
In-Situ Synthesis and Characterization of Nanocomposites in the Si-Ti-N and Si-Ti-C Systems

 , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
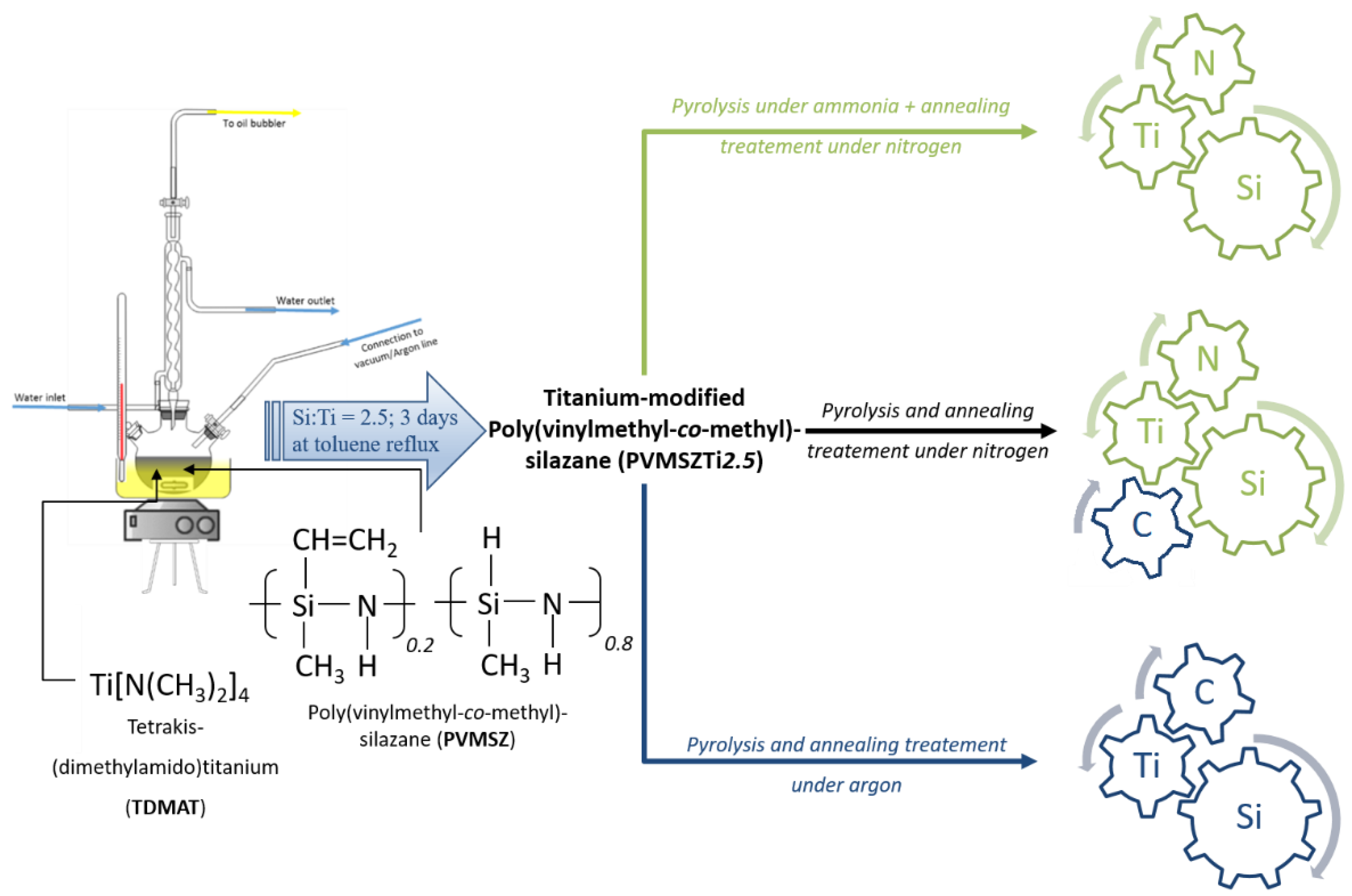
2.1. Polymer Synthesis and Characterization
- (i)
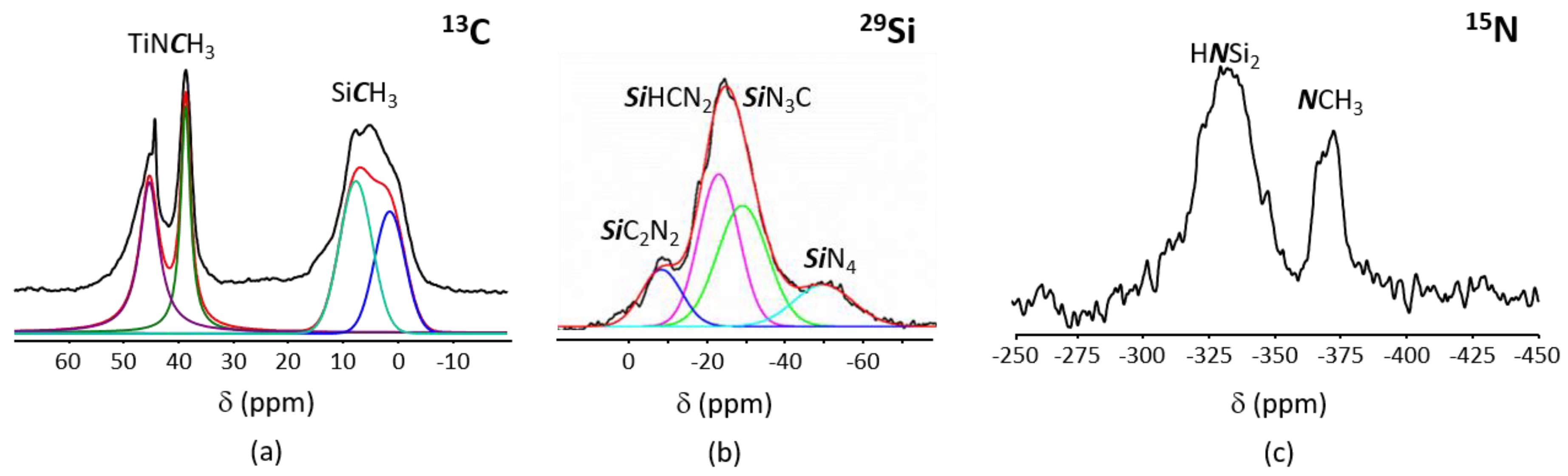
- those involving -NH units in PVMSZ and -N(CH3)2 groups in TDMAT to form -N-Ti[N(CH3)2]3 units releasing dimethylamine according to Equation (1). This reaction occurs probably majoritarly;
- (ii)
- those involving the silicon centers of PVMSZ and -N(CH3)2 groups in TDMAT causing the decrease of Si-H groups while forming -Si-N(CH3)-Ti- bridges in the obtained precursor and the concomitant evolution of methane according to Equation (2).
- (i)
- in SiCN3 environments (i.e., Si environment after reaction of the SiH groups in the monomeric unit [Si(CH3)(H)NH]0.8 with TDMAT,
- (ii)
- in SiCRN2 (R = H (i.e., Si environment in the monomeric unit [Si(CH3)(H)NH]0.8) or R = C (i.e., Si environment in the monomeric unit [Si(CH3)(CH=CH2)NH]0.2)); thus units present in PVMSZ that did not react upon chemical modification with TDMAT.
2.2. Ceramic Conversion
2.2.1. TG Experiments
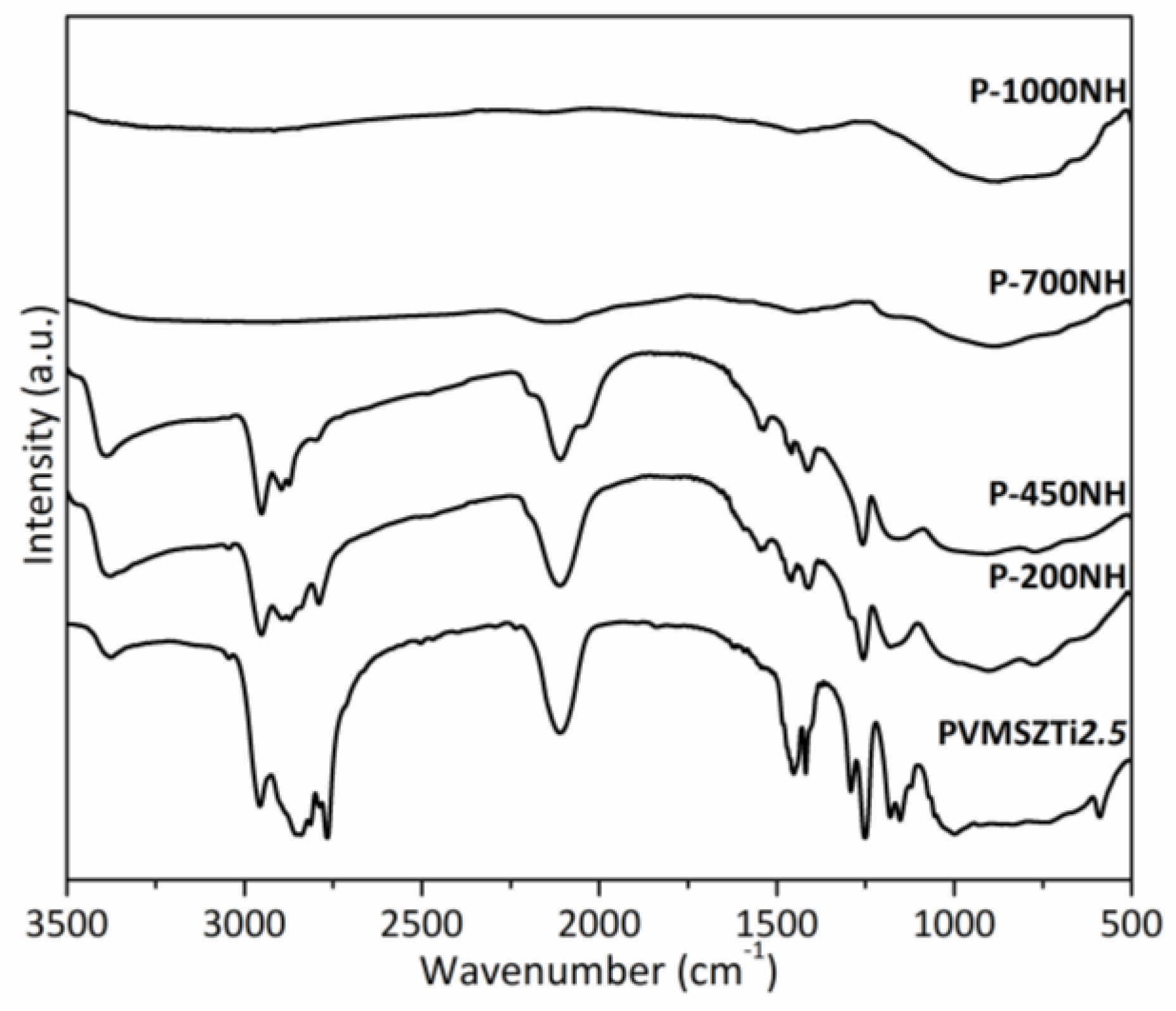
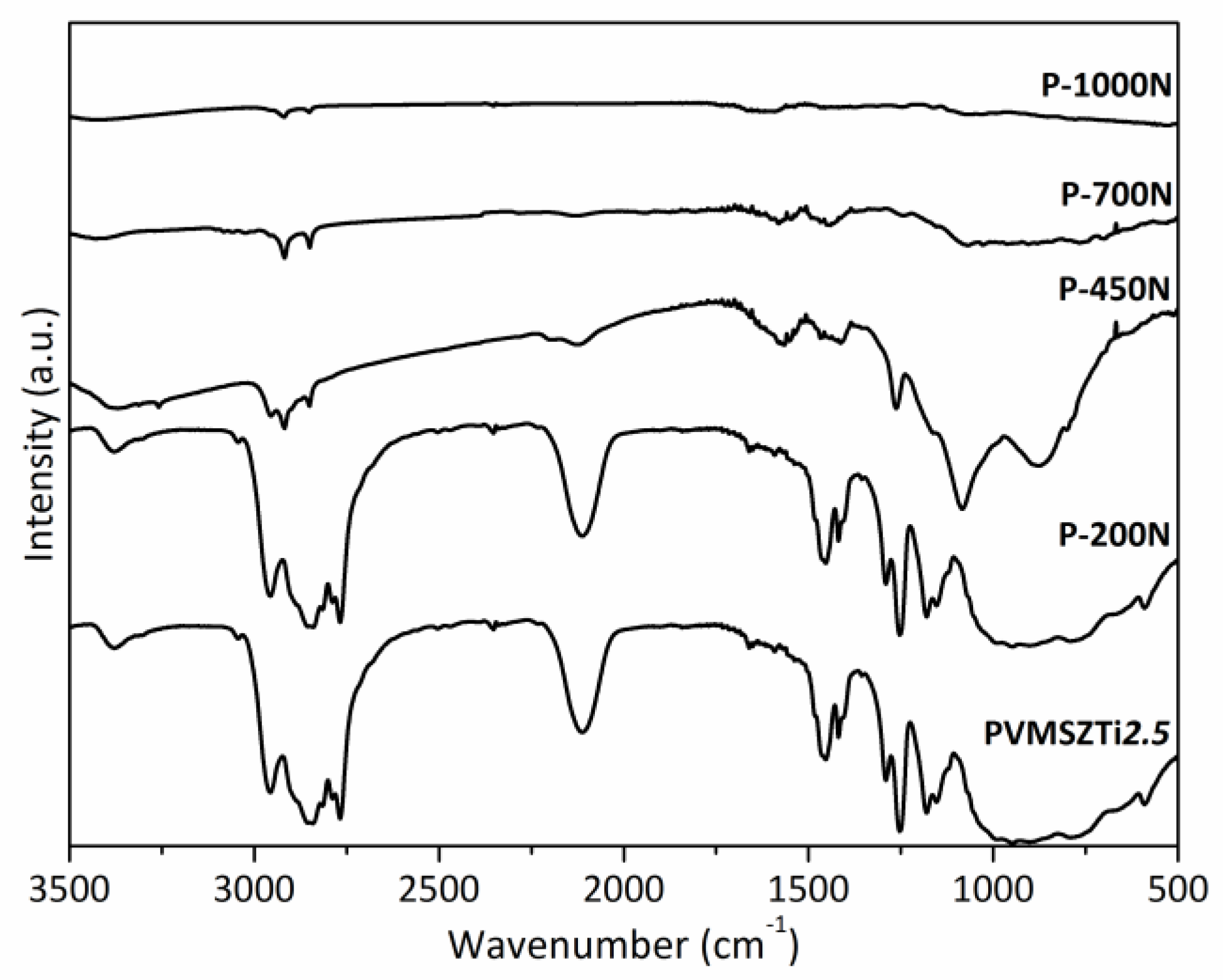
2.2.2. Pyrolysis in Flowing Ammonia
2.2.3. Pyrolysis in Flowing Nitrogen and Argon
- Pyrolysis in flowing nitrogen
- Pyrolysis in flowing argon
- (1)
- Below 300 °C, the precursor undergoes thermal cross-linking via condensation reactions involving both side groups of the type -N((CH3)2)x (x = 0, 1, 2, 3) and NH units (Equation (1)) and -Si-N(CH3)-Ti- that cleaves upon heating to abstract hydrogen releasing methylamine.
- (2)
- From 300 to 700 °C, mainly hydrocarbons such as methane and ethane/ethylene and hydrogen are released from the evolving system via radical reactions then H abstraction.
- (3)
- Above 700 °C, the remaining hydrogen atoms are gradually removed through homolytic cleavage of C-H bonds and then H abstraction to lead to a single-phase amorphous covalently bonded ceramic.
2.3. High-Temperature Phase and Microstructure Evolution of Single-Phase Amorphous Ceramics
- (i)
- the initial Si:Ti ratio (2.5) fixed in the polymer is retained in the derived ceramics,
- (ii)
- a strong tendency to decrease the carbon content from the sample treated under argon (P-1000A, carbon content of 16.4 wt%) to samples treated under ammonia (P-1000NH, carbon content of 1.5 wt%).
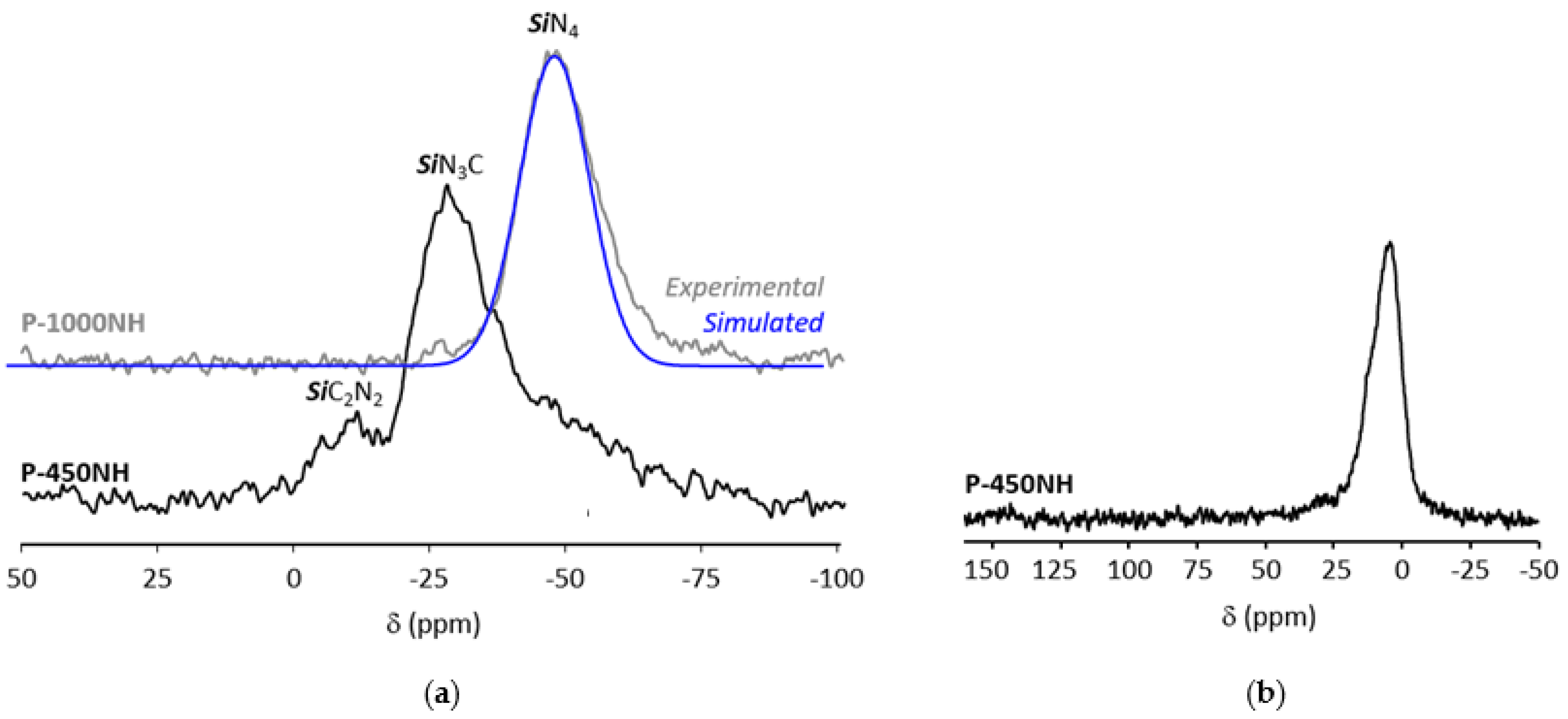
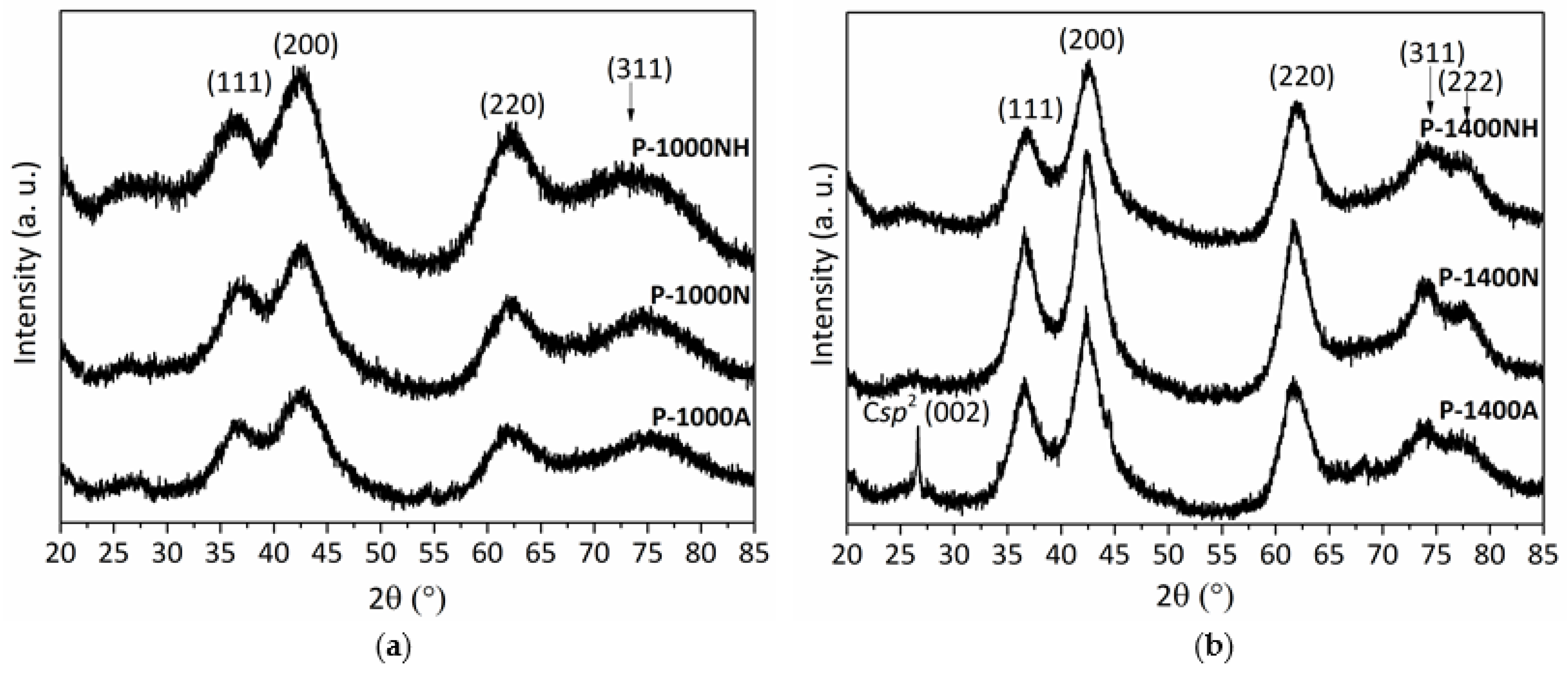
2.3.1. Ammonia-Treated Samples Heat-Treated in the Temperature Range 1500–1800 °C
2.3.2. Nitrogen and Argon-Treated Samples Heat-Treated in the Temperature Range 1500–1800 °C
3. Materials and Methods
3.1. General Comments
3.2. Polymer Synthesis
3.3. Synthesis of the Ceramic Materials
3.4. Material Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hnatko, M.; Hičák, M.; Labudová, M.; Galusková, D.; Sedláček, J.; Lenčéš, Z.; Šajgalík, P. Bioactive silicon nitride by surface thermal treatment. J. Eur. Ceram. Soc. 2020, 40, 1848–1858. [Google Scholar] [CrossRef]
- Sandra, F.; Ballestero, A.; NGuyen, V.L.; Tsampas, M.N.; Vernoux, P.; Balan, C.; Iwamoto, Y.; Demirci, U.B.; Miele, P.; Bernard, S. Silicon Carbide-based Membranes with High Filtration Efficiency, Durability and Catalytic Activity for CO/HC Oxidation and Soot Combustion. J. Membr. Sci. 2016, 501, 79–92. [Google Scholar] [CrossRef]
- Lale, A.; Wasan, A.; Kumar, R.; Miele, P.; Demirci, U.B.; Bernard, S. Organosilicon Polymer-Derived Mesoporous 3D Silicon Carbide, CarboNitride and Nitride Structures as Platinum Supports for Hydrogen Generation by Hydrolysis of Sodium Borohydride. Int. J. Hydro. Energy 2016, 41, 15477–15488. [Google Scholar] [CrossRef]
- Dong, L.; Wang, Y.; Tong, X.; Lei, T. Silicon carbide encapsulated graphite nanocomposites supported Pt nanoparticles as high-performance catalyst for methanol and ethanol oxidation reaction. Diam. Relat. Mater. 2020, 104, 107739–107744. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, Y.; Wen, Q.; Riedel, R.; Yu, Z. Dielectric Properties and Electromagnetic Wave Absorbing Performance of Single-Source-Precursor Synthesized Mo4.8Si3C0.6/SiC/Cfree Nanocomposites with an In Situ Formed Nowotny Phase. ACS Appl. Mater. Int. 2020, 12, 16912–16921. [Google Scholar] [CrossRef]
- Lale, A.; Mallmann, M.D.; Tada, S.; Bruma, A.; Özkar, S.; Kumar, R.; Haneda, M.; Machado, R.A.F.; Iwamoto, Y.; Demirci, U.B.; et al. Highly Active, Robust and Reusable Micro-/Mesoporous TiN/Si3N4 Nanocomposite-based Catalysts: Understanding the Key Role of TiN Nanoclusters and Amorphous Si3N4 Matrix in the Performance of the Catalyst System. Appl. Catal. B Environ. 2020, 272, 118975–118984. [Google Scholar] [CrossRef]
- Mera, G.; Gallei, M.; Bernard, S.; Ionescu, E. Ceramic Nanocomposites from Tailor-Made Preceramic Polymers. Nanocomposites 2015, 5, 468–540. [Google Scholar] [CrossRef]
- Eckardt, M.; Zaheer, M.; Kempe, R. Nitrogen-doped mesoporous SiC materials with catalytically active cobalt nanoparticles for the efficient and selective hydrogenation of nitroarenes. Sci. Rep. 2018, 8, 2567–2572. [Google Scholar] [CrossRef]
- Colombo, P.; Soraru, G.D.; Riedel, R.; Kleebe, H.-J. Polymer Derived Ceramics: From Nano-Structure to Applications; DEStech Publications: Lancaster, PA, USA, 2009. [Google Scholar]
- Bill, J.; Aldinger, F. Precursor-derived covalent ceramics. Adv. Mater. 1995, 7, 775–787. [Google Scholar] [CrossRef]
- Greil, P. Polymer derived engineering ceramics. Adv. Eng. Mater. 2000, 2, 339–348. [Google Scholar] [CrossRef]
- Colombo, P.; Mera, G.; Riedel, R.; Soraru, G.D. Polymer-derived ceramics: 40 years of research and innovation in advanced ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Ionescu, E.; Kleebe, H.-J.; Riedel, R. Silicon-containing polymer-derived ceramic nanocomposites (PDC-NCs): Preparative approaches and properties. Chem. Soc. Rev. 2012, 41, 5032–5052. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, M.; Schmalz, T.; Motz, G.; Kempe, R. Polymer derived non-oxide ceramics modified with late transition metals. Chem. Soc. Rev. 2012, 41, 5102–5116. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.; Miele, P. Polymer-derived boron nitride: A review on the chemistry, shaping and ceramic conversion of borazine derivatives. Materials 2014, 7, 7436–7459. [Google Scholar] [CrossRef] [PubMed]
- Stabler, C.; Ionescu, E.; Graczyk-Zajac, M.; Gonzalo-Juan, I.; Riedel, R. Silicon oxycarbide glasses and glass-ceramics: “All-Rounder” materials for advanced structural and functional applications. J. Am. Ceram. Soc. 2018, 101, 4817–4856. [Google Scholar] [CrossRef]
- Viard, A.; Fonblanc, D.; Lopez-Ferber, D.; Schmidt, M.; Lale, A.; Durif, C.; Balestrat, M.; Rossignol, F.; Weinmann, M.; Riedel, R.; et al. Polymer derived Si-B-C-N ceramics: 30 years of research. Adv. Eng. Mater. 2018, 20, 1800360–1800371. [Google Scholar] [CrossRef]
- Ionescu, E.; Bernard, S.; Lucas, R.; Kroll, P.; Ushakov, S.; Navrotsky, A.; Riedel, R. Ultrahigh temperature ceramics (UHTCs) and related materials—Syntheses from polymeric precursors and energetics. Adv. Eng. Mater. 2019, 21, 1900269–1900292. [Google Scholar] [CrossRef]
- Wen, Q.; Yu, Z.; Riedel, R. The fate and role of in situ formed carbon in polymer-derived ceramics. Prog. Mater. Sci. 2020, 109, 100623–100685. [Google Scholar] [CrossRef]
- Lale, A.; Schmidt, M.; Mallmann, M.D.; Bezerra, A.V.A.; Diz Acosta, E.; Machado, R.A.F.; Demirci, U.B.; Bernard, S. Polymer-derived ceramics with engineered mesoporosity: From design to application in catalysis. Surf. Coat. Technol. 2018, 350, 569–586. [Google Scholar] [CrossRef]
- Viard, A.; Miele, P.; Bernard, S. Polymer-derived ceramics route toward SiCN and SiBCN fibers: From chemistry of polycarbosilazanes to the design and characterization of ceramic fibers. J. Ceram. Soc. Jpn. 2016, 124, 967–980. [Google Scholar] [CrossRef]
- Flores, O.; Bordia, R.K.; Nestler, D.; Krenkel, W.; Motz, G. Ceramic fibers based on SiC and SiCN systems: Current research, development, and commercial status. Adv. Eng. Mater. 2014, 16, 621–636. [Google Scholar] [CrossRef]
- Barroso, G.; Li, Q.; Bordia, R.K.; Motz, G. Polymeric and ceramic silicon-based coatings—A review. J. Mater. Chem. A 2019, 7, 1936–1963. [Google Scholar] [CrossRef]
- Vakifahmetoglu, C.; Zeydanli, D.; Colombo, P. Porous polymer derived ceramics. Mater. Sci. Eng. R 2016, 106, 1–30. [Google Scholar] [CrossRef]
- Hotza, D.; Nishihora, R.; Machado, R.; Geffroy, P.-M.; Chartier, T.; Bernard, S. Tape casting of preceramic polymers towards advanced ceramics: A review. Int. J. Ceramic Eng. Sci. 2019, 1, 21–41. [Google Scholar] [CrossRef]
- Bechelany, M.C.; Proust, V.; Gervais, C.; Ghisleni, R.; Bernard, S.; Miele, P. In-situ controlled growth of titanium nitride in amorphous silicon nitride: A general route toward bulk non-oxide nitride nanocomposites with very high hardness. Adv. Mater. 2014, 26, 6548–6553. [Google Scholar] [CrossRef]
- Bechelany, M.C.; Proust, V.; Lale, A.; Miele, P.; Malo, S.; Gervais, C.; Bernard, S. Nanocomposites through chemistry of single-source precursors: Understanding the role of chemistry behind the design of monolith-type nanostructured titanium nitride/silicon nitride. Chem. Eur. J. 2017, 23, 832–845. [Google Scholar] [CrossRef]
- Lale, A.; Proust, V.; Bechelany, M.C.; Viard, A.; Malo, S.; Bernard, S. A comprehensive study on the influence of the polyorganosilazane chemistry and material shape on the high temperature behavior of titanium nitride/silicon nitride nanocomposites. J. Eur. Ceram Soc. 2017, 37, 5167–5175. [Google Scholar] [CrossRef]
- Yu, Z.; Min, H.; Zhan, J.; Yang, L. Preparation and dielectric properties of polymer-derived SiCTi ceramics. Ceram. Int. 2013, 39, 3999–4007. [Google Scholar] [CrossRef]
- Yu, Z.; Yang, L.; Min, H.; Zhang, P.; Zhou, C.; Riedel, R. Single-source-precursor synthesis of high temperature stable SiC/C/Fe nanocomposites from a processable hyperbranched polyferrocenylcarbosilane with high ceramic yield. J. Mater. Chem. C 2014, 2, 1057–1067. [Google Scholar] [CrossRef]
- Fonblanc, D.; Lopez-Ferber, D.; Wynn, M.; Lale, A.; Soleilhavoup, A.; Leriche, A.; Iwamoto, Y.; Rossignol, F.; Gervais, C.; Bernard, S. Crosslinking chemistry of poly(vinylmethyl-co-methyl)silazanes toward low-temperature formable preceramic polymers as precursors of functional aluminium-modified Si-C-N ceramics. Dalton Trans. 2018, 47, 14580–14593. [Google Scholar] [CrossRef]
- Viard, A.; Fonblanc, D.; Schmidt, M.; Lale, A.; Salameh, C.; Soleilhavoup, A.; Wynn, M.; Champagne, P.; Cerneaux, S.; Babonneau, F.; et al. Molecular chemistry and engineering of boron-modified polyorganosilazanes as new processable and functional SiBCN precursors. Chem. Eur. J. 2017, 23, 9076–9090. [Google Scholar] [CrossRef] [PubMed]
- Gibson, V.C.; Spitzmesser, S.K. Advances in non-metallocene olefin polymerization catalysis. Chem. Rev. 2003, 103, 283–316. [Google Scholar] [CrossRef] [PubMed]
- Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Functionalization of organic molecules by transition-metal-catalyzed C (sp3); H activation. Chem. A Eur. J. 2010, 16, 2654–2672. [Google Scholar] [CrossRef] [PubMed]
- Daugulis, O.; Do, H.-Q.; Shabashov, D. Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds. Acc. Chem. Res. 2009, 42, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Maugel, N.; Li, J.-J.; Wang, D.-H.; Breazzano, S.P.; Saunders, L.B.; Yu, J.-Q. Palladium-catalyzed methylation and arylation of sp2 and sp3 C-H bonds in simple carboxylic acids. J. Am. Chem. Soc. 2007, 129, 3510–3511. [Google Scholar] [CrossRef]
- Schmidt, M.; Durif, C.; Diz Acosta, E.; Salameh, C.; Plaisantin, H.; Miele, P.; Backov, R.; Machado, R.; Gervais, C.; Alauzun, J.G.; et al. Molecular-level processing of Si-(B)-C materials with tailored nano/microstructures. Chem. Eur. J. 2017, 23, 17103–17117. [Google Scholar] [CrossRef]
- Majoulet, O.; Alauzun, J.G.; Gottardo, L.; Gervais, C.; Schuster, M.E.; Bernard, S.; Miele, P. Ordered Mesoporous Silicoboron Carbonitride Ceramics from Boron-Modified Polysilazanes: Polymer Synthesis, Processing and Properties. Micro. Meso. Mater. 2011, 140, 40–50. [Google Scholar] [CrossRef]
- Berger, F.; Müller, A.; Aldinger, F.; Müller, K. Solid-state NMR investigations on Si-B-C-N ceramics derived from boron-modified poly (allylmethylsilazane). Z. Anorg. Allg. Chem. 2005, 631, 355–363. [Google Scholar] [CrossRef]
- Gottardo, L.; Bernard, S.; Gervais, C.; Inzenhofer, K.; Motz, G.; Weinmann, M.; Balan, C.; Miele, P. Chemistry, Structure and Processability of Boron-Modified Polysilazanes as Tailored Precursors of Ceramic Fibers. J. Mater. Chem. 2012, 22, 7739–7750. [Google Scholar] [CrossRef]
- Seitz, J.; Bill, J.; Egger, N.; Aldinger, F. Structural investigations of Si/C/N-ceramics from polysilazane precursors by nuclear magnetic resonance. J. Eur. Ceram. Soc. 1996, 16, 885–891. [Google Scholar] [CrossRef]
- Choong Kwet Yive, N.S.; Corriu, R.J.P.; Leclercq, D.; Mutin, P.H.; Vioux, A. Silicon carbonitride from polymeric precursors: Thermal cross-linking and pyrolysis of oligosilazane model compounds. Chem. Mater. 1992, 4, 141–146. [Google Scholar] [CrossRef]
- Choong Kwet Yive, N.S.; Corriu, R.J.P.; Leclercq, D.; Mutin, P.H.; Vioux, A. Thermogravimetric analysis/mass spectrometry investigation of the thermal conversion of organosilicon precursors into ceramics under argon and ammonia. 2. Poly (silazanes). Chem. Mater. 1992, 4, 1263–1271. [Google Scholar] [CrossRef]
- Allan, J.D.; Delia, A.E.; Coe, H.; Bower, K.N.; Alfarra, M.R.; Jimenez, J.L.; Middlebrook, A.M.; Drewnick, F.; Onasch, T.B.; Canagaratna, M.R.; et al. A generalized method for the extraction of chemically resolved mass spectra from aerodyne aerosol mass spectrometer data. J. Aerosol Sci. 2004, 35, 909–922. [Google Scholar] [CrossRef]
- Lavedrine, A.; Bahloul, D.; Goursat, P.; Choong Kwet Yive, N.S.; Corriu, R.; Leclerq, D.; Mutin, H.; Vioux, A. Pyrolysis of polyvinylsilazane precursors to silicon carbonitride. J. Eur. Ceram. Soc. 1991, 8, 221–227. [Google Scholar] [CrossRef]
- Bahloul, D.; Pereira, M.; Goursat, P.; Choong Kwet Yive, N.S.; Corriu, R.J.P. Preparation of silicon carbonitrides from an organosilicon polymer: I, thermal decomposition of the cross-linked polysilazane. J. Am. Ceram. Soc. 1993, 76, 1156–1162. [Google Scholar] [CrossRef]
- Bahloul, D.; Pereira, M.; Goursat, P. Preparation of silicon carbonitrides from an organosilicon polymer: II, thermal behavior at high temperature under argon. J. Am. Ceram. Soc. 1993, 76, 1163–1168. [Google Scholar] [CrossRef]
- Guilemany, J.; Alcobe, X.; Sanchiz, I. X-Ray Diffraction Analysis of Titanium Carbonitride 30/70 and 70/30 Solid Solutions. Powder Diffr. 1992, 7, 34–35. [Google Scholar] [CrossRef]
- Hering, N.; Schreiber, K.; Riedel, R.; Lichtenberger, O.; Woltersdorf, J. Synthesis of polymeric precursors for the formation of nanocrystalline Ti-C-N/amorphous Si-C-N composites. Appl. Organomet. Chem. 2001, 15, 879–886. [Google Scholar] [CrossRef]
- Cordoba, J.M.; Sayagues, M.J.; Alcalà, M.D.; Gotor, F.J. Synthesis of Titanium Carbonitride Phases by Reactive Milling of the Elemental Mixed Powders. J. Am. Ceram. Soc. 2005, 88, 1760–1764. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | (200) Peak Position (2θ, °) | d200 (nm) | Lattice Parameter a (nm) | Composition of the TiCxNy from XRD |
|---|---|---|---|---|
| P-1400NH | 42.6 | 0.212 | 0.4245 | TiN |
| P-1400N | 42.45 | 0.213 | 0.4264 | TiC0.2N0.8 |
| P-1400A | 42.3 | 0.214 | 0.4273 | TiC0.3N0.7 |
| P-1500NH | 42.6 | 0.212 | 0.4245 | TiN |
| P-1500N | 42.3 | 0.214 | 0.4273 | TiC0.3N0.7 |
| P-1500A | 42.25 | 0.214 | 0.4273 | TiC0.4N0.6 |
| P-1600NH | 42.6 | 0.212 | 0.4245 | TiN |
| P-1600N | 42.2 | 0.214 | 0.4283 | TiC0.4N0.6 |
| P-1600A | 42.1 | 0.215 | 0.4293 | TiC0.6N0.4 |
| P-1800NH | 42.6 | 0.212 | 0.4245 | TiN |
| P-1800N | 42.15 | 0.214 | 0.4288 | TiC0.5N0.5 |
| P-1800A | 41.9 | 0.216 | 0.4312 | TiC0.8N0.2 |
Sample Availability: Samples of the compounds P1000 to P1800 under argon and nitrogen are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balestrat, M.; Lale, A.; Bezerra, A.V.A.; Proust, V.; Awin, E.W.; Machado, R.A.F.; Carles, P.; Kumar, R.; Gervais, C.; Bernard, S. In-Situ Synthesis and Characterization of Nanocomposites in the Si-Ti-N and Si-Ti-C Systems. Molecules 2020, 25, 5236. https://doi.org/10.3390/molecules25225236
Balestrat M, Lale A, Bezerra AVA, Proust V, Awin EW, Machado RAF, Carles P, Kumar R, Gervais C, Bernard S. In-Situ Synthesis and Characterization of Nanocomposites in the Si-Ti-N and Si-Ti-C Systems. Molecules. 2020; 25(22):5236. https://doi.org/10.3390/molecules25225236
Chicago/Turabian StyleBalestrat, Maxime, Abhijeet Lale, André Vinícius Andrade Bezerra, Vanessa Proust, Eranezhuth Wasan Awin, Ricardo Antonio Francisco Machado, Pierre Carles, Ravi Kumar, Christel Gervais, and Samuel Bernard. 2020. "In-Situ Synthesis and Characterization of Nanocomposites in the Si-Ti-N and Si-Ti-C Systems" Molecules 25, no. 22: 5236. https://doi.org/10.3390/molecules25225236
APA StyleBalestrat, M., Lale, A., Bezerra, A. V. A., Proust, V., Awin, E. W., Machado, R. A. F., Carles, P., Kumar, R., Gervais, C., & Bernard, S. (2020). In-Situ Synthesis and Characterization of Nanocomposites in the Si-Ti-N and Si-Ti-C Systems. Molecules, 25(22), 5236. https://doi.org/10.3390/molecules25225236