
Naphthalenes and Quinolines by Domino Reactions of Morita–Baylis–Hillman Acetates



Abstract
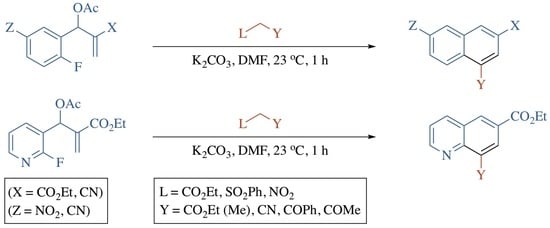
1. Introduction
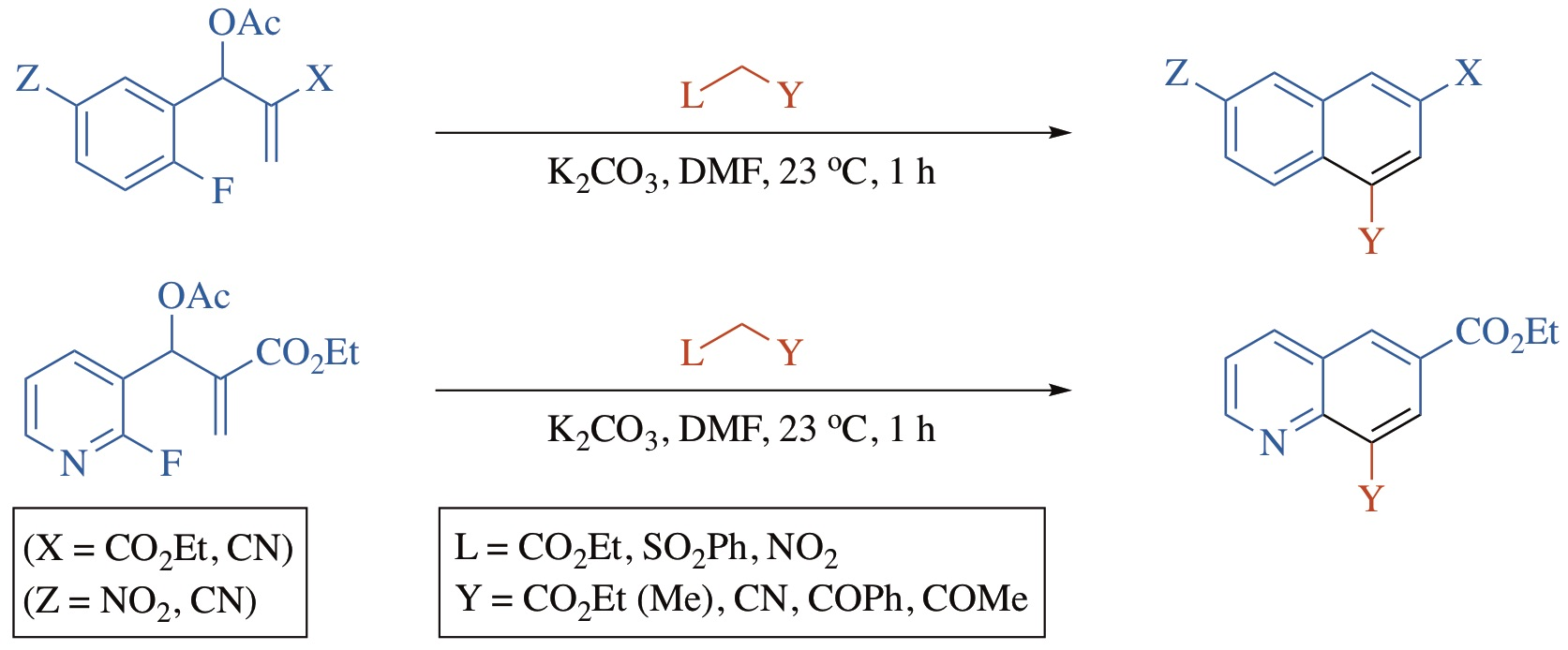
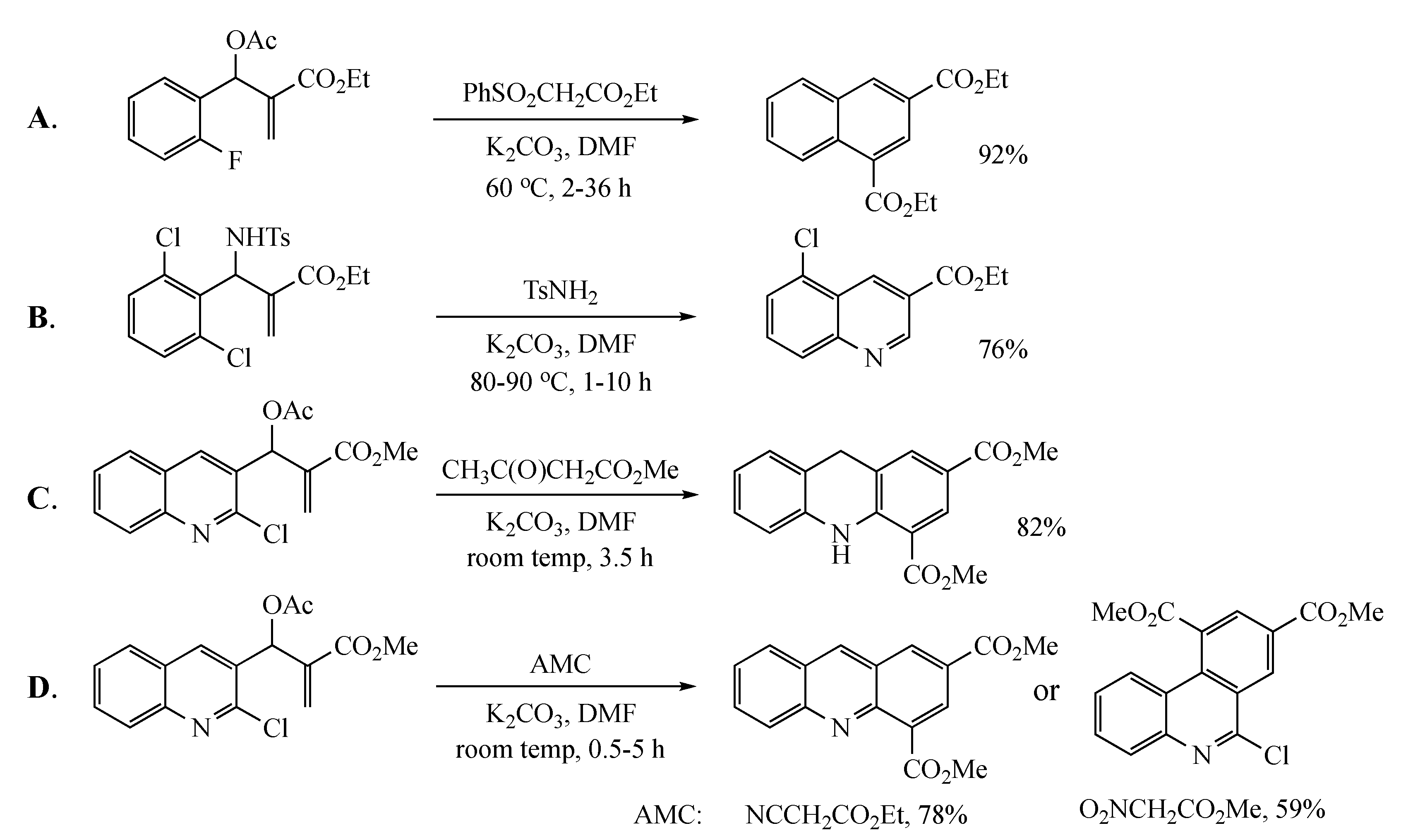
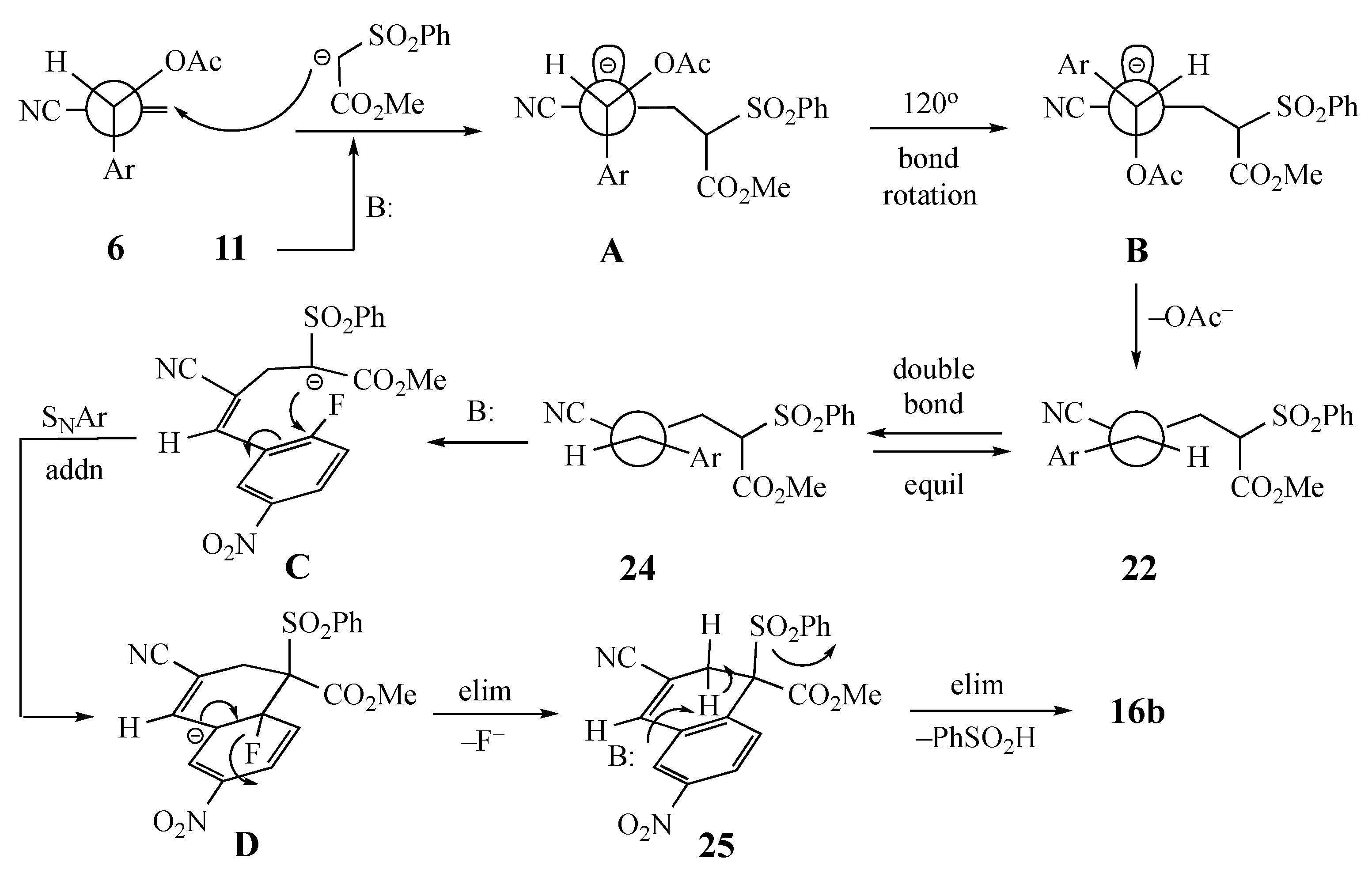
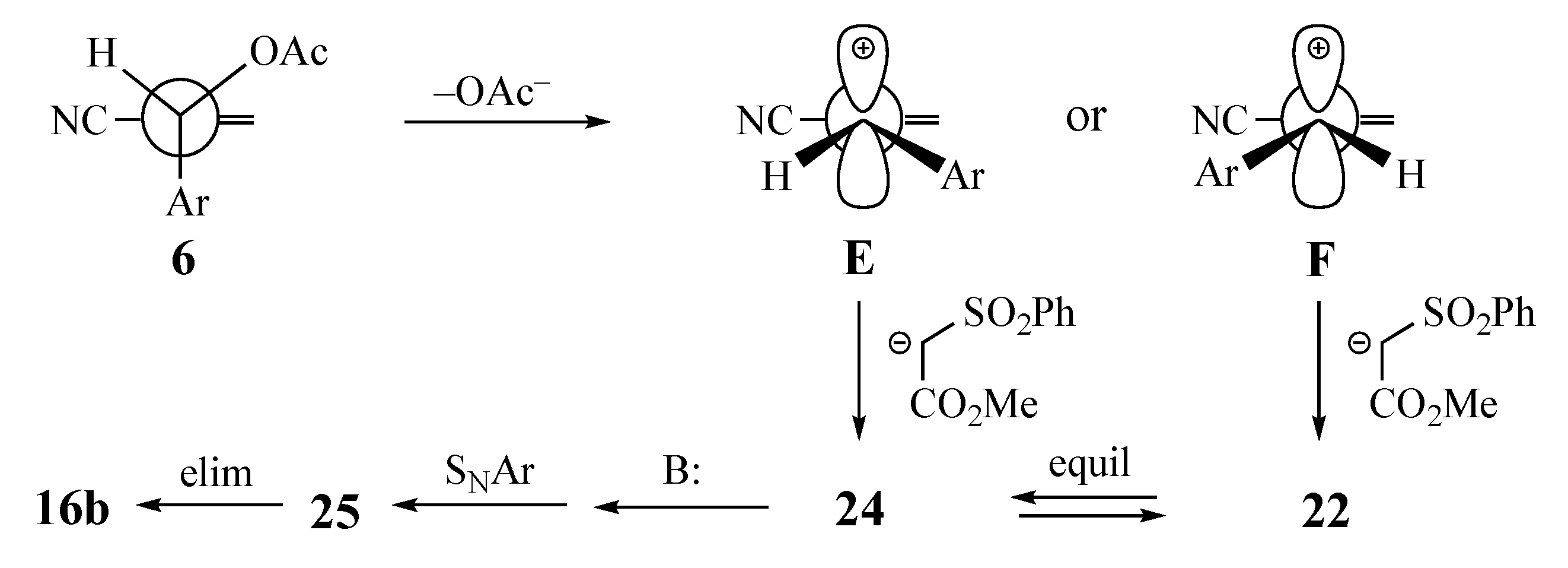
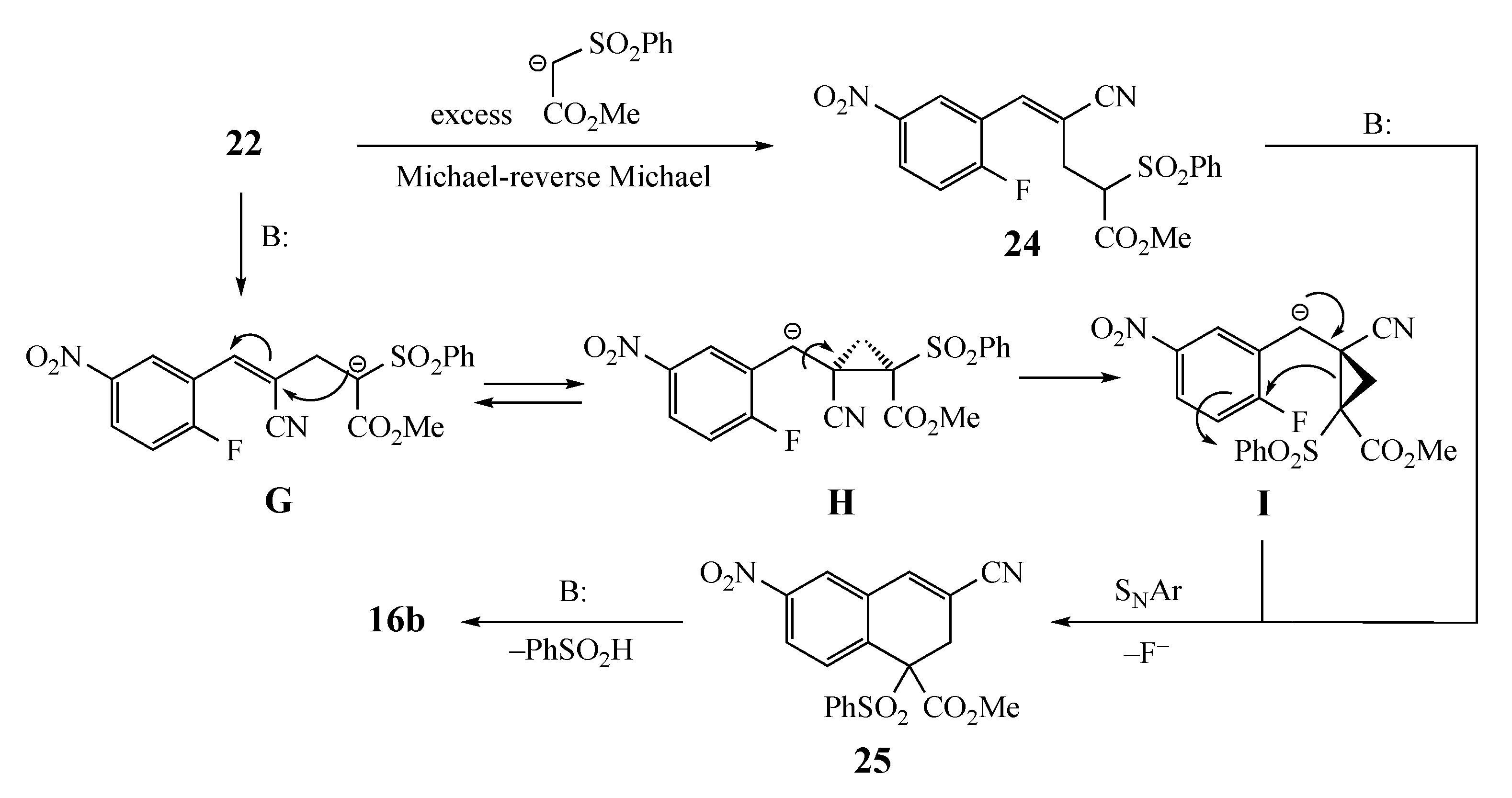
2. Results and Discussion
3. Material and Methods
3.1. General Methods
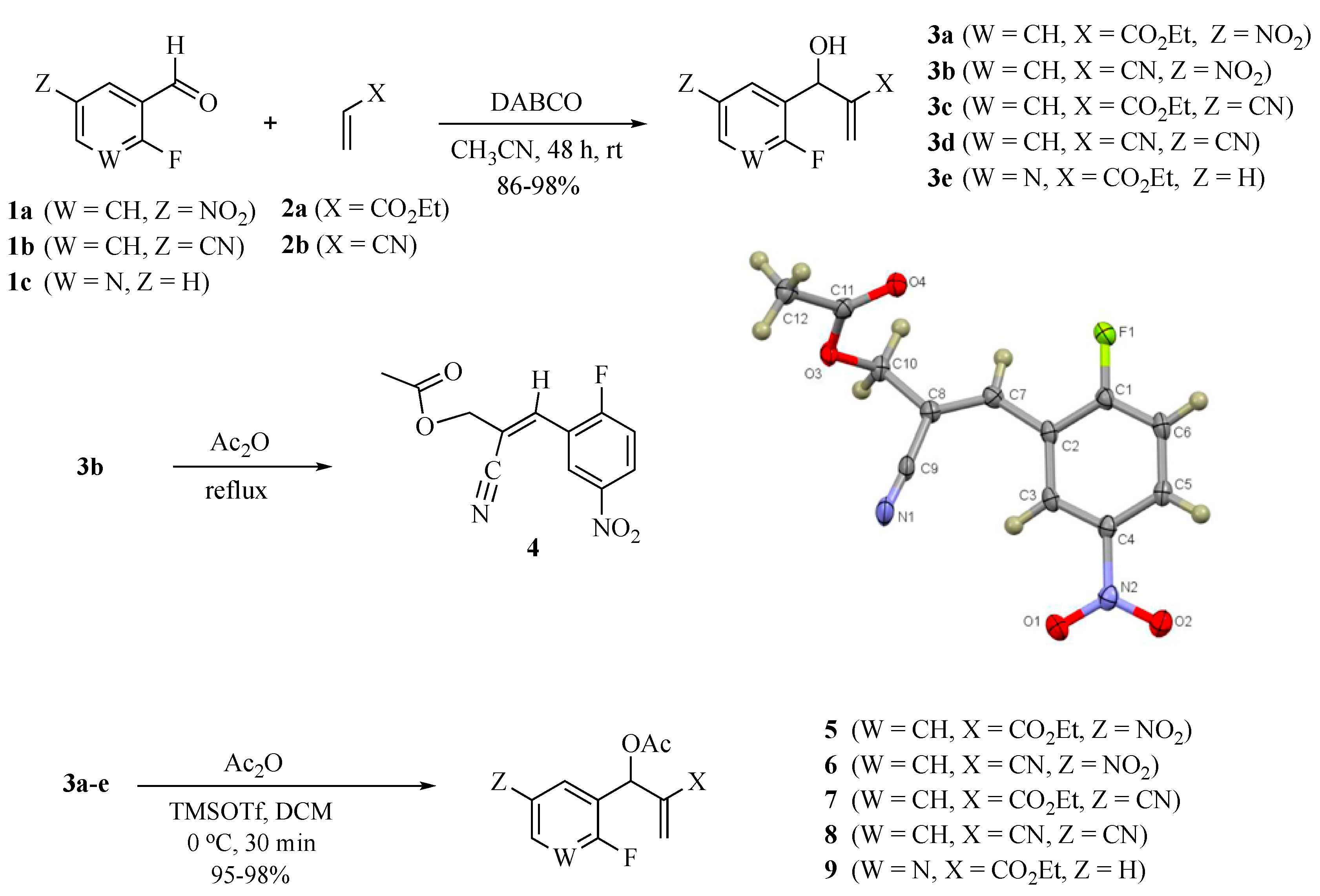
3.2. Representative Procedure for the Synthesis of MBH Alcohols (3a–e)
3.3. Synthesis of (E)-2-cyano-3-(2-fluoro-5-nitrophenyl)allyl acetate (4)
3.4. Representative procedure for the Synthesis of MBH Acetates (5–9)
3.4.1. Ethyl 2-(acetoxy (2-fluoro-5-nitrophenyl)methyl)acrylate (5)
3.4.2. 2-Cyano-1-(2-fluoro-5-nitrophenyl) allyl acetate (6)
3.4.3. Ethyl 2-(acetoxy(5-cyano-2-fluorophenyl)methyl)acrylate (7)
3.4.4. 2-Cyano-1-(5-cyano-2-fluorophenyl)allyl acetate (8)
3.4.5. Ethyl 2-(acetoxy(2-fluoropyridin-3-yl)methyl)acrylate (9)
3.5. Representative Procedure for the Synthesis of Naphthalene and Quinoline Analogs Using MBH Acetates and Active Methylene Compounds
3.5.1. Ethyl 4-cyano-7-nitro-2-naphthoate (15a) from 5 and ethyl cyanoacetate (10)
3.5.2. 3-Ethyl 1-methyl 6-nitronaphthalene-1,3-dicarboxylate (15b) from 5 and methyl phenylsulfonylacetate (11)
3.5.3. Diethyl 6-nitronaphthalene-1,3-dicarboxylate (15c) from 5 and ethyl nitroacetate (12)
3.5.4. Ethyl 4-benzoyl-7-nitro-2-naphthoate (15d) from 5 and 1-phenyl-2-(phenylsulfonyl)ethan-1-one (13)
3.5.5. Ethyl 4-acetyl-7-nitro-2-naphthoate (15e) from 5 and 1-(phenylsulfonyl)propan-2-one (14)
3.5.6. 6-Nitronaphthalene-1,3-dicarbonitrile (16a) from 6 and 10
3.5.7. Methyl 3-cyano-6-nitro-1-naphthoate (16b) from 6 and 11
3.5.8. Ethyl 3-cyano-6-nitro-1-naphthoate (16c) from 6 and 12
3.5.9. 4-Benzoyl-7-nitro-2-naphthonitrile (16d) from 6 and 13
3.5.10. 4-Acetyl-7-nitro-2-naphthonitrile (16e) from 6 and 14
3.5.11. Ethyl 4,7-dicyano-2-naphthoate (17a) from 7 and 10
3.5.12. 3-Ethyl 1-methyl 6-cyanonaphthalene-1,3-dicarboxylate (17b) from 7 and 11
3.5.13. Diethyl 6-cyanonaphthalene-1,3-dicarboxylate (17c) from 7 and 12
3.5.14. Ethyl 4-benzoyl-7-cyano-2-naphthoate (17d) from 7 and 13
3.5.15. Ethyl 4-acetyl-7-cyano-2-naphthoate (17e) from 7 and 14
3.5.16. Naphthalene-1,3,6-tricarbonitrile (18a) from 8 and 10
3.5.17. Methyl 3,6-dicyano-1-naphthoate (18b) from 8 and 11
3.5.18. Ethyl 3,6-dicyano-1-naphthoate (18c) from 8 and 12
3.5.19. 4-Benzoylnaphthalene-2,7-dicarbonitrile (18d) from 8 and 13
3.5.20. 4-Acetylnaphthalene-2,7-dicarbonitrile (18e) from 8 and 14
3.5.21. Ethyl 8-cyanoquinoline-6-carboxylate (19a) from 9 and 10
3.5.22. 6-Ethyl 8-methyl quinoline-6,8-dicarboxylate (19b) from 9 and 11
3.5.23. Ethyl 8-benzoylquinoline-6-carboxylate (19d) from 9 and 13
3.5.24. Ethyl 8-acetylquinoline-6-carboxylate (19e) from 9 and 14
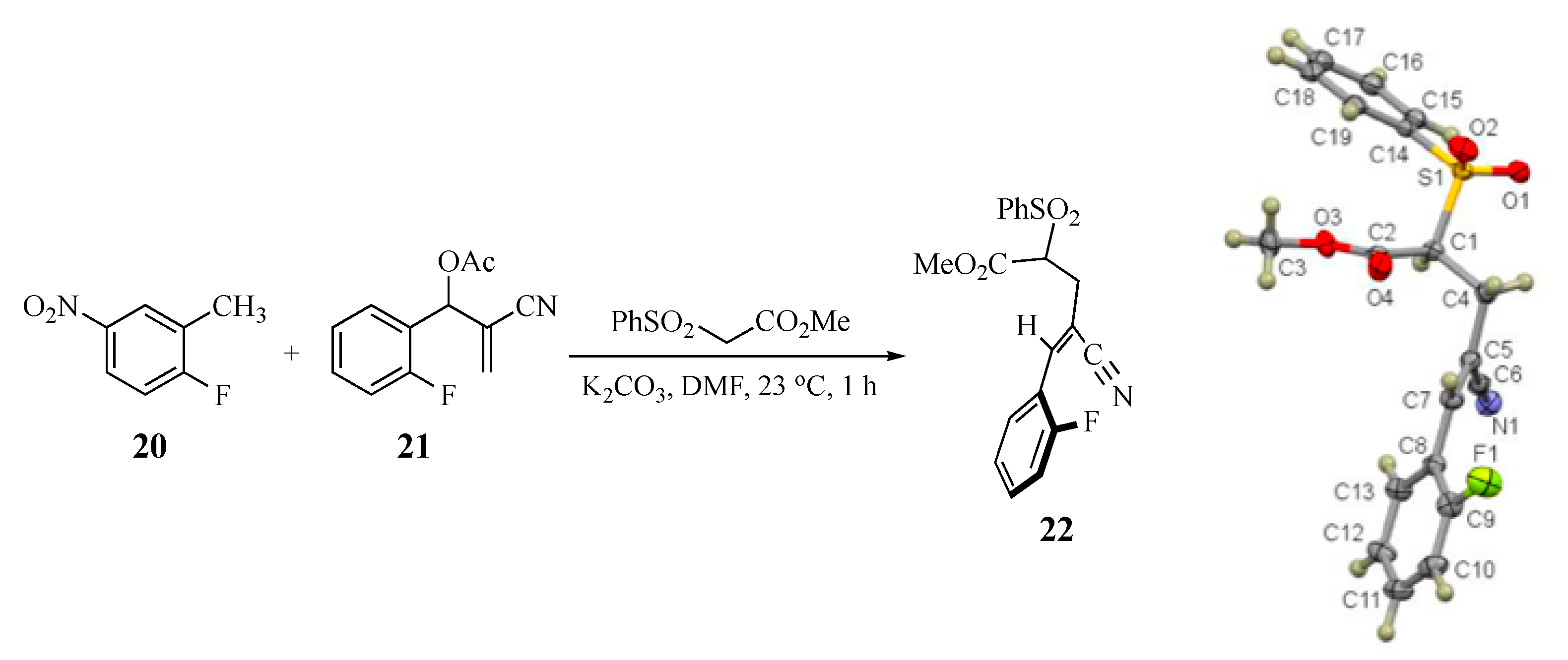
3.6. Competitive Reaction Control Experiment. Formation of Methyl (Z)-4-cyano-5-(2-fluorophenyl)-2-(phenylsulfonyl)-4-pentenoate (22)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Narendar Reddy, T.; Jayathirtha Rao, V. Importance of Baylis-Hillman adducts in modern drug discovery. Tetrahedron Lett. 2018, 59, 2859–2875. [Google Scholar] [CrossRef]
- Lima-Junior, C.G.; Vasconcellos, M.A.A. Morita Baylis-Hillman adducts: Biological activities and potentialities to the discovery of new cheaper drugs. Bioog. Med. Chem. 2012, 20, 3954–3971. [Google Scholar] [CrossRef] [PubMed]
- Makar, S.; Saha, T.; Singh, S.K. Naphthalene, a versatile platform in medicinal chemistry: Sky high perspective. Eur. J. Med. Chem. 2019, 161, 252–276. [Google Scholar] [CrossRef] [PubMed]
- Ofzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Beena; Rawat, D.S. Antituberculosis drug research: A critical review. Med. Res. Rev. 2013, 33, 693–764. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Chung, Y.M.; Gong, J.H.; Kim, J.N. Synthesis of naphthalenes from the reaction of Baylis-Hillman acetates and sulfonyl group-containing active methylene compounds. Bull. Korean Chem. Soc. 2002, 23, 787–788. [Google Scholar] [CrossRef][Green Version]
- Kim, J.N.; Lee, H.J.; Lee, K.Y.; Kim, H.S. Synthesis of 3-quinolinecarboxylic acid esters from the Baylis-Hillman adducts of 2-halobenzaldehyde N-tosylimines. Tetrahedron Lett. 2001, 42, 3737–3740. [Google Scholar] [CrossRef]
- Gupta, T.; Bharadwaj, K.C.; Singh, R.M. Cascade SN2’-SNAr, Elimination and 1,5-hydride shift reactions by acetylacetone/acetoacetic esters: Synthesis of 9,10-dihydroacridines. Eur. J. Org. Chem. 2016, 4981–4984. [Google Scholar] [CrossRef]
- Gupta, T.; Singh, J.B.; Mishra, K.; Singh, R.M. Active methylene compounds (AMCs)controlled facile synthesis of acridine and phenanthridine from morita Baylis-Hillman acetate. RSC Adv. 2017, 7, 54581–54585. [Google Scholar] [CrossRef]
- Morita, K.; Suzuki, Z.; Hirose, H. A tertiary phosphine-catalyzed reaction of acrylic compounds with aldehydes. Bull. Soc. Chem. Jpn. 1968, 41, 2815. [Google Scholar] [CrossRef]
- Baylis, A.B.; Hillman, M.E.D. Acrylic Compounds. German Patent 2155113, 1972. [Google Scholar]
- Hillman, M.E.D.; Baylis, A.B. The reaction of acrylic type compounds with aldehydes and certain ketones. U.S. Patent 3,743,669, 3 July 1973. [Google Scholar]
- Kürti, L.; Czakó, B. Strategic Applications of Named Reaction in Organic Chemistry; Elsevier: New York, NY, USA, 2005; pp. 48–49. [Google Scholar]
- Singh, B.; Chandra, A.; Singh, R.H. Base-free amination of BH acetates of 2-chloroquinolinyl-3- carboxaldehydes: A facile route to the synthesis of N-substituted-1,2-dihydrobenzo[b][1.8]naphthyridines. Tetrahedron 2011, 67, 2441–2446. [Google Scholar] [CrossRef]
- Procopiou, P.A.; Baugh, S.P.D.; Flack, S.S.; Inglis, G.G.A. An extremely powerful acylation reaction of alcohols with acid anhydrides catalyzed by trimethylsilyl trifluoromethanesulfonate. J. Org. Chem. 1998, 63, 2342–2347. [Google Scholar] [CrossRef]
- Love, B.E.; Raje, P.S. Preparation of 1-aryl-carbolines. J. Org. Chem. 1994, 59, 3219–3222. [Google Scholar] [CrossRef]
- Paredes, E.; Biolatta, B.; Kneetemen, M.; Mancini, P. One-step synthesis of 2,9-disubstituted phenanthrenes via Diels-Alder reactions using 1,4-disubstituted naphthalenes as dienophiles. Tetrahedron Lett. 2002, 43, 4601–4603. [Google Scholar] [CrossRef]
- Schenck, L.W.; Kuna, S.K.; Frank, W.; Alber, A.; Kucklaender, U. Dialkyl quinone-2,3-dicarboxylates in the Nenitzescu reaction. Tetrahedron 2005, 61, 9129–9139. [Google Scholar] [CrossRef]
- Kucukdisli, M.; Ferenc, D.; Heinz, M.; Wiebe, C.; Opatz, T. Simple two-step synthesis of 2,4-disubstituted pyrroles and 3,5-disubstituted pyrrole-2-carbonitriles from enones. BeilsteinJ. Org. Chem. 2014, 10, 466–470. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Fang, J.-M.; Wang, Y.-K.; Wang, M.-H.; Ko, T.-Y.; Cherng, Y.-J. Addition reactions of 2-amino-4-methoxypenta-2,4-dienenitrile with electrophiles containing electron-deficient multiple bonds. J. Chem. Soc., Perkin Trans. 1992, 1, 1209–1213. [Google Scholar] [CrossRef]
- Buchholz, R.; Hoffmann, H.M.R. α-Methylidene and α-Alkylidene-β-lactams from nonproteinogenic amino acids. Helv. Chim. Acta 1991, 74, 1213–1220. [Google Scholar] [CrossRef]
- Stohrer, W.-D. On the stereochemistry of the SN2’ reaction. Angew. Chem. Int. Ed. 1983, 22, 613–614. [Google Scholar] [CrossRef]
- Magid, R.M.; Fruchey, O.S. Stereochemistry of the SN2’ reaction of an acyclic allylic chloride with a secondary amine. J. Am. Chem. Soc. 1977, 99, 8368–8369. [Google Scholar] [CrossRef]
- Oritani, T.; Overton, K.H. Stereochemistry of the SN2’ reaction with acyclic allylic esters. J. Chem. Soc. Chem. Commun. 1978, 454–455. [Google Scholar] [CrossRef]
- Magid, R.M. Nucleophilic and organometallic displacement reactions of allylic compounds: Stereo- and regiochemistry. Tetrahedron 1980, 36, 1901–1930. [Google Scholar] [CrossRef]
- Stork, G.; Kreft III, A.F. Concerning the stereochemistry of the SN2’ reaction. “Concerted” allylic displacement in an acyclic system: Anti displacement with thiolate anion. J. Am. Chem. Soc. 1977, 99, 3851–3852. [Google Scholar] [CrossRef]
- Bach, R.D.; Wolber, G.J. Stereochemistry of the concerted SN2’ reaction of 3-chloropropene: A theoretical study. J. Am. Chem. Soc. 1985, 107, 1352–1357. [Google Scholar] [CrossRef]
- Bergman, E.D.; Ginsburg, D.; Pappo, R. The Michael Reaction. Org. React. 1959, 10, 179–555. [Google Scholar]
- Werner, R.M.; Williams, L.M.; Davis, J.T. The C-glycosyl analog of an N-linked glycoamino acid. Tetrahedron Lett. 1998, 39, 9135–9138. [Google Scholar] [CrossRef]
- Shao, H.; Ekthawatchai, S.; Chen, C.-S.; Wu, S.-H.; Zou, W. 1,2-Migration of 2’-oxoalkyl group and concomitant synthesis of 2-C-branched O-, S-glycosides and glycosyl azides via 1,2-cyclopropanated sugars. J. Org. Chem. 2005, 70, 4726–4734. [Google Scholar] [CrossRef]
- Henderson, A.P.; Bleasdale, C.; Delaney, K.; Lindstrom, A.B.; Rappaport, S.M.; Waidyanatha, S.; Watson, W.P.; Golding, B.T. Evidence for the formation of Michael adducts from reactions of [E,E]-muconaldehyde with glutathione and other thiols. Bioorg. Chem. 2005, 33, 363–373. [Google Scholar] [CrossRef]
- Hintermann, L.; Dittmer, C. Asymmetric ion-pairing catalysis of the reversible cyclization of 2’-hydroxychalcone to flavanone: Asymmetric catalysis of an equilibrating reaction. Eur. J. Org. Chem. 2012, 5573–5584. [Google Scholar] [CrossRef]
- McKenna, E.G.; Walker, B.J. Wittig reactions of ylide anions derived from stabilized ylides. J. Chem. Soc. Chem. Commun. 1989, 568–569. [Google Scholar] [CrossRef]
- Hoffmann, H.M.R.; Rabe, J. DABCO-catalyzed coupling of aldehydes with activated double bonds. 4. Stereoselective synthesis of trisubstituted olefins and terpenoid building blocks via 2-(hydroxyalkyl)-2-propenoic esters. J. Org. Chem. 1985, 50, 3849–3859. [Google Scholar] [CrossRef]
- Ameer, F.; Drewes, S.E.; Houston-McMillan, M.S.; Kaye, P.T. Necic acid synthons. Part 4. Regioselectivity in the reactions of chloro and iodo derivatives of selected 3-hydroxy-2-methylene-alkanoate esters with ethyl 3-methyl-3-oxobutanoate. J. Chem. Soc. Perkin Trans. 1985, 1, 1143–1145. [Google Scholar] [CrossRef]
- Basavaiah, D.; Pandiaraju, S.; Padmaja, K. The Friedel-Crafts chemistry: Acetates of the Baylis-Hillman adducts as novel stereodefined-electrophiles. Synlett 1996, 393–395. [Google Scholar] [CrossRef]
- Chavan, S.P.; Ethiraj, K.S.; Kamat, S.K. Facile synthesis of 2Z-2-chloromethyl aryl-2-enoates. Tetrahedron Lett. 1997, 38, 7415–7416. [Google Scholar] [CrossRef]
- Yadav, J.S.; Subba Reddy, B.V.; Madan, C. Montmorillonite clay-catalyzed stereoselective synthesis of aryl-substituted (E)- and (Z)-allyl iodides and bromides. New J. Chem. 2001, 25, 1114–1117. [Google Scholar] [CrossRef]
- Krishna, P.R.; Kannan, V.; Sharma, G.V.M. FeCl3 and Yb(OTf)3 mediated conversion of acetates of the Baylis-Hillman adducts into (Z) and (E) trisubstituted alkenes. Synth. Commun. 2004, 34, 55–64. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, Y. Direct and highly efficient synthesis of (Z)-allyl iodides from Baylis-Hillman adducts promoted by TMSCl/NaI system. Synlett 2005, 1039–1041. [Google Scholar] [CrossRef]
- Gohain, M.; Lin, S.; Bezuidenhoudt, B.C.B. Al(OTf)3-catalyzed SN2’ substitution of the-hydroxy group in Morita-Baylis-Hillman adducts with indoles. Tetrahedron Lett. 2015, 56, 2579–2582. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Substrate | AMC a | L b | X | Y | Z | Pdt (%Yield) |
|---|---|---|---|---|---|---|
 5 | 10 | CO2Et | CO2Et | CN | NO2 | 15a (90) |
| 11 | SO2Ph | CO2Et | CO2Me | NO2 | 15b (91) | |
| 12 | NO2 | CO2Et | CO2Et | NO2 | 15c (94) | |
| 13 | SO2Ph | CO2Et | COPh | NO2 | 15d (96) | |
| 14 | SO2Ph | CO2Et | COMe | NO2 | 15e (92) | |
 6 | 10 | CO2Et | CN | CN | NO2 | 16a (80) |
| 11 | SO2Ph | CN | CO2Me | NO2 | 16b (86) | |
| 12 | NO2 | CN | CO2Et | NO2 | 16c (88) | |
| 13 | SO2Ph | CN | COPh | NO2 | 16d (90) | |
| 14 | SO2Ph | CN | COMe | NO2 | 16e (88) | |
 7 | 10 | CO2Et | CO2Et | CN | CN | 17a (80) |
| 11 | SO2Ph | CO2Et | CO2Me | CN | 17b (82) | |
| 12 | NO2 | CO2Et | CO2Et | CN | 17c (83) | |
| 13 | SO2Ph | CO2Et | COPh | CN | 17d (85) | |
| 14 | SO2Ph | CO2Et | COMe | CN | 17e (84) | |
 8 | 10 | CO2Et | CN | CN | CN | 18a (75) |
| 11 | SO2Ph | CN | CO2Me | CN | 18b (78) | |
| 12 | NO2 | CN | CO2Et | CN | 18c (80) | |
| 13 | SO2Ph | CN | COPh | CN | 18d (80) | |
| 14 | SO2Ph | CN | COMe | CN | 18e (79) |

| Substrate | AMC a | L b | Y | Pdt (%Yield) |
|---|---|---|---|---|
 9 | 10 | CO2Et | CN | 19a (75) |
| 11 | SO2Ph | CO2Me | 19b (82) | |
| 12 | NO2 | CO2Et | ND c | |
| 13 | SO2Ph | COPh | 19d (80) | |
| 14 | SO2Ph | COMe | 19e (76) |
Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Annor-Gyamfi, J.K.; Ametsetor, E.; Meraz, K.; Bunce, R.A. Naphthalenes and Quinolines by Domino Reactions of Morita–Baylis–Hillman Acetates. Molecules 2020, 25, 5168. https://doi.org/10.3390/molecules25215168
Annor-Gyamfi JK, Ametsetor E, Meraz K, Bunce RA. Naphthalenes and Quinolines by Domino Reactions of Morita–Baylis–Hillman Acetates. Molecules. 2020; 25(21):5168. https://doi.org/10.3390/molecules25215168
Chicago/Turabian StyleAnnor-Gyamfi, Joel K., Ebenezer Ametsetor, Kevin Meraz, and Richard A. Bunce. 2020. "Naphthalenes and Quinolines by Domino Reactions of Morita–Baylis–Hillman Acetates" Molecules 25, no. 21: 5168. https://doi.org/10.3390/molecules25215168
APA StyleAnnor-Gyamfi, J. K., Ametsetor, E., Meraz, K., & Bunce, R. A. (2020). Naphthalenes and Quinolines by Domino Reactions of Morita–Baylis–Hillman Acetates. Molecules, 25(21), 5168. https://doi.org/10.3390/molecules25215168