An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation


 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
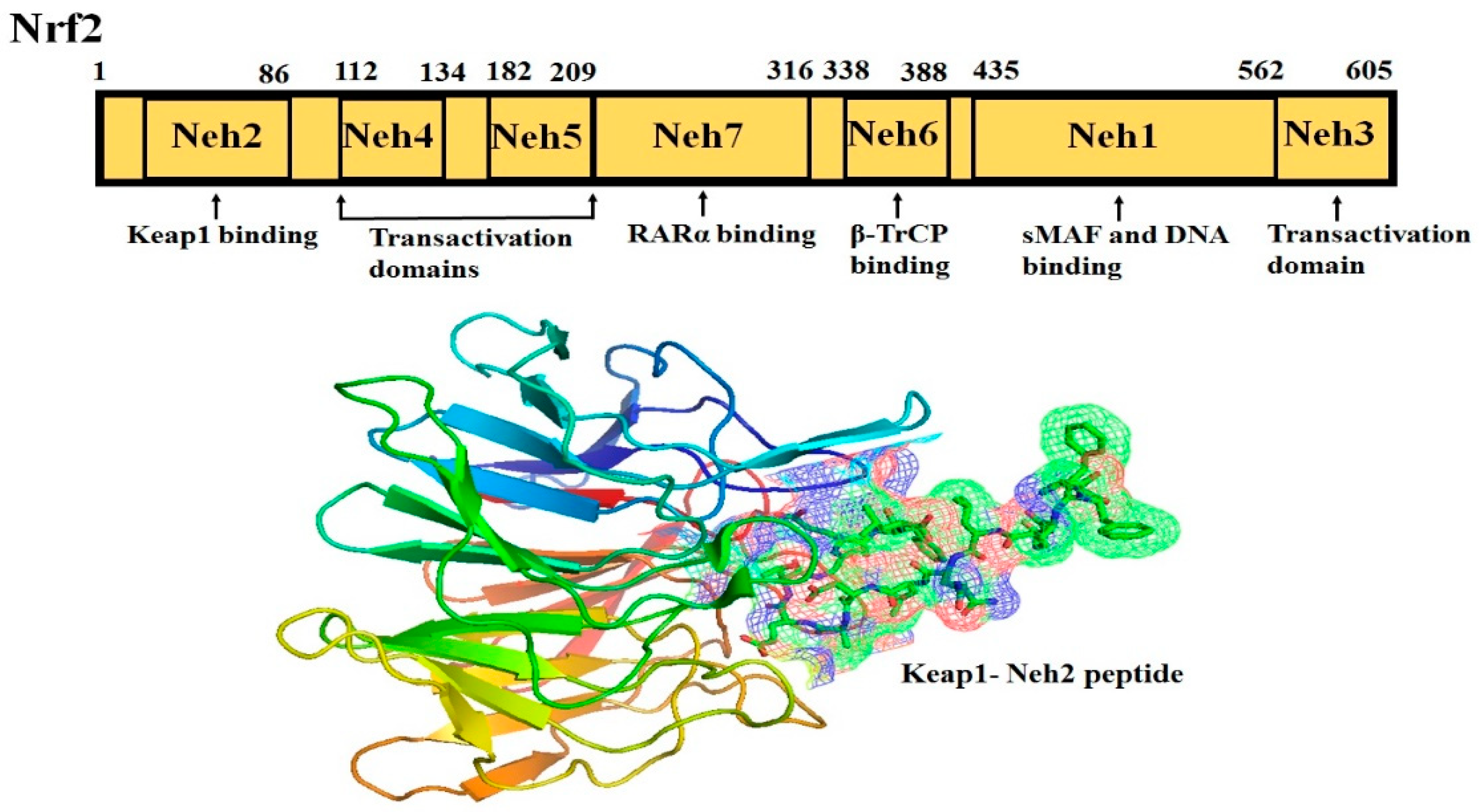
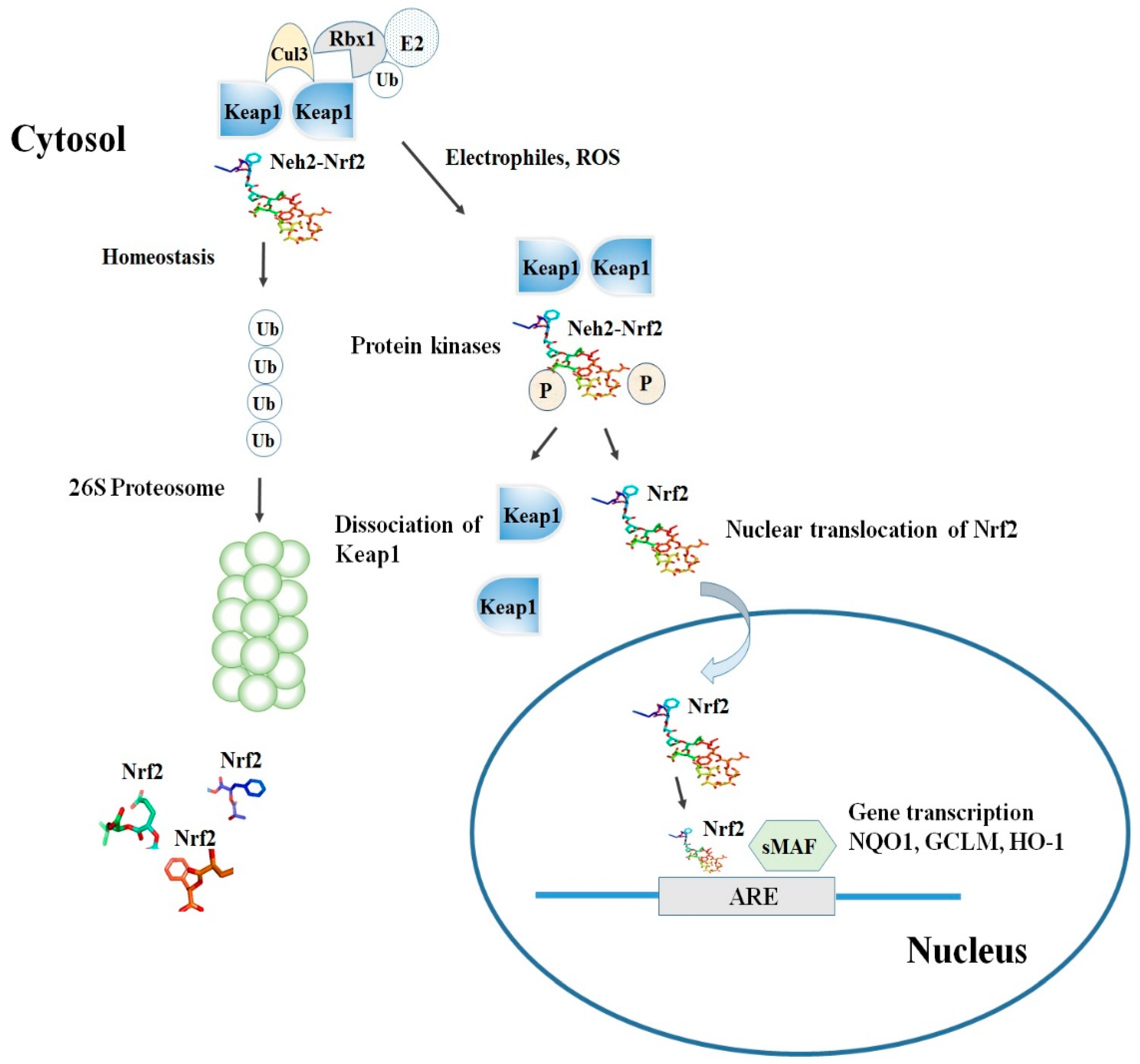
2. Nrf2-Keap1 Signaling Pathway
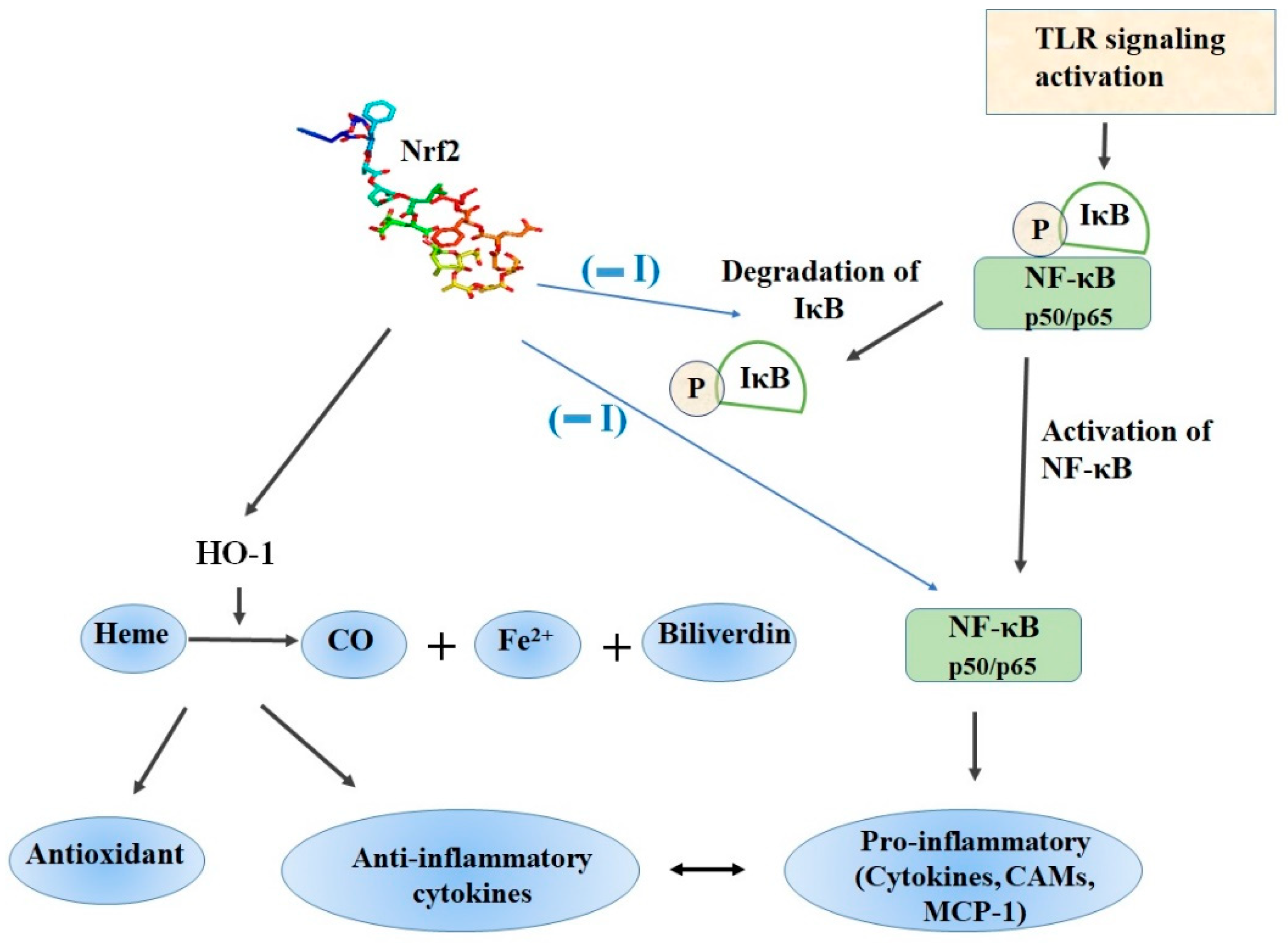
3. Nrf2 and NF-ĸB Interplay in the Regulation of Cellular Redox Pathway
4. Cross-Talk between Nrf2 and Inflammation
4.1. Role of Nrf2/HO-1 Axis in Inflammation
4.2. Functional Interaction of Nrf2 with Inflammatory Mediators
4.2.1. Role of Nrf2 in the Regulation of Cytokines
4.2.2. Role of Nrf2 in the Regulation of Cell Adhesion Molecules
4.2.3. Role of Nrf2 in the Regulation of Protease/Antiprotease Balance
5. Incongruity in the Role of Nrf2 in Inflammasome Signaling
6. Nrf2 Dependent Anti-Inflammatory Drugs
7. Polyphenols and Nrf2 Signaling Can Modulate Inflammation
8. Future Prospects
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Soehnlein, O.; Lindbon, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [PubMed]
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease; Garland Science: New York, NY, USA, 2005. [Google Scholar]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [PubMed]
- Noah, T.A.; Zachary, M.W.; Randy, J.N. Inflammation: Mechanisms, costs, and natural variation. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 385–406. [Google Scholar]
- Kaulmann, A.; Bohn, T. Carotenoids, inflammation, and oxidative stress–implications of cellular signaling pathways and relation to chronic disease prevention. Nutr. Res. 2014, 34, 907–929. [Google Scholar]
- Kaplan, M.H. STAT signaling in inflammation. JAKSTAT 2013, 2, e24198. [Google Scholar]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A. The Keap1-Nrf2pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell Biol. 2005, 25, 162–171. [Google Scholar]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar]
- Kansanen, E.; Kivel, A.M.; Levonen, A.L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Delta 12,14-prostaglandin J2. Free Radic. Biol. Med. 2009, 47, 1310–1317. [Google Scholar]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cell. 2011, 16, 123–140. [Google Scholar]
- Um, H.C.; Jang, J.H.; Kim, D.H.; Lee, C.; Surh, Y.J. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide 2011, 25, 161–168. [Google Scholar]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-Sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar]
- Kansanen, E.; Jyrkkanen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Gene Dev. 1999, 13, 76–86. [Google Scholar]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. [Google Scholar]
- Chowdhry, S.; Zhang, M.; McMahon, Y.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}- TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Sutter, T.R.; Kensler, T.W. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1,2-dithiole-3-thione. Mol. Med. 2001, 7, 135–145. [Google Scholar] [PubMed]
- Vollrath, V.; Wielandt, A.M.; Iruretagoyena, M.; Chianale, J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 2006, 395, 599–609. [Google Scholar] [PubMed]
- Gilmore, T. The Rel/NF-κB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [PubMed]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [PubMed]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar]
- Yerra, V.G.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar]
- Chen, L.G.; Zhang, Y.Q.; Wu, Z.Z.; Hsieh, C.W.; Chu, C.S.; Wung, B.S. Peanut arachidin-1 enhances Nrf2-mediated protective mechanisms against TNF-α-induced ICAM-1 expression and NF-κB activation in endothelial cells. Int. J. Mol. Med. 2018, 41, 541–547. [Google Scholar]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappab/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancer 2010, 2, 483–497. [Google Scholar]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar]
- Chuang, H.C.; Chang, C.W.; Chang, G.D.; Yao, T.P.; Chen, H. Histone deacetylase 3 binds to and regulates the GCMa transcription factor. Nucl. Acids Res. 2006, 34, 1459. [Google Scholar]
- Hung, H.L.; Kim, A.Y.; Hong, W.; Rakowski, C.; Blobel, G.A. Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J. Biol. Chem. 2001, 276, 10715. [Google Scholar]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868. [Google Scholar]
- Go, Y.M.; Gipp, J.J.; Mulcahy, R.T.; Jones, D.P. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J. Biol. Chem. 2004, 279, 5837. [Google Scholar]
- Carayol, N.; Chen, J.; Yang, F.; Jin, T.; Jin, L.; States, D.; Wang, C.Y. A dominant function of IKK/NF-kappaB signaling in global lipopolysaccharide-induced gene expression. J. Biol. Chem. 2006, 281, 31142. [Google Scholar]
- Hosoya, T.; Maruyama, A.; Kang, M.I.; Kawatani, Y.; Shibata, T.; Uchida, K.; Warabi, E.; Noguchi, N.; Itoh, K.; Yamamoto, M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J. Biol. Chem. 2005, 280, 27244–27250. [Google Scholar]
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408. [Google Scholar] [PubMed]
- Ahn, K.S.; Aggarwal, B.B. Transcription Factor NF-{kappa}B: A sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 2005, 1056, 218. [Google Scholar] [PubMed]
- Anwar, A.A.; Li, F.Y.; Leake, D.S.; Ishii, T.; Mann, G.E.; Siow, R.C. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: Role of mitogen-activated protein kinases and Nrf2. Free Radic. Biol. Med. 2005, 39, 227. [Google Scholar] [PubMed]
- Knorr-Wittmann, C.; Hengstermann, A.; Gebel, S.; Alam, J.; Muller, T. Characterization of Nrf2 activation and heme oxygenase-1 expression in NIH3T3 cells exposed to aqueous extracts of cigarette smoke. Free Radic. Biol. Med. 2005, 39, 1438. [Google Scholar] [PubMed]
- Lan, Q.; Mercurius, K.O.; Davies, P.F. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem. Biophys. Res. Commun. 1994, 201, 950. [Google Scholar] [PubMed]
- Arinze, I.J.; Kawai, Y. Transcriptional activation of the human Galphai2 gene promoter through nuclear factor-kappaB and antioxidant response elements. J. Biol. Chem. 2005, 280, 9786. [Google Scholar]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NFkappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar]
- Minelli, A.; Grottelli, S.; Mierla, A.; Pinnen, F.; Cacciatore, I.; Bellezza, I. Cyclo(His-Pro) exerts anti-inflammatory effects by modulating NF-κB and Nrf2 signalling. Int. J. Biochem. Cell Biol. 2012, 44, 525–535. [Google Scholar]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2- antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar]
- Iskander, K.; Li, J.; Han, S.; Zheng, B.; Jaiswal, A.K. NQO1 and NQO2 regulation of humoral immunity and autoimmunity. J. Biol. Chem. 2006, 281, 30917–30924. [Google Scholar] [PubMed]
- Das, K.C. c-Jun NH2-terminal kinase-mediated redox-dependent degradation of IkappaB: Role of thioredoxin in NF-kappaB activation. J. Biol. Chem. 2001, 276, 4662–4670. [Google Scholar] [PubMed]
- Freemerman, A.J.; Gallegos, A.; Powis, G. Nuclear factor kappaB transactivation is increased but is not involved in the proliferative effects of thioredoxin overexpression in MCF-7 breast cancer cells. Cancer Res. 1999, 59, 4090–4094. [Google Scholar] [PubMed]
- Hayashi, T.; Ueno, Y.; Okamoto, T. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J. Biol. Chem. 1993, 268, 11380–11388. [Google Scholar]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucl. Acids Res. 1992, 20, 3821–3830. [Google Scholar]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus: A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-kappaB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar]
- Yamauchi, K.; Nakano, Y.; Imai, T.; Takagi, T.; Tsuruma, K.; Shimazawa, M.; Iwama, T.; Hara, H. A novel nuclear factor erythroid 2-related factor 2 (Nrf2) activator RS9 attenuates brain injury after ischemia reperfusion in mice. Neuroscience 2016, 333, 302–310. [Google Scholar]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar]
- Chen, X.L.; Varner, S.E.; Rao, A.S.; Grey, J.Y.; Thomas, S.; Cook, C.K.; Wasserman, M.A.; Medford, R.M.; Jaiswal, A.K.; Kunsch, C. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: A novel anti-inflammatory mechanism. J. Biol. Chem. 2003, 278, 703–711. [Google Scholar]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Yla-Herttuala, S.; Tanila, H.; Levonen, A.L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [PubMed]
- Levonen, A.L.; Inkala, M.; Heikura, T.; Jauhiainen, S.; Jyrkkanen, H.K.; Kansanen, E.; Maatta, K.; Romppanen, E.; Turunen, P.; Rutanen, J.; et al. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 741–747. [Google Scholar] [PubMed]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [PubMed]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar]
- Jin, W.; Zhu, L.; Guan, Q.; Chen, G.; Wang, Q.F.; Yin, H.X.; Hang, C.H.; Shi, J.X.; Wang, H.D. Influence of Nrf2 genotype on pulmonary NF-kappaB activity and inflammatory response after traumatic brain injury. Ann. Clin. Lab. Sci. 2008, 38, 221–227. [Google Scholar] [PubMed]
- Cho, H.Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar]
- Rushworth, S.A.; Shah, S.; MacEwan, D.J. TNF mediates the sustained activation of Nrf2 in human monocytes. J. Immunol. 2011, 187, 702–707. [Google Scholar]
- Yang, H.; Magilnick, N.; Lee, C.; Kalmaz, D.; Ou, X.; Chan, J.Y.; Lu, S.C. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol. Cell Biol. 2005, 25, 5933–5946. [Google Scholar]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [PubMed]
- Brewer, A.C.; Murray, T.V.A.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A.M. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [PubMed]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [PubMed]
- Ross, D.; Siegel, D. Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Free Physiol. 2017, 8, 595. [Google Scholar]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar]
- Iskander, K.; Barrios, R.J.; Jaiswal, A.K. Disruption of NAD(P)H:quinone oxidoreductase 1 gene in mice leads to radiation induced myeloproliferative disease. Cancer Res. 2008, 68, 7915–7922. [Google Scholar]
- Li, X.; Liu, Z.; Zhang, A.; Han, C.; Shen, A.; Jiang, L.; Boothman, D.A.; Qiao, J.; Wang, Y.; Huang, X.; et al. NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat. Commun. 2019, 10, 3251. [Google Scholar]
- Das, A.; Kole, L.; Wang, L.; Barrios, R.; Moorthy, B.; Jaiswal, A.K. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic. Biol. Med. 2006, 40, 1843–1856. [Google Scholar]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Nat. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is upregulated by hypoxia/reoxygenation: Implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [PubMed]
- He, X.; Ma, Q. Redox regulation by nuclear factor erythroid 2-related factor 2: Gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol. Pharmacol. 2012, 82, 887–897. [Google Scholar] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Ann. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Rad. Biol. Med. 2009, 46, 443–453. [Google Scholar] [PubMed]
- Rundlof, A.K.; Carlsten, M.; Arner, E.S. The core promoter of human thioredoxin reductase 1: Cloning, transcriptional activity, and Oct-1, Sp1, and Sp3 binding reveal a housekeeping-type promoter for the AUrich element-regulated gene. J. Biol. Chem. 2004, 276, 30542–30551. [Google Scholar]
- Bae, S.H.; Sung, S.H.; Cho, E.J.; Lee, S.K.; Lee, H.E.; Woo, H.A.; Yu, D.Y.; Kil, I.S.; Rhee, S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology 2011, 53, 945–953. [Google Scholar]
- Moinova, H.R.; Mulcahy, R.T. An electrophile responsive element (EpRE) regulates betanaphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J. Biol. Chem. 1998, 273, 14683–14689. [Google Scholar]
- Hartl, D.; Starosta, V.; Maier, K.; Beck-Speier, I.; Rebhan, C.; Becker, B.F.; Latzin, P.; Fischer, R.; Ratjen, F.; Huber, R.M.; et al. Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic. Biol. Med. 2005, 39, 463–472. [Google Scholar]
- Manna, S.K.; Kuo, M.T.; Aggarwal, B.B. Overexpression of gamma-glutamylcysteine synthetase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappa B and activator protein-1. Oncogene 1999, 18, 4371–4382. [Google Scholar]
- Zhang, F.; Wang, X.; Wang, W.; Li, N.; Li, J. Glutamine reduces TNF-alpha by enhancing glutathione synthesis in lipopolysaccharide-stimulated alveolar epithelial cells of rats. Inflammation 2008, 31, 344–350. [Google Scholar]
- Sun, S.; Zhang, H.; Xue, B.; Wu, Y.; Wang, J.; Yin, Z.; Luo, L. Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats. Inflamm. Res. 2006, 55, 504–510. [Google Scholar] [PubMed]
- Peterson, J.D.; Herzenberg, L.A.; Vasquez, K.; Waltenbaugh, C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 3071–3076. [Google Scholar] [PubMed]
- Murata, Y.; Shimamura, T.; Hamuro, J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002, 14, 201–212. [Google Scholar]
- Kim, Y.C.; Masutani, H.; Yamaguchi, Y.; Itoh, K.; Yamamoto, M.; Yodoi, J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J. Biol. Chem. 2001, 276, 18399–18406. [Google Scholar] [PubMed]
- Jiménez-Osorio, A.S.; González-Reyes, S.; García-Niño, W.R.; Moreno-Macías, H.; Rodríguez-Arellano, M.E.; Vargas-Alarcón, G.; Zúñiga, J.; Barquera, R.; Pedraza-Chaverri, J. Association of Nuclear Factor-Erythroid 2-Related Factor 2, Thioredoxin interacting protein, and heme oxygenase-1 gene polymorphisms with diabetes and obesity in Mexican patients. Oxid. Med. Cell. Longev. 2016, 2016, 7367641. [Google Scholar] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar]
- Sato, H.; Fujiwara, K.; Sagara, J.; Bannai, S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem. J. 1995, 310, 547–551. [Google Scholar]
- Manandhar, S.; Cho, J.M.; Kim, J.A.; Kensler, T.W.; Kwak, M.K. Induction of Nrf2-regulated genes by 3H-1, 2-dithiole-3-thione through the ERK signaling pathway in murine keratinocytes. Eur. J. Pharmacol. 2007, 577, 17–27. [Google Scholar]
- Primiano, T.; Kensler, T.W.; Kuppusamy, P.; Zweier, J.L.; Sutter, T.R. Induction of hepatic heme oxygenase-1 and ferritin in rats by cancer chemopreventive dithiolethiones. Carcinogenesis 1996, 17, 2291–2296. [Google Scholar]
- Kerins, M.J.; Ooi, A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [PubMed]
- Gu, J.; Cheng, Y.; Wu, H.; Kong, L.; Wang, S.; Xu, Z.; Zhang, Z.; Tan, Y.; Keller, B.B.; Zhou, H.; et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes 2017, 66, 529–542. [Google Scholar] [PubMed]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nuc. Acids Res. 2007, 35, 7074–7086. [Google Scholar]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2020, 1, 54–61. [Google Scholar]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar]
- Wunder, C.; Potter, R.F. The heme oxygenase system: Its role in liver inflammation. Curr. Drug Target. Cardiovasc. Haematol. Disord. 2003, 3, 199–208. [Google Scholar]
- Filardi, E.S.; Vega, M.A.; Mateos, P.S.; Corbi, A.L.; Kroger, A.P. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar]
- Choi, K.M.; Kashyap, P.C.; Dutta, N.; Stoltz, G.J.; Ordog, T.; Donohue, T.S.; Bauer, A.J.; Linden, D.R.; Szurszewski, J.H.; Gibbons, S.J.; et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 2010, 138, 2399–2409. [Google Scholar]
- Nakamichi, I.; Habtezion, A.; Zhong, B.; Contag, C.H.; Butcher, E.C.; Omary, M.B. Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J. Clin. Investig. 2005, 115, 3007–3014. [Google Scholar]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar]
- Ho, F.M.; Kang, H.C.; Lee, S.T.; Chao, Y.; Chen, Y.C.; Huang, L.J.; Lin, W.-W. The anti-inflammatory actions of LCY-2-CHO, a carbazole analogue, in vascular smooth muscle cells. Biochem. Pharmacol. 2007, 74, 298–308. [Google Scholar] [PubMed]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO-1 pathway by Nardochinoid C inhibits inflammation and oxidative stress in lipopolysaccharide-stimulated macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [PubMed]
- Yet, S.F.; Perrella, M.A.; Layne, M.D.; Hsieh, C.M.; Maemura, K.; Kobzik, L.; Wiesel, P.; Christou, H.; Kourembanas, S.; Lee, M.E. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Investig. 1999, 103, R23–R29. [Google Scholar] [PubMed]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar]
- Chi, X.; Yao, W.; Xia, H.; Jin, Y.; Li, X.; Cai, J.; Hei, Z. Elevation of HO-1 expression mitigates intestinal ischemia-reperfusion injury and restores tight junction function in a rat liver transplantation model. Oxid. Med. Cell Longev. 2015, 2015, 986075. [Google Scholar]
- Kim, W.; Kim, H.U.; Lee, H.N.; Kim, S.H.; Kim, C.; Cha, Y.N.; Joe, Y.; Chung, H.T.; Jang, J.; Kim, K.; et al. Taurine chloramine stimulates efferocytosis through upregulation of Nrf2-mediated heme oxygenase-1 expression in murine macrophages: Possible involvement of carbon monoxide. Antioxid. Redox Signal. 2015, 23, 163–177. [Google Scholar]
- Lea, S.; Plumb, J.; Metcalfe, H.; Spicer, D.; Woodman, P.; Fox, J.C.; Singh, D. The effect of peroxysome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur. Respir. J. 2013, 43, 409–420. [Google Scholar]
- Prestera, T.; Talalay, P.; Alam, J.; Ahn, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: Regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1995, 1, 827–837. [Google Scholar]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Bilban, M.; Wagner, O.; Bach, F.H.; Smolen, J.S.; Scheinecker, C. Heme oxygenase-1 end-products carbon monoxide and biliverdin ameliorate murine collagen induced arthritis. Clin. Exp. Rheumatol. 2012, 30, 73–78. [Google Scholar]
- Patel, R.; Albadawi, H.; Steudel, W.; Hashmi, F.F.; Kang, J.; Yoo, H.J.; Watkins, M.T. Inhalation of carbon monoxide reduces skeletal muscle injury after hind limb ischemia-reperfusion injury in mice. Am. J. Surg. 2012, 203, 488–495. [Google Scholar] [PubMed]
- Liu, Y.; Zhu, B.; Wang, X.; Luo, L.; Li, P.; Paty, D.W.; Cynader, M.S. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: Implications for the role of oxidative stress in the development of multiple sclerosis. J. Neuroimmunol. 2003, 139, 27–35. [Google Scholar] [PubMed]
- Sawada, K.; Ohnishi, K.; Kosaka, T.; Chikano, S.; Egashira, A.; Okui, M.; Shintani, S.; Wada, M.; Nakasho, K.; Shimoyama, T. Exacerbated autoimmune hepatitis successfully treated with leukocytapheresis and bilirubin adsorption therapy. J. Gastroenterol. 1997, 32, 689–695. [Google Scholar]
- Kawamura, K.; Ishikawa, K.; Wada, Y.; Kimura, S.; Matsumoto, H.; Kohro, T.; Itabe, H.; Kodama, T.; Maruyama, Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 155–160. [Google Scholar]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taille, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892. [Google Scholar] [PubMed]
- Jun, Y.J.; Lee, M.; Shin, T.; Yoon, N.; Kim, J.H.; Kim, H.R. Eckol enhances heme oxygenase-1 expression through activation of Nrf2/JNK pathway in HepG2 cells. Molecules 2014, 19, 15638–15652. [Google Scholar]
- Lee, J.; Kim, S. Upregulation of heme oxygenase-1 expression by dehydrodiconiferyl alcohol (DHCA) through the AMPK-Nrf2 dependent pathway. Toxicol. Appl. Pharmacol. 2014, 281, 87–100. [Google Scholar]
- Choi, Y.H. The cytoprotective effect of isorhamnetin against oxidative stress is mediated by the upregulation of the Nrf2-dependent HO-1 expression in C2C12 myoblasts through scavenging reactive oxygen species and ERK inactivation. Gen. Physiol. Biophys. 2016, 35, 145–154. [Google Scholar]
- Zipper, L.M.; Mulcahy, R.T. ERK activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 Cells. Toxicol. Sci. 2003, 73, 124–134. [Google Scholar]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [PubMed]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The mediator subunit MED16 transduces NRF2-activating signals into antioxidant gene expression. Mol. Cell. Biol. 2015, 36, 407–420. [Google Scholar] [PubMed]
- Ziady, A.G.; Sokolow, A.; Shank, S.; Corey, D.; Myers, R.; Plafker, S.; Kelley, T.J. Interaction with CREB binding protein modulates the activities of Nrf2 and NF-κB in cystic fibrosis airway epithelial cells. Am. J. Phys. Lung Cell. Mol. Physiol. 2012, 302, L1221–L1231. [Google Scholar]
- Sharma, D.; Fondell, J.D. Ordered recruitment of histone acetyltransferases and the TRAP/Mediator complex to thyroid hormone-responsive promoters in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7934–7939. [Google Scholar] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [PubMed]
- Okuda, Y.; Sakoda, S.; Bernard, C.C.; Fujimura, H.; Saeki, Y.; Kishimoto, T.; Yanagihara, T. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 1998, 10, 703–708. [Google Scholar]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.G.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246. [Google Scholar]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar]
- Pae, H.O.; Oh, G.S.; Lee, B.S.; Rim, J.S.; Kim, Y.M.; Chung, H.T. 3-Hydroxyanthranilic acid, one of L-tryptophan metabolies, inhibits monocyte chemoattractant protein-1 secretion and vascular cell adhesion molecule-1 expression via heme oxygenase-1 induction in human umbilical vein endothelial cells. Atherosclerosis 2006, 187, 274–284. [Google Scholar] [PubMed]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T., Jr.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [PubMed]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [PubMed]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar]
- Shah, P.K. Inflammation, neointimal hyperplasia, and restenosis: As the leukocytes roll, the arteries thicken. Circulation 2003, 107, 2175–2177. [Google Scholar]
- Schiffrin, E.L. Beyond blood pressure: The endothelium and atherosclerosis progression. Am. J. Hypertens. 2002, 15, 115S–122S. [Google Scholar]
- Kim, J.H.; Choi, Y.K.; Lee, K.S.; Cho, D.H.; Baek, Y.Y.; Lee, D.K.; Ha, K.S.; Choe, J.; Wonc, M.H.; Jeoung, D.; et al. Functional dissection of Nrf2-dependent phase II genes in vascular inflammation and endotoxic injury using Keap1 siRNA. Free Radic. Biol. Med. 2012, 53, 629–640. [Google Scholar]
- Jin, W.; Wang, H.; Yan, W.; Xu, L.; Wang, X.; Zhao, X.; Yang, X.; Chen, G.; Ji, Y. Disruption of Nrf2 enhances upregulation of Nuclear Factor-κB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Med. Inflamm. 2008, 2008, 725174. [Google Scholar]
- Zakkar, M.; Heiden, K.V.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [PubMed]
- Zakkar, M.; Chaudhury, H.; Sandvik, G.; Enesa, K.; Luong, L.A.; Cuhlmann, S.; Mason, J.C.; Krams, R.; Clark, A.R.; Haskard, D.O.; et al. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ. Res. 2008, 103, 726–732. [Google Scholar] [PubMed]
- Yang, P.M.; Chen, H.Z.; Huang, Y.T.; Hsieh, C.W.; Wung, B.S. Lycopene inhibits NF-κB activation and adhesion molecule expression through Nrf2-mediated heme oxygenase-1 in endothelial cells. Int. J. Mol. Med. 2017, 39, 1533–1540. [Google Scholar]
- Koo, H.J.; Sohn, E.H.; Pyo, S.; Woo, H.G.; Park, D.W.; Ham, Y.M.; Jang, S.A.; Park, S.Y.; Kang, S.C. An ethanol root extract of Cynanchum wilfordii containing acetophenones suppresses the expression of VCAM-1 and ICAM-1 in TNF-α-stimulated human aortic smooth muscle cells through the NF-κB pathway. Int. J. Mol. Med. 2015, 35, 915–924. [Google Scholar] [PubMed]
- Sarah, A.; Struck, B.; Genrich, G.; Helm, O.; Sipos, B.; Sebens, S.; Schäfer, H. The crosstalk between Nrf2 and TGF-β1 in the epithelial-mesenchymal transition of pancreatic duct epithelial cells. PLoS ONE 2015, 10, e0132978. [Google Scholar]
- Coppini, R.; Santini, L.; Palandri, C.; Sartiani, L.; Cerbai, E.; Raimondi, L. Pharmacological inhibition of serine proteases to reduce cardiac inflammation and fibrosis in atrial fibrillation. Front. Pharmacol. 2019, 10, 1420. [Google Scholar] [PubMed]
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Izuka, T.I.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor Nrf2 plays a pivotal role in protection against Elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975. [Google Scholar]
- Jin, F.Y.; Nathan, C.; Radzioch, D.; Ding, A. Secretory leukocyte protease inhibitor a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell 1997, 88, 417–426. [Google Scholar]
- Sallenave, J.M.; Shulmann, J.; Crossley, J.; Jordana, M.; Gauldie, J. Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am. J. Respir. Cell Mol. Biol. 1994, 11, 733–741. [Google Scholar]
- Meyer, M.; Kesic, M.J.; Clarke, J.; Ho, E.; Simmen, R.C.; Diaz-Sanchez, D.; Noah, T.L.; Jaspers, I. Sulforaphane induces SLPI secretion in the nasal mucosa. Respir. Med. 2012, 107, 472–475. [Google Scholar]
- Meyer, M.; Jaspers, I. Respiratory protease/antiprotease balance determines susceptibility to viral infection and can be modified by nutritional antioxidants. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1189–L1201. [Google Scholar] [PubMed]
- Saw, C.L.L.; Yang, A.Y.; Huang, M.T.; Liu, Y.; Lee, J.H.; Khor, T.O.; Su, Z.Y.; Shu, L.; Lu, Y.; Conney, A.H.; et al. Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci. 2014, 4, 39. [Google Scholar] [PubMed]
- Chaiprasongsuk, A.; Lohakul, J.; Soontrapa, K.; Sampattavanich, S.; Akarasereenont, P.; Panich, U. Activation of Nrf2 Reduces UVA-Mediated MMP-1 upregulation via MAPK/AP-1 signaling cascades: The photoprotective effects of Sulforaphane and Hispidulins. J. Pharmacol. Exp. Ther. 2017, 360, 388–398. [Google Scholar] [PubMed]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [PubMed]
- Kim, B.C.; Jeon, W.K.; Hong, H.Y.; Jeon, K.B.; Hahn, J.H.; Kim, Y.M.; Numazawa, S.; Yosida, T.; Park, E.H.; Lim, C.J. The anti-inflammatory activity of Phellinus linteus (Berk. & M.A. Curt.) is mediated through the PKCdelta/Nrf2/ARE signaling to upregulation of heme oxygenase-1. J. Ethnopharmacol. 2007, 113, 240–247. [Google Scholar] [PubMed]
- Lee, S.H.; Sohn, D.H.; Jin, X.Y.; Kim, S.W.; Choi, S.C.; Seo, G.S. 2’,4’,6’-tris(methoxymethoxy) chalcone protects against trinitrobenzene sulfonic acid-induced colitis and blocks tumor necrosis factor-alpha-induced intestinal epithelial inflammation via heme oxygenase 1-dependent and independent pathways. Biochem. Pharmacol. 2007, 74, 870–880. [Google Scholar]
- Mao, L.; Wang, H.; Qiao, L.; Wang, X. Disruption of Nrf2 enhances the upregulation of Nuclear Factor-kappaB activity, Tumor Necrosis Factor-α, and Matrix Metalloproteinase-9 after spinal cord injury in mice. Med. Inflamm. 2010, 2010, 238321. [Google Scholar]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Art. Res. Ther. 2015, 17, 269. [Google Scholar]
- Long, M.; Vega, M.R.; Wen, Q.; Bharara, M.; Jiang, T.; Zhang, R.; Zhou, S.; Wong, P.K.; Wondrak, G.T.; Zheng, H.; et al. An essential role of NRF2 in diabetic wound healing. Diabetes 2016, 65, 780–793. [Google Scholar]
- Nicholson, A.C.; Febbraio, M.; Han, J.; Silverstein, R.L.; Hajjar, D.P. CD36 in atherosclerosis: The role of a class B macrophage scavenger receptor. Ann. N. Y. Acad. Sci. 2000, 902, 128–131. [Google Scholar]
- Yoshida, H.; Quehenberger, O.; Kondratenko, N.; Green, S.; Steinberg, D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 794–802. [Google Scholar] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages. Circul. Res. 2004, 94, 609–616. [Google Scholar]
- Takabe, W.; Warabi, E.; Noguchi, N. Anti-atherogenic effect of laminar shear stress via Nrf2 activation. Antioxid. Redox Signal. 2011, 15, 1415–1426. [Google Scholar]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [PubMed]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar]
- Zhao, X.; Sun, G.; Ting, S.M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar]
- Kundu, J.K.; Surh, Y.J. Nrf2-Keap1 signaling as a potential target for chemoprevention of inflammation-associated carcinogenesis. Pharm. Res. 2010, 27, 999–1013. [Google Scholar]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12,14-prostaglandin J2. Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [PubMed]
- Mochizuki, M.; Ishii, Y.; Itoh, K.; Iizuka, T.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Matsuno, Y.; Hegab, A.E.; Nomura, A.; et al. Role of 15-deoxy-Δ12,14-prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am. J. Respir. Crit. Care Med. 2005, 171, 1260–1266. [Google Scholar] [PubMed]
- Gong, P.; Stewart, D.; Hu, B.; Li, N.; Cook, J.; Nel, A.; Alam, J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Δ12,14-prostaglandin J2 is mediated by the stress response elements and transcription factor Nrf2. Antioxid. Redox Signal. 2002, 4, 249–257. [Google Scholar]
- Levonen, A.L.; Landar, A.; Ramachandran, A.; Ceaser, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signaling: Role of cysteine modification in controlling antioxidant defenses in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar]
- Hsieh, C.Y.; Hsiao, H.Y.; Wu, W.Y.; Liu, C.A.; Tsai, Y.C.; Chao, Y.J.; Wang, D.L.; Hsieh, H.-J. Regulation of shear-induced nuclear translocation of the Nrf2 transcription factor in endothelial cells. J. Biomed. Sci. 2009, 16, 12. [Google Scholar]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome- activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species- induced NLRP3 priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [PubMed]
- Li, H.Y.; Zhong, Y.F.; Wu, S.Y.; Shi, N. NF-E2 related factor 2 activation and heme oxygenase-1 induction by tert-butylhydroquinone protect against deltamethrin-mediated oxidative stress in PC12 cells. Chem. Res. Toxicol. 2007, 20, 1242–1251. [Google Scholar] [PubMed]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [PubMed]
- Ka, S.M.; Lin, J.C.; Lin, T.J.; Liu, F.C.; Chao, L.K.; Ho, C.L.; Yeh, L.T.; Sytwu, H.K.; Hua, K.F.; Chen, A. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arth. Res. Ther. 2015, 17, 331. [Google Scholar]
- Liu, X.; Wang, T.; Cai, L.; Qi, J.; Zhang, P.; Li, Y. Biochanin A protects lipopolysaccharide/ D-galactosamine-induced acute liver injury inmice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2016, 38, 324–331. [Google Scholar] [PubMed]
- Liu, Q.; Lv, H.; Wen, Z.; Ci, X.; Peng, L. Isoliquiritigenin activates Nuclear Factor Erythroid-2 Related Factor 2 to suppress the NOD-Like Receptor Protein 3 inflammasome and inhibits the NF-κB pathway in macrophages and in acute lung injury. Front. Immunol. 2017, 8, 1518. [Google Scholar] [PubMed]
- He, L.; Peng, X.; Zhu, J.; Chen, X.; Liu, H.; Tang, C.; Dong, Z.; Liu, F.; Peng, Y. Mangiferin attenuate sepsis-induced acute kidney injury via antioxidant and anti-inflammatory effects. Am. J. Nephrol. 2014, 40, 441–450. [Google Scholar]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar]
- An, Y.W.; Jhang, K.A.; Woo, S.Y.; Kang, J.L.; Chong, Y.H. Sulforaphane exerts its anti-inflammatory effect against amyloid-β peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol. Aging 2016, 38, 1–10. [Google Scholar]
- Lv, H.; Liu, Q.; Wen, Z.; Feng, H.; Deng, X.; Ci, X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox Biol. 2017, 12, 311–324. [Google Scholar]
- Dong, Z.; Shang, H.; Chen, Y.Q.; Pan, L.L.; Bhatia, M.; Sun, J. Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2- mediated oxidative stress and NLRP3 inflammatory pathway. Oxid. Med. Cell Longev. 2016, 2016, 7864150. [Google Scholar]
- Zhao, C.; Gillette, D.D.; Li, X.; Zhang, Z.; Wen, H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 2014, 289, 17020–17029. [Google Scholar] [PubMed]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [PubMed]
- Jhang, J.J.; Cheng, Y.T.; Ho, C.Y.; Yen, G.C. Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell Mol. Immunol. 2015, 12, 424–434. [Google Scholar] [PubMed]
- Reddy, N.M.; Suryanarayana, V.; Kalvakolanu, D.V.; Yamamoto, M.; Kensler, T.W.; Hassoun, P.M.; Kleeberger, S.R.; Reddy, S.P. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J. Immunol. 2009, 183, 4601–4608. [Google Scholar]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar]
- Schjerning, A.; McGettigan, P.; Gislason, G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat. Rev. Cardiol. 2020, 17, 574–584. [Google Scholar]
- Jian, Z.; Tang, L.; Yi, X.; Liu, B.; Zhang, Q.; Zhu, G.; Wang, G.; Gao, T.; Li, C. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2O2-induced oxidative stress. J. Cell Mol. Med. 2016, 20, 1307–1318. [Google Scholar]
- Wang, W.; Chen, S.; Zhou, Z.; Dai, H.; Wang, H.; Li, Y.; Zhou, K.; Shen, Z.; Guo, Y.; Liu, C.; et al. Aspirin suppresses neuronal apoptosis, reduces tissue inflammation, and restrains astrocyte activation by activating the Nrf2/HO-1 signaling pathway. Neuroreport 2018, 29, 524–531. [Google Scholar]
- Wang, J.S.; Ho, F.M.; Kang, H.C.; Lin, W.W.; Huang, K.C. Celecoxib induces heme oxygenase-1 expression in macrophages and vascular smooth muscle cells via ROS-dependent signaling pathway. Naunyn. Schmiedebergs Arch. Pharmacol. 2011, 383, 159–168. [Google Scholar] [PubMed]
- Al-Rashed, F.; Calay, D.; Lang, M.; Thornton, C.C.; Bauer, A.; Kiprianos, A.; Haskard, D.O.; Seneviratne, A.; Boyle, J.J.; Schönthal, A.H.; et al. Celecoxib exerts protective effects in the vascular endothelium via COX-2-independent activation of AMPK-CREB-Nrf2 signalling. Sci. Rep. 2018, 8, 6271. [Google Scholar] [PubMed]
- Bao, S.; Nie, X.; Ou, R.; Wang, C.; Ku, P.; Li, K. Effects of diclofenac on the expression of Nrf2 and its downstream target genes in mosquito fish (Gambusia affinis). Aquat. Toxicol. 2017, 188, 43–53. [Google Scholar] [PubMed]
- Yoshinaga, N.; Arimura, N.; Otsuka, H.; Kawahara, K.; Hashiguchi, T.; Maruyama, I.; Sakamoto, T. NSAIDs inhibit neovascularization of choroid through HO-1-dependent pathway. Lab. Investig. 2011, 91, 1277–1290. [Google Scholar] [PubMed]
- Lee, H.J.; Han, Y.M.; Kim, E.H.; Kim, Y.J.; Hahm, K.B. A possible involvement of Nrf2-mediated heme oxygenase-1 up-regulation in protective effect of the proton pump inhibitor pantoprazole against indomethacin-induced gastric damage in rats. BMC Gastroenterol. 2012, 12, 143. [Google Scholar]
- Dunlap, T.; Chandrasena, R.E.; Wang, Z.; Sinha, V.; Thatcher, G.R.J. Quinone formation as a chemoprevention strategy for hybrid drugs: Balancing cytotoxicity and cytoprotection. Chem. Res. Toxicol. 2007, 20, 1903–1912. [Google Scholar] [PubMed]
- Gao, J.; Kashfi, K.; Liu, X.; Rigas, B. NO-donating aspirin induces phase II enzymes in vitro and in vivo. Carcinogenesis 2006, 27, 803–810. [Google Scholar]
- Hulsman, N.; Medema, J.P.; Bos, C.; Jongejan, A.; Leurs, R.; Smit, M.J.; de Esch, I.J.; Richel, D.; Wijtmans, M. Chemical insights in the concept of hybrid drugs: The antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem. 2007, 50, 2424–2431. [Google Scholar]
- Dunlap, T.; Piyankarage, S.C.; Wijewickrama, G.T.; Abdul-Hay, S.; Vanni, M.; Litosh, V.; Luo, J.; Thatcher, G.R.J. Quinone-induced activation of Keap1/Nrf2 signaling by aspirin prodrugs masquerading as nitric oxide. Chem. Res. Toxicol. 2012, 25, 2725–2736. [Google Scholar]
- Zhu, H.; Jia, Z.; Li, Y.R. Nrf2 signaling in macrophages. React. Oxy. Sp. (Apex) 2016, 2, 417–420. [Google Scholar]
- Arellano-Buendía, A.S.; Tostado-González, M.; García-Arroyo, F.E.; Cristóbal-García, M.; Loredo-Mendoza, M.L.; Tapia, E.; Sánchez-Lozada, L.G.; Osorio-Alonso, H. Anti-Inflammatory therapy modulates Nrf2-Keap1 in kidney from rats with diabetes. Oxid. Med. Cell. Longev. 2016, 2016, 4693801. [Google Scholar] [PubMed]
- Cheng, X.; Gao, D.X.; Song, J.J.; Ren, F.Z.; Mao, X.Y. Casein glycomacropeptide hydrolysate exerts cytoprotection against H2O2-induced oxidative stress in RAW 264.7 macrophages via ROS-dependent heme oxygenase-1 expression. RSC Adv. 2015, 5, 4511–4523. [Google Scholar]
- Li, T.; Chen, B.; Du, M.; Song, J.; Cheng, X.; Wang, X.; Mao, X. Casein Glycomacropeptide hydrolysates exert cytoprotective effect against cellular oxidative stress by up-regulating HO-1 expression in HepG2 cells. Nutrients 2017, 9, 31. [Google Scholar]
- Quinti, L.; Naidu, S.D.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [PubMed]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2- and NRF2-targeting thiazole- containing compound with therapeutic activity in Huntington’s disease models. Cell Chem. Biol. 2016, 23, 849–861. [Google Scholar]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar]
- Gold, R.; Linker, R.A.; Stangel, M. Fumaric acid and its esters: An emerging treatment for multiple sclerosis with antioxidative mechanism of action. Clin. Immunol. 2012, 142, 44–48. [Google Scholar]
- Mrowietz, U.; Christophers, E.; Altmeyer, P. Treatment of psoriasis with fumaric acid esters: Results of a prospective multicentre study. German multicentre study. Br. J. Dermatol. 1998, 138, 456–460. [Google Scholar]
- Ma, Y.; Kosinska-Cagnazzo, A.; Kerr, W.L.; Amarowicz, R.; Swanson, R.B.; Pegg, R.B. Separation and characterization of soluble esterified and glycoside-bound phenolic compounds in dry-blanched peanut skins by liquid chromatography-electrospray ionization mass spectrometry. J. Agric. Food Chem. 2014, 62, 11488–11504. [Google Scholar]
- Rose, P.; Won, Y.K.; Ong, C.N.; Whiteman, M. Beta-phenylethyl and 8-methylsulphinyloctyl isothiocyanates, constituents of watercress, suppress LPS induced production of nitric oxide and prostaglandin E2 in RAW 264.7 macrophages. Nitric Oxide 2005, 12, 237–243. [Google Scholar] [PubMed]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [PubMed]
- Kim, J.S.; Oh, J.M.; Choi, H.; Kim, S.W.; Kim, S.W.; Kim, B.G.; Cho, J.H.; Lee, J.; Lee, D.C. Activation of the Nrf2/HO-1 pathway by curcumin inhibits oxidative stress in human nasal fibroblasts exposed to urban particulate matter. BMC Comp. Med. Ther. 2020, 20, 101. [Google Scholar]
- Jiang, C.; Luo, P.; Li, X.; Liu, P.; Li, Y.; Xu, J. Nrf2/ARE is a key pathway for curcumin-mediated protection of TMJ chondrocytes from oxidative stress and inflammation. Cell Stress Chaperones 2020, 25, 395–406. [Google Scholar]
- Kim, A.N.; Jeon, W.K.; Lee, J.J.; Kim, B.C. Up-regulation of heme oxygenase-1 expression through CaMKII-ERK1/2-Nrf2 signaling mediates the anti-inflammatory effect of bisdemethoxycurcumin in LPS-stimulated macrophages. Free Radic. Biol. Med. 2010, 49, 323–331. [Google Scholar]
- Pugazhenthi, S.; Akhov, L.; Selvaraj, G.; Wang, M.; Alam, J. Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am. J. Phys. Endocrinol. Metabol. 2007, 293, E645–E655. [Google Scholar]
- Li, J.; Sapper, T.N.; Mah, E.; Rudraiah, S.; Schill, K.E.; Chitchumroonchokchai, C.; Moller, M.V.; McDonald, J.D.; Rohrer, P.R.; Manautou, J.E.; et al. Green tea extract provides extensive Nrf2-independent protection against lipid accumulation and NFkappaB pro-inflammatory responses during nonalcoholic steatohepatitis in mice fed a high-fat diet. Mol. Nutr. Food Res. 2016, 60, 858–870. [Google Scholar]
- Zhu, G.F.; Guo, H.J.; Huang, Y.; Wu, C.T.; Zhang, X.F. Eriodictyol, a plant flavonoid, attenuates LPS-induced acute lung injury through its antioxidative and anti-inflammatory activity. Exp. Ther. Med. 2016, 10, 2259–2266. [Google Scholar]
- Li, C.Z.; Jin, H.H.; Sun, H.X.; Zhang, Z.Z.; Zheng, J.X.; Li, S.H.; Han, S.H. Eriodictyol attenuates cisplatin-induced kidney injury by inhibiting oxidative stress and inflammation. Eur. J. Pharmacol. 2016, 772, 124–130. [Google Scholar]
- Boyanapalli, S.S.; Paredes-Gonzalez, X.; Fuentes, F.; Zhang, C.; Guo, Y.; Pung, D.; Saw, C.L.; Kong, A.N. Nrf2 knockout attenuates the anti-inflammatory effects of phenethyl isothiocyanate and curcumin. Chem. Res. Toxicol. 2016, 27, 2036–2043. [Google Scholar]
- Leonardo, C.C.; Dore, S. Dietary flavonoids are neuroprotective through Nrf2-coordinated induction of endogenous cytoprotective proteins. Nutr. Neurosci. 2016, 14, 226–236. [Google Scholar]
- Wang, Y.; Wang, B.; Du, F.; Su, X.; Sun, G.; Zhou, G.; Bian, X.; Liu, N. Epigallocatechin-3-Gallate attenuates oxidative stress and inflammation in obstructive nephropathy via NF-κB and Nrf2/HO-1 signalling pathway regulation. Basic Clin. Pharmacol. Tocicol. 2015, 117, 164–172. [Google Scholar]
- Kanlaya, R.; Khamchun, S.; Kapincharanon, C.; Thongboonkerd, V. Protective effect of epigallocatechin-3-gallate (EGCG) via Nrf2 pathway against oxalate-induced epithelial mesenchymal transition (EMT) of renal tubular cells. Sci. Rep. 2016, 6, 30233. [Google Scholar] [PubMed]
- Shanmugam, T.; Selvaraj, M.; Poomalai, S. Epigallocatechin gallate potentially abrogates fluoride induced lung oxidative stress, inflammation via Nrf2/Keap1 signaling pathway in rats: An in-vivo and in-silico study. Int. Immunopharmacol. 2016, 39, 128–139. [Google Scholar]
- Sun, W.; Liu, X.; Zhang, H.; Song, Y.; Li, T.; Liu, X.; Liu, Y.; Guo, L.; Wang, F.; Yang, T.; et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic. Biol. Med. 2017, 108, 840–857. [Google Scholar]
- Ruiz-Miyazawa, K.W.; Pinho-Ribeiro, F.A.; Borghi, S.M.; Staurengo-Ferrari, L.; Fattori, V.; Amaral, F.A.; Teixeira, M.M.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; et al. Hesperidin methylchalcone suppresses experimental gout arthritis in mice by inhibiting NF-kappaB activation. J. Agric. Food Chem. 2018, 66, 6269–6280. [Google Scholar]
- Zhang, Y.; Liu, B.; Chen, X.; Zhang, N.; Li, G.; Zhang, L.H.; Tan, L.Y. Naringenin ameliorates behavioral dysfunction and neurological deficits in a d-galactose-induced aging mouse model through activation of PI3K/Akt/Nrf2 pathway. Rejuvenation Res. 2017, 20, 462–472. [Google Scholar]
- Calixto-Campos, C.; Corrêa, M.P.; Carvalho, T.T.; Zarpelon, A.C.; Hohmann, M.S.N.; Rossaneis, A.C.; Coelho-Silva, L.; Pavanelli, W.R.; Pinge-Filho, P.; Crespigio, J.; et al. Quercetin reduces ehrlich tumor-induced cancer pain in mice. Anal. Cell. Pathol. (Amst.) 2015, 2015, 285708. [Google Scholar]
- Borghi, S.M.; Mizokami, S.S.; Pinho-Ribeiro, F.A.; Fattori, V.; Crespigio, J.; Clemente-Napimoga, J.T.; Napimoga, M.H.; Pitol, D.L.; Issa, J.P.M.; Fukada, S.Y.; et al. The flavonoid quercetin inhibits titanium dioxide (TiO2)-induced chronic arthritis in mice. J. Nutr. Biochem. 2017, 53, 81–95. [Google Scholar]
- Anantharaju, P.G.; Gowda, P.C.; Vimalambike, M.G.; Madhunapantula, S.V. An overview on the role of dietary phenolics for the treatment of cancers. Nutr. J. 2016, 15, 99. [Google Scholar]
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates beta-amyloid-induced oxidative stress via Akt/GSK-3beta/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic. Biol. Med. 2018, 120, 114–123. [Google Scholar] [PubMed]
- Kim, K.H.; Sadikot, R.T.; Joo, M. Therapeutic effect of ent-kaur-16-en-19-oic acid on neutrophilic lung inflammation and sepsis is mediated by Nrf2. Biochem. Biophys. Res. Commun. 2016, 474, 534–540. [Google Scholar] [PubMed]
- Kim, Y.M.; Kim, H.J.; Chang, K.C. Glycyrrhizin reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7 cells and endotoxemic mice by p38/Nrf2-dependent induction of HO-1. Int. Immunopharmacol. 2015, 26, 112–118. [Google Scholar] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. https://doi.org/10.3390/molecules25225474
Saha S, Buttari B, Panieri E, Profumo E, Saso L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules. 2020; 25(22):5474. https://doi.org/10.3390/molecules25225474
Chicago/Turabian StyleSaha, Sarmistha, Brigitta Buttari, Emiliano Panieri, Elisabetta Profumo, and Luciano Saso. 2020. "An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation" Molecules 25, no. 22: 5474. https://doi.org/10.3390/molecules25225474
APA StyleSaha, S., Buttari, B., Panieri, E., Profumo, E., & Saso, L. (2020). An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules, 25(22), 5474. https://doi.org/10.3390/molecules25225474