1.40 | 4.15 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabouel, I.; Richy, N.; Amar, A.; Boucekkine, A.; Roisnel, T.; Mongin, O.; Humphrey, M.G.; Paul, F. 1,3,5-Triaryl-1,3,5-Triazinane-2,4,6-Trithiones: Synthesis, Electronic Structure and Linear Optical Properties. Molecules 2020, 25, 5475. https://doi.org/10.3390/molecules25225475
Rabouel I, Richy N, Amar A, Boucekkine A, Roisnel T, Mongin O, Humphrey MG, Paul F. 1,3,5-Triaryl-1,3,5-Triazinane-2,4,6-Trithiones: Synthesis, Electronic Structure and Linear Optical Properties. Molecules. 2020; 25(22):5475. https://doi.org/10.3390/molecules25225475
Chicago/Turabian StyleRabouel, Ismaël, Nicolas Richy, Anissa Amar, Abdou Boucekkine, Thierry Roisnel, Olivier Mongin, Mark G. Humphrey, and Frédéric Paul. 2020. "1,3,5-Triaryl-1,3,5-Triazinane-2,4,6-Trithiones: Synthesis, Electronic Structure and Linear Optical Properties" Molecules 25, no. 22: 5475. https://doi.org/10.3390/molecules25225475
APA StyleRabouel, I., Richy, N., Amar, A., Boucekkine, A., Roisnel, T., Mongin, O., Humphrey, M. G., & Paul, F. (2020). 1,3,5-Triaryl-1,3,5-Triazinane-2,4,6-Trithiones: Synthesis, Electronic Structure and Linear Optical Properties. Molecules, 25(22), 5475. https://doi.org/10.3390/molecules25225475