
“What Doesn’t Kill You Makes You Stronger”: Future Applications of Amyloid Aggregates in Biomedicine

 , ,
, ,
Abstract
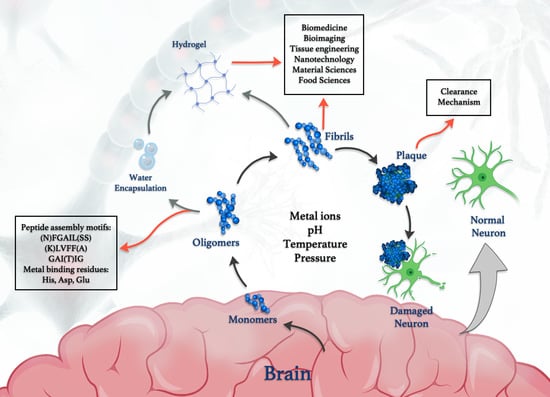
1. Introduction
2. Amyloid Core Peptides as Model Systems to Study Amyloidogenesis
3. Factors Influencing Aggregation
4. Metal Binding Sites in Amyloid Oligomers and Their Polymerization “Switching” Character
5. Functional Metal/Amyloid Complexes
6. Biomedical Applications of Amyloid Peptides
6.1. Antiviral Activity of Amyloid Peptides against Human Viral Infections
6.2. Cell Penetrating Amyloid Peptides and Their Applications
6.3. Amyloid in Bioimaging: Bioorganic Nanodots
7. Amyloid Clearance Mechanisms
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Su, R.; Liang, M.; Huang, R.; Wang, M.; Qi, W.; He, Z. Physicochemical strategies for inhibition of amyloid fibril formation: An overview of recent advances. Curr. Med. Chem. 2012, 19, 4157–4174. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.-I.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D. A primer of amyloid nomenclature. Amyloid 2007, 14, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.-I.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D. Amyloid: Toward terminology clarification report from the nomenclature committee of the international society of amyloidosis. Amyloid 2005, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar]
- Recchia, A.; Debetto, P.; Negro, A.; Guidolin, D.; Skaper, S.D.; Giusti, P. α-Synuclein and Parkinson’s disease. FASEB J. 2004, 18, 617–626. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Quan, Z.; Wong, W.; Guo, J.; Zhang, R.; Yang, Q.; Dai, R.; McGeer, P.L.; Qing, H. Epigallocatechin gallate (EGCG) inhibits alpha-synuclein aggregation: A potential agent for Parkinson’s disease. Neurochem. Res. 2016, 41, 2788–2796. [Google Scholar] [CrossRef]
- Barracchia, C.G.; Tira, R.; Parolini, F.; Munari, F.; Bubacco, L.; Spyroulias, G.A.; D’Onofrio, M.; Assfalg, M. Unsaturated Fatty Acid-Induced Conformational Transitions and Aggregation of the Repeat Domain of Tau. Molecules 2020, 25, 2716. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Liberski, P.P.; Sikorska, B.; Brown, P. Kuru: The first prion disease. In Neurodegenerative Diseases; Springer: New York, NY, USA, 2012; pp. 143–153. [Google Scholar]
- Alsiary, R.A.; Alghrably, M.; Saoudi, A.; Al-Ghamdi, S.; Jaremko, L.; Jaremko, M.; Emwas, A.H. Using NMR spectroscopy to investigate the role played by copper in prion diseases. Neurol. Sci. 2020, 41, 2389–2406. [Google Scholar] [CrossRef]
- Alghrably, M.; Czaban, I.; Jaremko, Ł.; Jaremko, M. Interaction of amylin species with transition metals and membranes. J. Inorg. Biochem. 2019, 191, 69–76. [Google Scholar] [CrossRef]
- Despa, F.; Goldstein, L.B.; Biessels, G.J. Amylin as a Potential Link between Type 2 Diabetes and Alzheimer Disease. Ann. Neurol. 2020, 87, 486. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Valenti-Azcarate, R.; Amat-Villegas, I.; Riverol, M.; Marcilla, I.; de Andrea, C.E.; Sánchez-Arias, J.A.; del Mar Carmona-Abellan, M.; Marti, G.; Erro, M.E. Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann. Neurol. 2019, 86, 539–551. [Google Scholar] [CrossRef]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef]
- Gazit, E. The “correctly folded” state of proteins: Is it a metastable state? Angew. Chem. Int. Ed. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Brovchenko, I.; Oleinikova, A.; Winter, R. Peptide aggregation in finite systems. Biophys. J. 2008, 95, 3208–3221. [Google Scholar] [CrossRef] [PubMed]
- Poulson, B.G.; Szczepski, K.; Lachowicz, J.I.; Jaremko, L.; Emwas, A.-H.; Jaremko, M. Aggregation of biologically important peptides and proteins: Inhibition or acceleration depending on protein and metal ion concentrations. RSC Adv. 2020, 10, 215–227. [Google Scholar] [CrossRef]
- Wang, W.; Roberts, C.J. Non-Arrhenius protein aggregation. AAPS J. 2013, 15, 840–851. [Google Scholar] [CrossRef]
- Alghrably, M.; Dudek, D.; Emwas, A.H.; Jaremko, Ł.; Jaremko, M.; Rowińska-Zyrek, M. Copper (II) and Amylin Analogues: A Complicated Relationship. Inorg. Chem. 2020, 59, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Zapadka, K.L.; Becher, F.J.; Gomes Dos Santos, A.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef]
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 2007, 17, 48–57. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils: Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Murphy, R.M. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 1923–1934. [Google Scholar] [CrossRef][Green Version]
- Ghosh, P.; De, P. Modulation of Amyloid Protein Fibrillation by Synthetic Polymers: Recent Advances in the Context of Neurodegenerative Diseases. ACS Appl. Bio Mater. 2020, 3, 6598–6625. [Google Scholar] [CrossRef]
- Kodaka, M. Interpretation of concentration-dependence in aggregation kinetics. Biophys. Chem. 2004, 109, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Hofrichter, J.; Ross, P.D.; Eaton, W.A. Kinetics and mechanism of deoxyhemoglobin S gelation: A new approach to understanding sickle cell disease. Proc. Natl. Acad. Sci. USA 1974, 71, 4864–4868. [Google Scholar] [CrossRef]
- Wei, G.; Su, Z.; Reynolds, N.P.; Arosio, P.; Hamley, I.W.; Gazit, E.; Mezzenga, R. Self-assembling peptide and protein amyloids: From structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Z.; Su, Z.; Wei, G. Design, fabrication, and biomedical applications of bioinspired peptide–inorganic nanomaterial hybrids. J. Mater. Chem. B 2017, 5, 1130–1142. [Google Scholar] [CrossRef]
- Lee, S.; Trinh, T.H.; Yoo, M.; Shin, J.; Lee, H.; Kim, J.; Hwang, E.; Lim, Y.-B.; Ryou, C. Self-assembling peptides and their application in the treatment of diseases. Int. J. Mol. Sci. 2019, 20, 5850. [Google Scholar] [CrossRef]
- Michiels, E.; Roose, K.; Gallardo, R.; Khodaparast, L.; Khodaparast, L.; van der Kant, R.; Siemons, M.; Houben, B.; Ramakers, M.; Wilkinson, H. Reverse engineering synthetic antiviral amyloids. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kokotidou, C.; Jonnalagadda, S.V.R.; Orr, A.A.; Vrentzos, G.; Kretsovali, A.; Tamamis, P.; Mitraki, A. Designer Amyloid Cell-Penetrating Peptides for Potential Use as Gene Transfer Vehicles. Biomolecules 2020, 10, 7. [Google Scholar] [CrossRef]
- Lapshina, N.; Shishkin, I.I.; Nandi, R.; Noskov, R.E.; Barhom, H.; Joseph, S.; Apter, B.; Ellenbogen, T.; Natan, A.; Ginzburg, P. Bioinspired Amyloid Nanodots with Visible Fluorescence. Adv. Opt. Mater. 2019, 7, 1801400. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J. Clearance of Alzheimer’s amyloid-β 1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Massey, A.; KUO, Y.M.; Roher, A.E. Cerebral amyloid angiopathy: Accumulation of Aβ in interstitial fluid drainage pathways in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 903, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Landreh, M.; Sawaya, M.R.; Hipp, M.S.; Eisenberg, D.S.; Wüthrich, K.; Hartl, F.U. The formation, function and regulation of amyloids: Insights from structural biology. J. Intern. Med. 2016, 280, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Stroo, E.; Koopman, M.; Nollen, E.A.; Mata-Cabana, A. Cellular regulation of amyloid formation in aging and disease. Front. Neurosci. 2017, 11, 64. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Sawaya, M.R.; Wall, J.S.; Eisenberg, D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA 2009, 106, 18990–18995. [Google Scholar] [CrossRef]
- Zanuy, D.; Nussinov, R. The sequence dependence of fiber organization. A comparative molecular dynamics study of the islet amyloid polypeptide segments 22–27 and 22–29. J. Mol. Biol. 2003, 329, 565–584. [Google Scholar] [CrossRef]
- Tenidis, K.; Waldner, M.; Bernhagen, J.; Fischle, W.; Bergmann, M.; Weber, M.; Merkle, M.-L.; Voelter, W.; Brunner, H.; Kapurniotu, A. Identification of a penta-and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 2000, 295, 1055–1071. [Google Scholar] [CrossRef]
- Azriel, R.; Gazit, E. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide an experimental support for the key role of the phenylalanine residue in amyloid formation. J. Biol. Chem. 2001, 276, 34156–34161. [Google Scholar] [CrossRef] [PubMed]
- Hauser, C.; Deng, R.; Mishra, A.; Loo, Y.; Khoe, U.; Zhuang, F.; Cheong, D.W.; Accardo, A.; Sullivan, M.B.; Riekel, C.; et al. Natural tri-to hexapeptides self-assemble in water to amyloid β-type fiber aggregates by unexpected α-helical intermediate structures. Proc. Natl. Acad. Sci. USA 2011, 108, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.; Cheong, D.W.; Accardo, A.; Di Fabrizio, E.; Riekel, C.; Hauser, C.A. Aliphatic peptides show similar self-assembly to amyloid core sequences, challenging the importance of aromatic interactions in amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. Self-assembled peptide nanostructures: The design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 2007, 36, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Snell, J.M.; Sheftic, S.R.; Patil, S.M.; Daniels, S.B.; Kolling, F.W.; Alexandrescu, A.T. pH dependence of amylin fibrillization. Biochemistry 2014, 53, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.; Chatani, E.; Kihara, M.; Ban, T.; Sakai, M.; Hasegawa, K.; Naiki, H.; Rao, C.M.; Goto, Y. Critical balance of electrostatic and hydrophobic interactions is required for β2-microglobulin amyloid fibril growth and stability. Biochemistry 2005, 44, 1288–1299. [Google Scholar] [CrossRef]
- Cromwell, M.E.; Hilario, E.; Jacobson, F. Protein aggregation and bioprocessing. AAPS J. 2006, 8, E572–E579. [Google Scholar] [CrossRef]
- Engelhardt, K.; Lexis, M.; Gochev, G.; Konnerth, C.; Miller, R.; Willenbacher, N.; Peukert, W.; Braunschweig, B.r. pH effects on the molecular structure of β-lactoglobulin modified air–water interfaces and its impact on foam rheology. Langmuir 2013, 29, 11646–11655. [Google Scholar] [CrossRef]
- Chi, E.Y.; Krishnan, S.; Randolph, T.W.; Carpenter, J.F. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 2003, 20, 1325–1336. [Google Scholar] [CrossRef]
- Li, R.; Wu, Z.; Wang, Y.; Ding, L.; Wang, Y. Role of pH-induced structural change in protein aggregation in foam fractionation of bovine serum albumin. Biotechnol. Rep. 2016, 9, 46–52. [Google Scholar] [CrossRef]
- Wilkening, A.; Rüb, C.; Sylvester, M.; Voos, W. Analysis of heat-induced protein aggregation in human mitochondria. J. Biol. Chem. 2018, 293, 11537–11552. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.; Roberts, C.J.; Rodrigues, M.A. Connecting high-temperature and low-temperature protein stability and aggregation. PLoS ONE 2017, 12, e0176748. [Google Scholar] [CrossRef]
- Speed, M.A.; King, J.; Wang, D.I. Polymerization mechanism of polypeptide chain aggregation. Biotechnol. Bioeng. 1997, 54, 333–343. [Google Scholar] [CrossRef]
- Zlateva, T.; Boteva, R.; Salvato, B.; Tsanev, R. Factors affecting the dissociation and aggregation of human interferon gamma. Int. J. Biol. Macromol. 1999, 26, 357–362. [Google Scholar] [CrossRef]
- Vermeer, A.W.; Norde, W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multi-domain protein. Biophys. J. 2000, 78, 394–404. [Google Scholar] [CrossRef]
- John, R.J.S.; Carpenter, J.F.; Randolph, T.W. High pressure fosters protein refolding from aggregates at high concentrations. Proc. Natl. Acad. Sci. USA 1999, 96, 13029–13033. [Google Scholar] [CrossRef] [PubMed]
- Foguel, D.; Silva, J.L. New insights into the mechanisms of protein misfolding and aggregation in amyloidogenic diseases derived from pressure studies. Biochemistry 2004, 43, 11361–11370. [Google Scholar] [CrossRef]
- Randolph, T.W.; Seefeldt, M.; Carpenter, J.F. High hydrostatic pressure as a tool to study protein aggregation and amyloidosis. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 2002, 1595, 224–234. [Google Scholar] [CrossRef]
- Niraula, T.N.; Konno, T.; Li, H.; Yamada, H.; Akasaka, K.; Tachibana, H. Pressure-dissociable reversible assembly of intrinsically denatured lysozyme is a precursor for amyloid fibrils. Proc. Natl. Acad. Sci. USA 2004, 101, 4089–4093. [Google Scholar] [CrossRef]
- Torrent, J.; Alvarez-Martinez, M.T.; Harricane, M.C.; Heitz, F.; Liautard, J.P.; Balny, C.; Lange, R. High pressure induces scrapie-like prion protein misfolding and amyloid fibril formation. Biochemistry 2004, 43, 7162–7170. [Google Scholar] [CrossRef]
- Foguel, D.; Suarez, M.C.; Ferrão-Gonzales, A.D.; Porto, T.C.; Palmieri, L.; Einsiedler, C.M.; Andrade, L.R.; Lashuel, H.A.; Lansbury, P.T.; Kelly, J.W. Dissociation of amyloid fibrils of α-synuclein and transthyretin by pressure reveals their reversible nature and the formation of water-excluded cavities. Proc. Natl. Acad. Sci. USA 2003, 100, 9831–9836. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Grudzielanek, S.; Dzwolak, W.; Winter, R. High pressure promotes circularly shaped insulin amyloid. J. Mol. Biol. 2004, 338, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Foguel, D.; Silva, J.L. Pressure–temperature folding landscape in proteins involved in neurodegenerative diseases and cancer. Biophys. Chem. 2013, 183, 9–18. [Google Scholar] [CrossRef]
- Kim, Y.S.; Randolph, T.W.; Seefeldt, M.B.; Carpenter, J.F. High-pressure studies on protein aggregates and amyloid fibrils. Methods Enzymol. 2006, 413, 237–253. [Google Scholar]
- Wang, Y.; Pu, J.; An, B.; Lu, T.K.; Zhong, C. Emerging paradigms for synthetic design of functional amyloids. J. Mol. Biol. 2018, 430, 3720–3734. [Google Scholar] [CrossRef] [PubMed]
- Näslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Tomic, J.L.; Pensalfini, A.; Head, E.; Glabe, C.G. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 2009, 35, 352–358. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Tabner, B.J.; Mayes, J.; Allsop, D. Hypothesis: Soluble Aβ oligomers in association with redox-active metal ions are the optimal generators of reactive oxygen species in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2011, 2011, 546380. [Google Scholar] [CrossRef]
- Valensin, D.; Migliorini, C.; Valensin, G.; Gaggelli, E.; La Penna, G.; Kozlowski, H.; Gabbiani, C.; Messori, L. Exploring the reactions of β-Amyloid (Aβ) peptide 1–28 with AlIII and FeIII Ions. Inorg. Chem. 2011, 50, 6865–6867. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.E.; Smith, D.G.; Bush, A.I.; Masters, C.L.; Barnham, K.J. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-β peptide with membrane lipid. J. Biol. Chem. 2003, 278, 2977–2982. [Google Scholar] [CrossRef]
- Syme, C.D.; Nadal, R.C.; Rigby, S.E.; Viles, J.H. Copper Binding to the Amyloid-β (Aβ) Peptide Associated with Alzheimer’s Disease folding, coordination geometry, pH dependence, stoichiometry, and affinity of Aβ-(1–28): Insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 2004, 279, 18169–18177. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Titmuss, S.J.; Epa, V.C.; Barnham, K.J.; Masters, C.L.; Varghese, J.N. The structure of the amyloid-β peptide high-affinity copper II binding site in Alzheimer disease. Biophys. J. 2008, 95, 3447–3456. [Google Scholar] [CrossRef] [PubMed]
- Drew, S.C.; Masters, C.L.; Barnham, K.J. Alanine-2 carbonyl is an oxygen ligand in Cu2+ coordination of Alzheimer’s disease amyloid-β peptide− relevance to N-terminally truncated forms. J. Am. Chem. Soc. 2009, 131, 8760–8761. [Google Scholar] [CrossRef]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Ségalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural changes of region 1–16 of the Alzheimer disease amyloid β-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Minicozzi, V.; Stellato, F.; Comai, M.; Dalla Serra, M.; Potrich, C.; Meyer-Klaucke, W.; Morante, S. Identifying the minimal copper-and zinc-binding site sequence in amyloid-β peptides. J. Biol. Chem. 2008, 283, 10784–10792. [Google Scholar] [CrossRef] [PubMed]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron (II) binding to amyloid-β, the Alzheimer’s peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef]
- Ricchelli, F.; Drago, D.; Filippi, B.; Tognon, G.; Zatta, P. Aluminum-triggered structural modifications and aggregation of β-amyloids. Cell. Mol. Life Sci. CMLS 2005, 62, 1724–1733. [Google Scholar] [CrossRef]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Interaction of α-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef]
- Binolfi, A.; Valiente-Gabioud, A.A.; Duran, R.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Exploring the structural details of Cu (I) binding to α-synuclein by NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 194–196. [Google Scholar] [CrossRef]
- Verdone, G.; Corazza, A.; Viglino, P.; Pettirossi, F.; Giorgetti, S.; Mangione, P.; Andreola, A.; Stoppini, M.; Bellotti, V.; Esposito, G. The solution structure of human β2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2002, 11, 487–499. [Google Scholar] [CrossRef]
- Salamekh, S.; Brender, J.R.; Hyung, S.-J.; Nanga, R.P.R.; Vivekanandan, S.; Ruotolo, B.T.; Ramamoorthy, A. A two-site mechanism for the inhibition of IAPP amyloidogenesis by zinc. J. Mol. Biol. 2011, 410, 294–306. [Google Scholar] [CrossRef]
- He, L.; Zhu, D.; Zhao, C.; Jia, X.; Wang, X.; Du, W. Effects of gold complexes on the assembly behavior of human islet amyloid polypeptide. J. Inorg. Biochem. 2015, 152, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Xu, J.; Du, W. Assembly behavior of amylin fragment hIAPP19–37 regulated by Au (III) complexes. J. Inorg. Biochem. 2019, 201, 110807. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, X.; Zhao, C.; Wang, H.; Du, W. Ruthenium complexes as novel inhibitors of human islet amyloid polypeptide fibril formation. Metallomics 2013, 5, 1599–1603. [Google Scholar] [CrossRef]
- Gong, G.; Wang, W.; Du, W. Binuclear ruthenium complexes inhibit the fibril formation of human islet amyloid polypeptide. RSC Adv. 2017, 7, 18512–18522. [Google Scholar] [CrossRef]
- Soragni, A.; Zambelli, B.; Mukrasch, M.D.; Biernat, J.; Jeganathan, S.; Griesinger, C.; Ciurli, S.; Mandelkow, E.; Zweckstetter, M. Structural characterization of binding of Cu (II) to tau protein. Biochemistry 2008, 47, 10841–10851. [Google Scholar] [CrossRef]
- Ahmadi, S.; Ebralidze, I.I.; She, Z.; Kraatz, H.B. Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s Disease. Electrochim. Acta 2017, 236, 384–393. [Google Scholar] [CrossRef]
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.-B. Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation. J. Inorg. Biochem. 2019, 194, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8531–8535. [Google Scholar] [CrossRef]
- Kozlowski, H.; Janicka-Klos, A.; Stanczak, P.; Valensin, D.; Valensin, G.; Kulon, K. Specificity in the Cu2+ interactions with prion protein fragments and related His-rich peptides from mammals to fishes. Coord. Chem. Rev. 2008, 252, 1069–1078. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.-M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s ß-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef]
- Bush, A.I.; Multhaup, G.; Moir, R.D.; Williamson, T.G.; Small, D.H.; Rumble, B.; Pollwein, P.; Beyreuther, K.; Masters, C. A novel zinc (II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J. Biol. Chem. 1993, 268, 16109–16112. [Google Scholar] [PubMed]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.E.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic aggregation of Alzheimer Aβ by Cu (II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s disease amyloid-β binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef]
- Miura, T.; Suzuki, K.; Kohata, N.; Takeuchi, H. Metal binding modes of Alzheimer’s amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry 2000, 39, 7024–7031. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-T.; Howlett, G.; Barrow, C.J. Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the Aβ peptide of Alzheimer’s disease. Biochemistry 1999, 38, 9373–9378. [Google Scholar] [CrossRef]
- Yang, D.S.; McLaurin, J.; Qin, K.; Westaway, D.; Fraser, P.E. Examining the zinc binding site of the amyloid-β peptide. Eur. J. Biochem. 2000, 267, 6692–6698. [Google Scholar] [CrossRef]
- Dong, J.; Shokes, J.E.; Scott, R.A.; Lynn, D.G. Modulating amyloid self-assembly and fibril morphology with Zn (II). J. Am. Chem. Soc. 2006, 128, 3540–3542. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Mihara, H. Peptide and protein mimetics inhibiting amyloid β-peptide aggregation. Acc. Chem. Res. 2008, 41, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Stellato, F.; Menestrina, G.; Dalla Serra, M.; Potrich, C.; Tomazzolli, R.; Meyer-Klaucke, W.; Morante, S. Metal binding in amyloid β-peptides shows intra-and inter-peptide coordination modes. Eur. Biophys. J. 2006, 35, 340–351. [Google Scholar] [CrossRef]
- Morgan, D.M.; Dong, J.; Jacob, J.; Lu, K.; Apkarian, R.P.; Thiyagarajan, P.; Lynn, D.G. Metal switch for amyloid formation: Insight into the structure of the nucleus. J. Am. Chem. Soc. 2002, 124, 12644–12645. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Aβ aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef]
- Karr, J.W.; Kaupp, L.J.; Szalai, V.A. Amyloid-β binds Cu2+ in a mononuclear metal ion binding site. J. Am. Chem. Soc. 2004, 126, 13534–13538. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Miura, T.; Takeuchi, H. Inhibitory effect of copper (II) on zinc (II)-induced aggregation of amyloid β-peptide. Biochem. Biophys. Res. Commun. 2001, 285, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-H.; Sun, X.; Yu, Y.-P.; Hu, J.; Zhao, L.; Liu, Q.; Zhao, Y.-F.; Li, Y.-M. TiO2 nanoparticles promote β-amyloid fibrillation in vitro. Biochem. Biophys. Res. Commun. 2008, 373, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Grumezescu, A.M. Antimicrobial Nanoarchitectonics: From Synthesis to Applications; William Andrew: Bucharest, Romania, 2017. [Google Scholar]
- Benzinger, T.L.; Gregory, D.M.; Burkoth, T.S.; Miller-Auer, H.; Lynn, D.G.; Botto, R.E.; Meredith, S.C. Propagating structure of Alzheimer’s β-amyloid (10–35) is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA 1998, 95, 13407–13412. [Google Scholar] [CrossRef]
- Emwas, A.H.M.; Al-Talla, Z.A.; Guo, X.; Al-Ghamdi, S.; Al-Masri, H.T. Utilizing NMR and EPR spectroscopy to probe the role of copper in prion diseases. Magn. Reson. Chem. 2013, 51, 255–268. [Google Scholar] [CrossRef]
- Millhauser, G.L. Copper binding in the prion protein. Acc. Chem. Res. 2004, 37, 79–85. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Walter, E.D.; Newell, D.J.; Jackson, P.J.; Aronoff-Spencer, E.; Peisach, J.; Gerfen, G.J.; Bennett, B.; Antholine, W.E.; Millhauser, G.L. The octarepeat domain of the prion protein binds Cu (II) with three distinct coordination modes at pH 7.4. J. Am. Chem. Soc. 2005, 127, 12647–12656. [Google Scholar] [CrossRef] [PubMed]
- Morante, S.; González-Iglesias, R.; Potrich, C.; Meneghini, C.; Meyer-Klaucke, W.; Menestrina, G.; Gasset, M. Inter-and Intra-Octarepeat Cu (II) Site Geometries in the Prion Protein Implications in Cu (II) Binding Cooperativity and Cu (II)-Mediated Assemblies. J. Biol. Chem. 2004, 279, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Antonyuk, S.; Elam, J.S.; Hough, M.A.; Strange, R.W.; Doucette, P.A.; Rodriguez, J.A.; Hayward, L.J.; Valentine, J.S.; Hart, P.J.; Hasnain, S.S. Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci. 2005, 14, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Hart, P.J. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 3617–3622. [Google Scholar] [CrossRef]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.-S.; Li, L. Oxidative modifications and aggregation of Cu, Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Structural characterization of copper (II) binding to α-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef]
- Borsarelli, C.D.; Falomir-Lockhart, L.J.; Ostatna, V.; Fauerbach, J.A.; Hsiao, H.-H.; Urlaub, H.; Paleček, E.; Jares-Erijman, E.A.; Jovin, T.M. Biophysical properties and cellular toxicity of covalent crosslinked oligomers of α-synuclein formed by photoinduced side-chain tyrosyl radicals. Free Radic. Biol. Med. 2012, 53, 1004–1015. [Google Scholar] [CrossRef]
- Song, M.; Sun, Y.; Luo, Y.; Zhu, Y.; Liu, Y.; Li, H. Exploring the mechanism of inhibition of Au nanoparticles on the aggregation of amyloid-β (16–22) peptides at the atom level by all-atom molecular dynamics. Int. J. Mol. Sci. 2018, 19, 1815. [Google Scholar] [CrossRef]
- Ma, Q.; Wei, G.; Yang, X. Influence of Au nanoparticles on the aggregation of amyloid-β-(25–35) peptides. Nanoscale 2013, 5, 10397–10403. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, C.; Zhu, D.; Gong, G.; Du, W. Inhibition of amyloid peptide fibril formation by gold–sulfur complexes. J. Inorg. Biochem. 2017, 171, 1–9. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, X.; He, L.; Zhu, D.; Wang, B.; Du, W. Influence of gold–bipyridyl derivants on aggregation and disaggregation of the prion neuropeptide PrP106–126. Metallomics 2014, 6, 2117–2125. [Google Scholar] [CrossRef]
- Seker, U.O.S.; Chen, A.Y.; Citorik, R.J.; Lu, T.K. Synthetic biogenesis of bacterial amyloid nanomaterials with tunable inorganic–organic interfaces and electrical conductivity. ACS Synth. Biol. 2017, 6, 266–275. [Google Scholar] [CrossRef]
- Luong, T.Q.; Erwin, N.; Neumann, M.; Schmidt, A.; Loos, C.; Schmidt, V.; Fändrich, M.; Winter, R. Hydrostatic pressure increases the catalytic activity of amyloid fibril enzymes. Angew. Chem. Int. Ed. 2016, 55, 12412–12416. [Google Scholar] [CrossRef]
- Makin, O.S.; Serpell, L.C. Structures for amyloid fibrils. FEBS J. 2005, 272, 5950–5961. [Google Scholar] [CrossRef]
- Song, R.; Wu, X.; Xue, B.; Yang, Y.; Huang, W.; Zeng, G.; Wang, J.; Li, W.; Cao, Y.; Wang, W. Principles governing catalytic activity of self-assembled short peptides. J. Am. Chem. Soc. 2018, 141, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stöhr, J.; Smith, T.A.; Hu, X.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Al-Garawi, Z.; McIntosh, B.; Neill-Hall, D.; Hatimy, A.; Sweet, S.; Bagley, M.; Serpell, L. The amyloid architecture provides a scaffold for enzyme-like catalysts. Nanoscale 2017, 9, 10773–10783. [Google Scholar] [CrossRef]
- Monasterio, O.; Nova, E.; Diaz-Espinoza, R. Development of a novel catalytic amyloid displaying a metal-dependent ATPase-like activity. Biochem. Biophys. Res. Commun. 2017, 482, 1194–1200. [Google Scholar] [CrossRef]
- Cao, Y.; Mezzenga, R. Food protein amyloid fibrils: Origin, structure, formation, characterization, applications and health implications. Adv. Colloid Interface Sci. 2019, 269, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Zappone, B.; De Santo, M.P.; Labate, C.; Rizzuti, B.; Guzzi, R. Catalytic activity of copper ions in the amyloid fibrillation of β-lactoglobulin. Soft Matter 2013, 9, 2412–2419. [Google Scholar] [CrossRef]
- Guzzi, R.; Rizzuti, B.; Labate, C.; Zappone, B.; De Santo, M.P. Ferric ions inhibit the amyloid fibrillation of β-lactoglobulin at high temperature. Biomacromolecules 2015, 16, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.E.; Miti, T.; Richmond, T.; Muschol, M. Spatial extent of charge repulsion regulates assembly pathways for lysozyme amyloid fibrils. PLoS ONE 2011, 6, e18171. [Google Scholar] [CrossRef] [PubMed]
- Waku, T.; Tanaka, N. Recent advances in nanofibrous assemblies based on β-sheet-forming peptides for biomedical applications. Polym. Int. 2017, 66, 277–288. [Google Scholar] [CrossRef]
- Mahmoud, A. New vaccines: Challenges of discovery. Microb. Biotechnol. 2016, 9, 549–552. [Google Scholar] [CrossRef]
- Enquist, L.W. Virology in the 21st Century. J. Virol. 2009, 83, 5296–5308. [Google Scholar] [CrossRef]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1009–1026. [Google Scholar]
- De Clercq, E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004, 30, 115–133. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral drugs: Current state of the art. J. Clin. Virol. 2001, 22, 73–89. [Google Scholar] [CrossRef]
- Agier, J.; Efenberger, M.; Brzezińska-Błaszczyk, E. Cathelicidin impact on inflammatory cells. Cent.-Eur. J. Immunol. 2015, 40, 225. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Silva, O.N.; Lu, T.K.; Franco, O.L. Antimicrobial peptides: Role in human disease and potential as immunotherapies. Pharmacol. Ther. 2017, 178, 132–140. [Google Scholar] [CrossRef]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human antimicrobial peptides as therapeutics for viral infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef]
- Sokolov, Y.; Mirzabekov, T.; Martin, D.W.; Lehrer, R.I.; Kagan, B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta (BBA)-Biomembr. 1999, 1420, 23–29. [Google Scholar] [CrossRef]
- Kagan, B.L.; Selsted, M.E.; Ganz, T.; Lehrer, R.I. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1990, 87, 210–214. [Google Scholar] [CrossRef]
- Thundimadathil, J.; Roeske, R.W.; Guo, L. A synthetic peptide forms voltage-gated porin-like ion channels in lipid bilayer membranes. Biochem. Biophys. Res. Commun. 2005, 330, 585–590. [Google Scholar] [CrossRef]
- Thundimadathil, J.; Roeske, R.W.; Jiang, H.Y.; Guo, L. Aggregation and porin-like channel activity of a β sheet peptide. Biochemistry 2005, 44, 10259–10270. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Ma, B.; Lal, R.; Nussinov, R. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic alzheimer β-amyloid ion channels. Biophys. J. 2008, 95, 4631–4642. [Google Scholar] [CrossRef] [PubMed]
- Boas, L.C.P.V.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Chinchar, V.; Bryan, L.; Silphadaung, U.; Noga, E.; Wade, D.; Rollins-Smith, L. Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology 2004, 323, 268–275. [Google Scholar] [CrossRef]
- Altmann, S.E.; Brandt, C.R.; Jahrling, P.B.; Blaney, J.E. Antiviral activity of the EB peptide against zoonotic poxviruses. Virol. J. 2012, 9, 6. [Google Scholar] [CrossRef]
- Falco, A.; Mas, V.; Tafalla, C.; Perez, L.; Coll, J.; Estepa, A. Dual antiviral activity of human alpha-defensin-1 against viral haemorrhagic septicaemia rhabdovirus (VHSV): Inactivation of virus particles and induction of a type I interferon-related response. Antivir. Res. 2007, 76, 111–123. [Google Scholar] [CrossRef]
- Crack, L.; Jones, L.; Malavige, G.; Patel, V.; Ogg, G. Human antimicrobial peptides LL-37 and human β-defensin-2 reduce viral replication in keratinocytes infected with varicella zoster virus. Clin. Exp. Dermatol. Exp. Dermatol. 2012, 37, 534–543. [Google Scholar] [CrossRef]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The interaction of antimicrobial peptides with membranes. Adv. Colloid Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic α helical antimicrobial peptides. A systematic study of the effects of structural and physical properties on biological activity. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Verma, A.; Kim, E.J.; White, M.R.; Hartshorn, K.L. LL-37 modulates human neutrophil responses to influenza A virus. J. Leukoc. Biol. 2014, 96, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 2011, 6, e25333. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Lin, Y.; McCullers, J.A.; Samarasinghe, A.E. Antimicrobial peptides alter early immune response to influenza A virus infection in C57BL/6 mice. Antivir. Res. 2016, 133, 208–217. [Google Scholar] [CrossRef]
- Abe, K.i.; Nozaki, A.; Tamura, K.; Ikeda, M.; Naka, K.; Dansako, H.; Hoshino, H.o.; Tanaka, K.; Kato, N. Tandem Repeats of Lactoferrin-Derived Anti-Hepatitis C Virus Peptide Enhance Antiviral Activity in Cultured Human Hepatocytes. Microbiol. Immunol. 2007, 51, 117–125. [Google Scholar] [CrossRef]
- Jenssen, H. Anti herpes simplex virus activity of lactoferrin/lactoferricin–an example of antiviral activity of antimicrobial protein/peptide. Cell. Mol. Life Sci. Cmls 2005, 62, 3002–3013. [Google Scholar] [CrossRef]
- Jaishankar, D.; Yakoub, A.M.; Bogdanov, A.; Valyi-Nagy, T.; Shukla, D. Characterization of a proteolytically stable D-peptide that suppresses herpes simplex virus 1 infection: Implications for the development of entry-based antiviral therapy. J. Virol. 2015, 89, 1932–1938. [Google Scholar] [CrossRef]
- Lee, N.; Walker, E.; Egerer, L.; Bunnell, B.A.; Mondal, D.; von Laer, D.; Braun, S.E. 476. The Therapeutic Potential of Secreted Antiviral Entry Inhibitor (SAVE) Peptides Expressed By Transduced MSCs To Block HIV Infection. Mol. Ther. 2014, 22, 1. [Google Scholar]
- Pessi, A.; Ingallinella, P.; Bianchi, E.; Wang, Y.-J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.; Moore, J.; Miller, M. Dramatic Increase of Antiviral Potency of An HIV Peptide Fusion Inhibitor by Targeting to Lipid Rafts Via Addition of A Cholesterol Group. In Biopolymers; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2009; p. 302. [Google Scholar]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef]
- Liu, T.; She, R.; Wang, K.; Bao, H.; Zhang, Y.; Luo, D.; Hu, Y.; Ding, Y.; Wang, D.; Peng, K. Effects of rabbit sacculus rotundus antimicrobial peptides on the intestinal mucosal immunity in chickens. Poult. Sci. 2008, 87, 250–254. [Google Scholar] [CrossRef]
- Skalickova, S.; Heger, Z.; Krejcova, L.; Pekarik, V.; Bastl, K.; Janda, J.; Kostolansky, F.; Vareckova, E.; Zitka, O.; Adam, V. Perspective of use of antiviral peptides against influenza virus. Viruses 2015, 7, 5428–5442. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef]
- Mulder, K.; Lima, L.A.; Miranda, V.; Dias, S.C.; Franco, O.L. Current scenario of peptide-based drugs: The key roles of cationic antitumor and antiviral peptides. Front. Microbiol. 2013, 4, 321. [Google Scholar] [CrossRef]
- Barlow, P.G.; Findlay, E.G.; Currie, S.M.; Davidson, D.J. Antiviral potential of cathelicidins. Future Microbiol. 2014, 9, 55–73. [Google Scholar] [CrossRef]
- Findlay, E.G.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef]
- Matusevich, O.; Egorov, V.; Gluzdikov, I.; Titov, M.; Zarubaev, V.; Shtro, A.; Slita, A.; Dukov, M.; Shurygina, A.-P.; Smirnova, T. Synthesis and antiviral activity of PB1 component of the influenza A RNA polymerase peptide fragments. Antivir. Res. 2015, 113, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Zabrodskaya, Y.A.; Lebedev, D.V.; Egorova, M.A.; Shaldzhyan, A.A.; Shvetsov, A.V.; Kuklin, A.I.; Vinogradova, D.S.; Klopov, N.V.; Matusevich, O.V.; Cheremnykh, T.A. The amyloidogenicity of the influenza virus PB1-derived peptide sheds light on its antiviral activity. Biophys. Chem. 2018, 234, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Veloria, J.R.; Chen, L.; Li, L.; Breen, G.A.; Lee, J.; Goux, W.J. Novel cell-penetrating-amyloid peptide conjugates preferentially kill cancer cells. Medchemcomm 2018, 9, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.; McCarthy, D.; Hart, S.L.; Tagalakis, A.D. Systematic Comparisons of Formulations of Linear Oligolysine Peptides with si RNA and Plasmid DNA. Chem. Biol. Drug Des. 2016, 87, 747–763. [Google Scholar] [CrossRef]
- Vaughan, E.E.; Dean, D.A. Intracellular trafficking of plasmids during transfection is mediated by microtubules. Mol. Ther. 2006, 13, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.; Hart, S.L. Comparative structural and functional studies of nanoparticle formulations for DNA and siRNA delivery. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 210–219. [Google Scholar] [CrossRef]
- Papanikolopoulou, K.; Schoehn, G.; Forge, V.; Forsyth, V.T.; Riekel, C.; Hernandez, J.-F.; Ruigrok, R.W.; Mitraki, A. Amyloid fibril formation from sequences of a natural β-structured fibrous protein, the adenovirus fiber. J. Biol. Chem. 2005, 280, 2481–2490. [Google Scholar] [CrossRef]
- Jonnalagadda, S.V.R.; Kokotidou, C.; Orr, A.A.; Fotopoulou, E.; Henderson, K.J.; Choi, C.-H.; Lim, W.T.; Choi, S.J.; Jeong, H.-K.; Mitraki, A. Computational Design of Functional Amyloid Materials with Cesium Binding, Deposition, and Capture Properties. J. Phys. Chem. B 2018, 122, 7555–7568. [Google Scholar] [CrossRef] [PubMed]
- Raucher, D.; Ryu, J.S. Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med. 2015, 21, 560–570. [Google Scholar] [CrossRef]
- Yolamanova, M.; Meier, C.; Shaytan, A.K.; Vas, V.; Bertoncini, C.W.; Arnold, F.; Zirafi, O.; Usmani, S.M.; Müller, J.A.; Sauter, D. Peptide nanofibrils boost retroviral gene transfer and provide a rapid means for concentrating viruses. Nat. Nanotechnol. 2013, 8, 130–136. [Google Scholar] [CrossRef]
- Li, D.; Jones, E.M.; Sawaya, M.R.; Furukawa, H.; Luo, F.; Ivanova, M.; Sievers, S.A.; Wang, W.; Yaghi, O.M.; Liu, C. Structure-based design of functional amyloid materials. J. Am. Chem. Soc. 2014, 136, 18044–18051. [Google Scholar] [CrossRef] [PubMed]
- Munch, J.; Rucker, E.; Standker, L.; Adermann, K.; Goffinet, C.; Schindler, M.; Wildum, S.; Chinnadurai, R.; Rajan, D.; Specht, A.; et al. Semen-derived amyloid 561 fibrils drastically enhance HIV infection. Cell 2007, 131, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Kirti, S.; Patel, K.; Das, S.; Shrimali, P.; Samanta, S.; Kumar, R.; Chatterjee, D.; Ghosh, D.; Kumar, A.; Tayalia, P. Amyloid fibrils with positive charge enhance retroviral transduction in mammalian cells. ACS Biomater. Sci. Eng. 2018, 5, 126–138. [Google Scholar] [CrossRef]
- Henning-Knechtel, A.; Kumar, S.; Wallin, C.; Król, S.; Wärmländer, S.K.; Jarvet, J.; Esposito, G.; Kirmizialtin, S.; Gräslund, A.; Hamilton, A.D. Designed cell-penetrating peptide inhibitors of amyloid-beta aggregation and cytotoxicity. Cell Rep. Phys. Sci. 2020, 1, 100014. [Google Scholar] [CrossRef]
- Österlund, N.; Luo, J.; Wärmländer, S.K.; Gräslund, A. Membrane-mimetic systems for biophysical studies of the amyloid-β peptide. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 492–501. [Google Scholar]
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.L.; Strzelec, A.; Kiessling, L.L.; Murphy, R.M. Structure-function relationships for inhibitors of β-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry 2001, 40, 7882–7889. [Google Scholar] [CrossRef]
- Ni, M.; Tresset, G.; Iliescu, C.; Hauser, C.A. Ultrashort Peptide Theranostic Nanoparticles by Microfluidic-Assisted Rapid Solvent Exchange. IEEE Trans. Nanobiosci. 2020, 19, 627–632. [Google Scholar] [CrossRef]
- Wolfbeis, O.S. An overview of nanoparticles commonly used in fluorescent bioimaging. Chem. Soc. Rev. 2015, 44, 4743–4768. [Google Scholar] [CrossRef]
- Sharma, A.; Gadly, T.; Neogy, S.; Ghosh, S.K.; Kumbhakar, M. Molecular origin and self-assembly of fluorescent carbon nanodots in polar solvents. J. Phys. Chem. Lett. 2017, 8, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Schnermann, M.J. Chemical biology: Organic dyes for deep bioimaging. Nature 2017, 551, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.T.; Schierle, G.S.K.; Kumita, J.R.; Bertoncini, C.W.; Dobson, C.M.; Kaminski, C.F. Protein amyloids develop an intrinsic fluorescence signature during aggregation. Analyst 2013, 138, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Tcherkasskaya, O. Photo-activity induced by amyloidogenesis. Protein Sci. 2007, 16, 561–571. [Google Scholar] [CrossRef]
- Bekard, I.B.; Dunstan, D.E. Tyrosine autofluorescence as a measure of bovine insulin fibrillation. Biophys. J. 2009, 97, 2521–2531. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Mukherjee, S.; Sharma, S.; Agrawal, V.; Kishan, K.R.; Guptasarma, P. A novel UV laser-induced visible blue radiation from protein crystals and aggregates: Scattering artifacts or fluorescence transitions of peptide electrons delocalized through hydrogen bonding? Arch. Biochem. Biophys. 2004, 428, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Del Mercato, L.L.; Pompa, P.P.; Maruccio, G.; Della Torre, A.; Sabella, S.; Tamburro, A.M.; Cingolani, R.; Rinaldi, R. Charge transport and intrinsic fluorescence in amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2007, 104, 18019–18024. [Google Scholar] [CrossRef]
- Sharpe, S.; Simonetti, K.; Yau, J.; Walsh, P. Solid-State NMR characterization of autofluorescent fibrils formed by the elastin-derived peptide GVGVAGVG. Biomacromolecules 2011, 12, 1546–1555. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Portaccio, M.; Irace, G.; Sirangelo, I. Insights into insulin fibril assembly at physiological and acidic pH and related amyloid intrinsic fluorescence. Int. J. Mol. Sci. 2017, 18, 2551. [Google Scholar] [CrossRef]
- Grisanti, L.; Pinotsi, D.; Gebauer, R.; Schierle, G.S.K.; Hassanali, A.A. A computational study on how structure influences the optical properties in model crystal structures of amyloid fibrils. Phys. Chem. Chem. Phys. 2017, 19, 4030–4040. [Google Scholar] [CrossRef]
- Pinotsi, D.; Grisanti, L.; Mahou, P.; Gebauer, R.; Kaminski, C.F.; Hassanali, A.; Kaminski Schierle, G.S. Proton transfer and structure-specific fluorescence in hydrogen bond-rich protein structures. J. Am. Chem. Soc. 2016, 138, 3046–3057. [Google Scholar] [CrossRef]
- Sirangelo, I.; Borriello, M.; Irace, G.; Iannuzzi, C. Intrinsic blue-green fluorescence in amyloyd fibrils. Aims Biophys. 2018, 5, 155. [Google Scholar] [CrossRef]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Kirkegaard, J.B.; Arosio, P.; Michaels, T.C.; Vendruscolo, M.; Dobson, C.M.; Linse, S.; Knowles, T.P. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016, 11, 252–272. [Google Scholar] [CrossRef]
- Handelman, A.; Kuritz, N.; Natan, A.; Rosenman, G. Reconstructive phase transition in ultrashort peptide nanostructures and induced visible photoluminescence. Langmuir 2016, 32, 2847–2862. [Google Scholar] [CrossRef] [PubMed]
- Di Fabrizio, E.; Schlücker, S.; Wenger, J.; Regmi, R.; Rigneault, H.; Calafiore, G.; West, M.; Cabrini, S.; Fleischer, M.; Van Hulst, N.F. Roadmap on biosensing and photonics with advanced nano-optical methods. J. Opt. 2016, 18, 063003. [Google Scholar] [CrossRef]
- Ni, M.; Zhuo, S.; Iliescu, C.; So, P.T.; Mehta, J.S.; Yu, H.; Hauser, C.A. Self-assembling amyloid-like peptides as exogenous second harmonic probes for bioimaging applications. J. Biophotonics 2019, 12, e201900065. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Yoon, S.-S.; Jo, S.A. Mechanisms of amyloid-β peptide clearance: Potential therapeutic targets for Alzheimer’s disease. Biomol. Ther. 2012, 20, 245. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Beckett, C.; Belyaev, N.D.; Turner, A.J. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 2012, 120, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Leong, Y.Q.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab. Brain Dis. 2020, 35, 11–30. [Google Scholar] [CrossRef]
- Bu, G.; Cam, J.; Zerbinatti, C. LRP in Amyloid-β Production and Metabolism. Ann. N. Y. Acad. Sci. 2006, 1086, 35–53. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Ferreira, S.T. β-amyloid production, aggregation, and clearance as targets for therapy in Alzheimer’s disease. Cell. Mol. Neurobiol. 2002, 22, 545–563. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Umans, L.; Van Leuven, F.; Van Den Berghe, H. Study of the synthesis and secretion of normal and artificial mutants of murine amyloid precursor protein (APP): Cleavage of APP occurs in a late compartment of the default secretion pathway. J. Cell Biol. 1993, 121, 295–304. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Ward, P.; Henriksson, T.; Beattie, E.; Neve, R.; Lieberburg, I.; Fritz, L. The Alzheimer amyloid precursor protein. Identification of a stable intermediate in the biosynthetic/degradative pathway. J. Biol. Chem. 1990, 265, 4492–4497. [Google Scholar]
- Brown, R.C.; Cascio, C.; Papadopoulos, V. Pathways of neurosteroid biosynthesis in cell lines from human brain: Regulation of dehydroepiandrosterone formation by oxidative stress and β-amyloid peptide. J. Neurochem. 2000, 74, 847–859. [Google Scholar] [CrossRef]
- Hernández-Zimbrón, L.F.; Gorostieta-Salas, E.; Díaz-Hung, M.L.; Pérez-Garmendia, R.; Gevorkian, G.; Quiroz-Mercado, H. Beta amyloid peptides: Extracellular and intracellular mechanisms of clearance in Alzheimer’s disease. In Update on Dement; Morettied, D., Ed.; Hindawi: London, UK, 2016. [Google Scholar]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Clearance of amyloid-beta in Alzheimer’s disease: Progress, problems and perspectives. Drug Discov. Today 2006, 11, 931–938. [Google Scholar] [CrossRef]

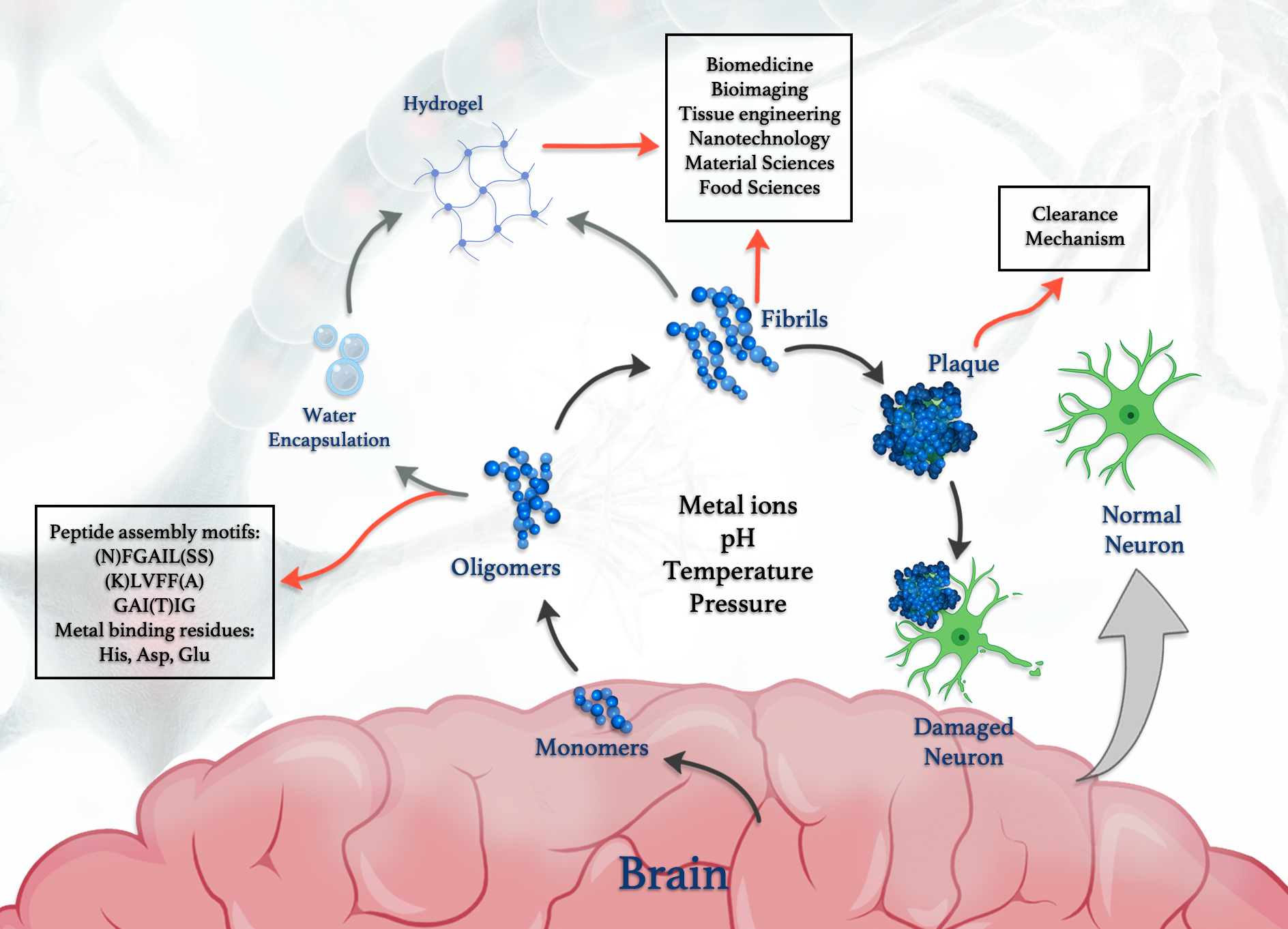{kind=link}
| Protein | Peptide | pH | Metal | Binding Sites | Ref |
|---|---|---|---|---|---|
| Aβ | (Rat) Aβ1–28 monomer | 7.5 | Cu(II) | Asp1, His6, Glu11, His14 | [81] |
| Aβ1–40/Aβ1–42 monomer | 5.5–7.5 | Cu(II) | His6, His13, His14, Tyr10 | [82] | |
| Aβ1–16/Aβ1–28 monomer | 7.4 | Cu(II) | Asp1, His6, His13, His14 | [83] | |
| Aβ1–16/Aβ1–40 | 7.4 | Cu(II) | His6, Glu11, His13, His14 | [84] | |
| Aβ3–40/42 | 6.3–8 | Cu(II) | Ala2, His6, His13, His14 | [85] | |
| Aβ1–16 monomer | 6.5–7.4 | Zn(II) | His6, Glu11, His13, His14 | [86] | |
| Aβ1–28 monomer | 7.5 | Zn(II) | Asp1, His6, Glu11, His14 | [81] | |
| Aβ1–28 monomer | 7.5 | Zn(II) | Asp1, His6, Glu11, His13, His14 | [81] | |
| Aβ1–40 Two monomers | 7.4 | Zn(II) | His13 and His14 of two adjacent A peptides | [87] | |
| Aβ1–28 monomer | 5.3–8.0 | Fe(III) | No significant binding | [81] | |
| Aβ1–16/Aβ1–40 | physiological | Fe(II) | Asp1, Glu3, His6, His13, His14 | [88] | |
| Aβ1–28 monomer | 5.3–8.0 | Al(III) | No significant binding | [81] | |
| Aβ1–40/Aβ1–42 | No data | Al(III) | sequence 1–16 and sequence 20–35 | [89] | |
| α-Synuclein (α-S) | α-S1–140 | 6.5 | Mn(II), Fe(II), Co(II) and Ni(II) | Asp119, Pro120, Asp121, Asn122, and Glu123 | [90] |
| α-S1–140 | 7.2–7.4 | Cu(II) | amino acids 3–9 and 49–52 (STRONGER); amino acids 20–24 and 39–44 (WEAKER); His50 | [91] | |
| Human islet amyloid polypeptide (hIAPP, amylin) | IAPP14−22/IAPP15−22 | 7.5 | Cu(II) | His18, Ser19, Ser20 | [91] |
| IAPP1−19 | 6.5 | Cu(II) | His13, His31 | [92] | |
| IAPP1−19 | 7.45 | Zn(II) | His18 | [92,93] | |
| hIAPP19−37 | No data | Au(I) | possible coordination between the gold and the histidine residue | [94,95] | |
| hIAPP1–37 | 7.5 | Ru(II) | C-terminal of the hIAPP could be involved in the binding | [96,97] | |
| Tau | Human Tau40 isoform (441 aa), K32 comprising residues (Met)Ser198-Tyr394, K32Δcys with Cys291, and Cys322 replaced by Ala | 6.5 | Cu(II) | 287VQSKCGS293 and 310YKPVDLSKVTSKCGS324 | [98] |
| Tau-410, 2N3R (n-tau) | 7.4 | Fe(II)/Fe(III) | clear iron/tau binding and Fe(II)/Fe(III) redox reaction | [99] | |
| hTau40 and phosphorylated hTau40 | 7.4 | Fe(II)/Fe(III), Cu(II), Zn(II) | hTau40/metal interactions; phosphorylated hTau40/metal no interactions/weak Zn(II) interactions | [100] | |
| Prion | human PrP91–231 in either the oxidized α-form or the reduced β-form; PrP52−98 | 8.0 | Cu(II), Ni(II), | Cu(II)/hPrP91–231 and Ni(II)/hPrP91–231 interactions; low affinity to Zn(II) and Mn(II) | [101] |
| Octa-repeat region of N-terminal (PHGGGWGQ) | 7–8 | Cu(II) | Coordination mode {Nimid, nN−} | [102] | |
| PrP106–126 | 7–8 | Cu(II), Zn(II), Mn(II) | Strong coordination of Cu(II) and weak coordination of Zn(II) ions by hydrophobic tail PrP112−126; Mn(II) coordinated by the His111, Gly124, and Leu125 residues | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelrahman, S.; Alghrably, M.; Lachowicz, J.I.; Emwas, A.-H.; Hauser, C.A.E.; Jaremko, M. “What Doesn’t Kill You Makes You Stronger”: Future Applications of Amyloid Aggregates in Biomedicine. Molecules 2020, 25, 5245. https://doi.org/10.3390/molecules25225245
Abdelrahman S, Alghrably M, Lachowicz JI, Emwas A-H, Hauser CAE, Jaremko M. “What Doesn’t Kill You Makes You Stronger”: Future Applications of Amyloid Aggregates in Biomedicine. Molecules. 2020; 25(22):5245. https://doi.org/10.3390/molecules25225245
Chicago/Turabian StyleAbdelrahman, Sherin, Mawadda Alghrably, Joanna Izabela Lachowicz, Abdul-Hamid Emwas, Charlotte A. E. Hauser, and Mariusz Jaremko. 2020. "“What Doesn’t Kill You Makes You Stronger”: Future Applications of Amyloid Aggregates in Biomedicine" Molecules 25, no. 22: 5245. https://doi.org/10.3390/molecules25225245
APA StyleAbdelrahman, S., Alghrably, M., Lachowicz, J. I., Emwas, A.-H., Hauser, C. A. E., & Jaremko, M. (2020). “What Doesn’t Kill You Makes You Stronger”: Future Applications of Amyloid Aggregates in Biomedicine. Molecules, 25(22), 5245. https://doi.org/10.3390/molecules25225245