Abstract
A library of amine, oxime, ether, epoxy and acyl derivatives of the benzobicyclo[3.2.1]octene were synthesized and evaluated as inhibitors of both human acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). The majority of the tested compounds exhibited higher selectivity for BChE. Structural adjustment for AChE seems to have been achieved by acylation, and the furan ring opening of furo-benzobicyclo[3.2.1]octadiene results for compound 51 with the highest AChE affinity (IC50 = 8.3 µM). Interestingly, its analogue, an oxime ether with a benzobicyclo[3.2.1]-skeleton, compound 32 was one of the most potent BChE inhibitors in this study (IC50 = 31 µM), but not as potent as endo-43, an ether derivative of the benzobicyclo[3.2.1]octene with an additional phenyl substituent (IC50 = 17 µM). Therefore, we identified several cholinesterase inhibitors with a potential for further development as potential drugs for the treatment of neurodegenerative diseases.
1. Introduction
Acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8), as they control the acetylcholine ambient in the synapsis, are pharmacologically relevant enzyme targets in neurodegenerative disorders [1,2]. AChE has an essential physiological role in the body as it controls the transmission of nerve impulses in the cholinergic synapses of the central and peripheral nervous system by hydrolysis of the neurotransmitter acetylcholine. Loss of neurons results in a significant reduction in AChE and an increase in BChE levels in the brain [3,4]. BChE, even though it is not physiologically essential, serves as a co-regulator of cholinergic neurotransmission capable of catalyzing the hydrolysis of acetylcholine [5]. Except for cholinergic activity, cholinesterases are associated with amyloid plaques and neurofibrillary tangles which together with the aggregation of toxic amyloid-β peptide lead to the dysfunction and apoptosis of neurons [6,7,8,9,10]. Therefore, compounds acting as inhibitors of cholinesterase could be considered as potential drugs in Alzheimer’s disease (AD) treatment, other neurological disorders and therapeutics [11,12,13,14,15]. Due to the composition of their active site, two enzymes can differ in selectivity and specificity for inhibitor binding [16,17,18]. Moreover, it has been well-documented that BChE reacts with a wider range of ligands than AChE and is less selective [19,20,21,22,23,24,25,26].
Recently we prepared photoproducts, shown in Figure 1, and probed the cholinesterase inhibition [27,28]. Compounds endo-1 and endo-2 were the most potent inhibitors of AChE (IC50 = 17.5 µM) and BChE (IC50 = 8.8 µM), respectively, among the tested compounds. All of the tested photoproducts exhibited a binding preference for BChE. In comparison to huperzine A, a selective BChE inhibitor (with similar basic methano-bridged aryl-bicyclo[3.3.1]octadiene skeleton), compound endo-2 was about five times more potent as a BChE inhibitor [10].
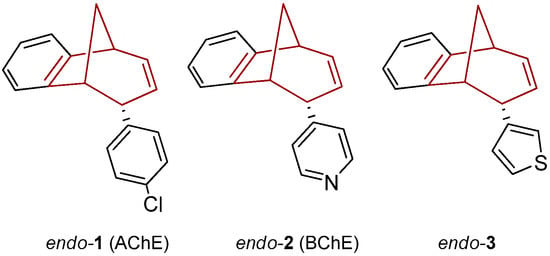
Figure 1.
Photoproducts as potent inhibitors of BChE and AChE [24].
The present study focused on the additional functionalization of the primary photoproducts to obtain new amines, epoxides, alcohols and ethers (Figure 2A,B), or to find new oximes, oxime ethers or acyl derivatives (Figure 2C,D). The mandatory postulate was high yield/productivity in the photochemical reaction. That strategy resulted in a wide range of related compounds, four groups of molecules with different functionalities and a common methano-bridged benzobicyclo[3.2.1]octadiene skeleton found in huperzine A, many biologically active natural sesquiterpenoids [29] or similar to bicyclo[3.2.1]octane benzylamines proven as potential neurokinin–1 antagonist drugs [30]. Compounds were evaluated as inhibitors of both human AChE and BChE.
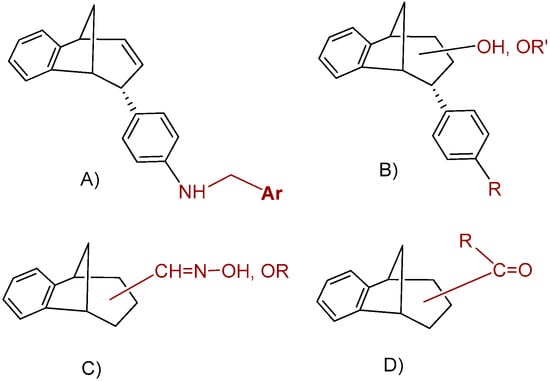
Figure 2.
Functionalization of the benzobicyclo[3.2.1]octadiene skeleton employed in this study: (A) amines; (B) epoxides, alcohols and ethers; (C) oximes and oxime ethers; and (D) acyl derivatives.
2. Results and Discussion
2.1. Benzylic Amines with the Benzobicyclo[3.2.1]-Skeleton
Chloro-substituted endo-1 (Scheme 1) was chosen as a suitable substrate for the Buchwald−Hartwig amination reaction. Compound endo-1 was obtained in one photochemical step according to the described method from the previously prepared o-vinylphenyl substituted butadienes [31]. The Buchwald−Hartwig amination successfully gave amino substituted benzobicyclo[3.2.1]octadienes endo-2–18 (Scheme 1) at 21–88% yields [32].
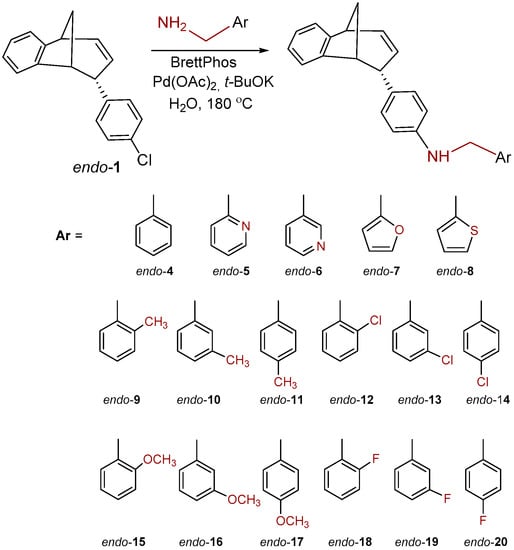
Scheme 1.
Synthesis of benzobicyclo[3.2.1]octadiene benzylamines, endo-4–20.
The benzylamines obtained in good yields were tested as potential cholinesterase inhibitors as well as 1, 9, 17 and 20, rearranged products obtained during amination of endo-1 with different benzylamines (Figure 3).
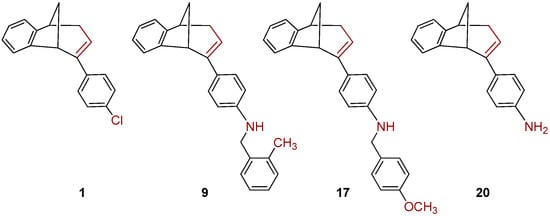
Figure 3.
The rearranged products 1, 9, 17 and 20 obtained during amination of endo-1.
We tested 18 compounds described above as reversible inhibitors of human AChE and BChE (Table 1), but it seems that benzobicyclo[3.2.1]octadiene benzylamines were poor inhibitors of both cholinesterases. The exception was endo-10, a compound with methyl on the benzylamine substituent in meta position, which seems important for the inhibition and binding interactions between BChE and benzylamine. The rearranged products (1, 9, 17 and 20; Figure 3) were more potent BChE inhibitors than endo-benzylamines of the bicyclo[3.2.1]octane skeleton. However, none of the compounds (≤100 µM) showed potency to inhibit AChE. Unfortunately, in the case of AChE, due to the solvent interference, we could not use a higher concentration.

Table 1.
Inhibition of BChE by benzobicyclo[3.2.1]octadiene benzylamines expressed as IC50 ± SEM.
2.2. Oxime Derivatives Possessing Benzobicyclo[3.2.1]-Skeleton
Further functionalization of the basic skeleton (Scheme 2) was performed according to the synthesis of the formyl derivative [33], and a previously published study that combined docking and density functional theory [34]. From the formyl derivative 21, the corresponding bicyclic oxime 22 was synthesized as a crucial substrate for further synthesis of oxime ethers 22′. Along with the expected oxime 22 and oxime ethers 26–30, due to opening of the furan ring two classes were formed—opened oximes 23–25 and oxime ethers 31–35 (Figure 4).
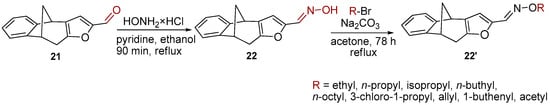
Scheme 2.
Synthetic pathway toward a new oxime and oxime ethers 22 and 22′.
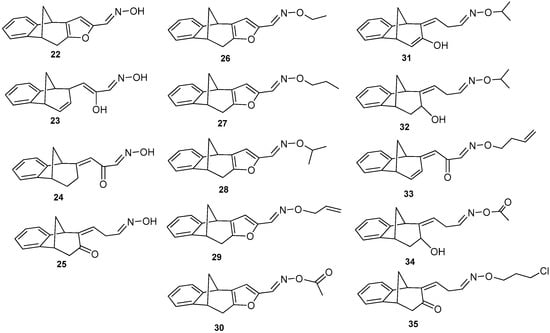
Figure 4.
Obtained oximes 22–25 and oxime ethers 26–35.
Oxime derivatives possessing a benzobicyclo[3.2.1]-skeleton (Figure 4) were prepared at sufficient amounts and were tested as cholinesterase inhibitors. According to the results given in Table 2, three different functionalities of compounds including oximes, oxime ethers with/without furan ring seem highly related to inhibition potency. The lead inhibitors could be detected in each group, oxime 22 (IC50 = 25 ± 6 µM), oxime ether 28 with isopropyl group and furan ring (IC50 = 24 ± 3 µM) and oxime ether 32 with an opened furan ring and isopropyl functionality (IC50 = 31 ± 4 µM). As for the presence of the furan ring, it seems important only for the inhibition of BChE with oxime derivatives 22, 23, 24, and 25. In other words, IC50 for the opened oxime ether compounds was in the same range like IC50 for furan-containing oxime ethers, as shown for compounds 28 and 32. However, IC50 decreased for compounds with more saturated bicyclic moiety, e.g., octadiene 31 was a weaker inhibitor than octene analogue 32. Compounds 27 and 29 differ only in the saturation of their chain substituent and again propenyl derivative 29 inhibited slightly less potently than its propyl analogue 27. Moreover, out of opened oxime ether derivatives (compounds 31–35), butenyl derivative 33 was the less potent inhibitor. Therefore, the saturation of a compound can maybe play a crucial role for their beneficial interaction inside of the BChE active gorge. In case of AChE, none of the compounds were able to inhibit more than 20% of enzyme with 200 µM concentration.
Table 2.
Inhibition of BChE by oximes and oxime ethers with benzobicyclo[3.2.1]-skeleton expressed as IC50 ± SEM.
2.3. Epoxides, Alcohols and Ethers with Benzobicyclo[3.2.1]-Skeleton
Further functionalization of the benzobicyclo[3.2.1]octadiene skeleton was made through the synthesis of novel epoxy derivatives, suitable substrates for novel alcohols and corresponding ethers. It is worth mentioning that functionalization utilized the addition reaction to the free double bond on the basic methano-bridged bicyclic skeleton. As it is known, epoxides represent three-membered cyclic ethers of high reactivity and one way of synthesizing them is by epoxidation using peroxy acids, which have an electrophilic oxygen atom, in a suitable solvent. This reaction represents a stereospecific syn-addition to the double bond [35,36,37]. In this study, the reaction pathway to novel epoxides, alcohols and ethers with the specific geometry (Scheme 3) began by obtaining corresponding epoxides of photoproducts endo-36 and endo-37, and via alcohols the pathway led to desired ethers with a benzobicyclo[3.2.1]octadiene structure. More specifically, the reaction of endo-36 and endo-37 with meta-chloroperbenzoic acid (m-CPBA) in dichloromethane (DCM) as solvent gave the corresponding epoxides endo-38 (61%) and endo-39 (50%). Although two stereoisomers could be obtained regarding the oxygen position at the three-membered ring relative to the plane of the larger ring in the bridged bicyclic system (Figure 5), we were unable to determine the exact stereochemistry of endo-38 and endo-39 by spectroscopic analysis (see experimental and SI, Figures S1–S19). Therefore, the stereochemistry was predicted by DFT calculations (See SI).
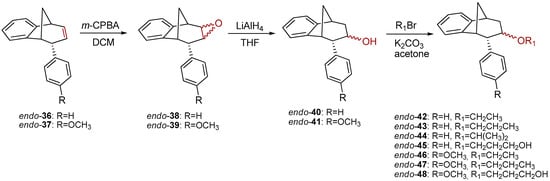
Scheme 3.
Synthetic path to novel epoxides, alcohols and ethers.
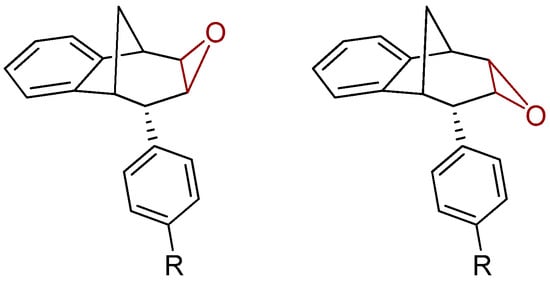
Figure 5.
Representation of the stereoisomers of the obtained epoxides endo-38 (R = H) and endo-39 (R = OCH3).
The ring opening was carried out by reacting epoxides endo-38 and endo-39 and LiAlH4 in tetrahydrofuran (THF; Scheme 3) as it is known that epoxides due to tension of the three-membered ring are highly reactive. The resulting products of the reaction with reducing agent were alcohols that can theoretically have four isomers: two regioisomers, and within them two more stereoisomers. According to the literature, the most likely isomer, resulting from the epoxide opening product, is a regioisomer with an hydroxy (OH) group on the middle carbon atom of the larger ring in the bridged bicycle system [38]. Finally, the conversion of alcohol into different ethers was carried out by a simple reaction of the alcohol with corresponding alkyl bromides and potassium carbonate in acetone (Scheme 3). The isolated products (20%–50% yields) were characterized by spectroscopic methods (see experimental and SI, Figures S1–S19).
Results on the biological activity of obtained epoxides, alcohols and ethers with a benzobicyclo[3.2.1]-skeleton in Table 3 show higher efficiency in the inhibition of both AChE and BChE than their counterparts. Epoxide endo-38 as a highly reactive compound has proven to be a potent inhibitor for AChE even though its potency was still 3-fold lower than for BChE. An opening of the epoxide ring resulted in derivatives with higher selectivity of BChE, but the IC50 was only possible to evaluate for ether, endo-43, or alcohol, endo-45, compounds without a substituent on the endo-oriented benzene ring. It seems that a methoxy group in the para position on the phenyl substituent (endo-46 and endo-47) disturbed beneficial interactions for inhibition by a methoxy group as an electron-donating group. Interestingly, a study by Mohammadi-Farani et al. with similar compounds containing a methoxy group in the meta position on the phenyl ring, reported an increase of the inhibition potency for AChE [39]. Therefore, in the case of endo-46 and endo-47, their para-methoxy group probably could not achieve effective stabilization in the active site.
Table 3.
Inhibition of human AChE and BChE by epoxides, alcohols and ethers possessing benzobicyclo[3.2.1]-skeleton expressed as IC50 ± SEM.
The Modelling of an Epoxide Ring Opening
To predict with more certainty which of the four possible alcohol isomers is dominant among the products, we computationally modelled the reaction of an epoxide opening in molecule endo-38. Calculations were performed at the (SMD)M06-2X/6-31G(d) level of theory in tetrahydrofuran as a solvent, using Gaussian09 programme package [40]. The stationary points of reactions were localized at the potential energy surface and vibrational analysis was performed to verify the minima and transition states on the PES for all structures (See SI).
The epoxide opening step occurs upon the nucleophilic attack by a hydride anion at one of two carbons that belong to the epoxide ring: the middle carbon of the larger ring in the bridged bicycle system, denoted as B in Scheme 4, or the neighboring carbon atom denoted as A. A total of four reactions may occur, two starting from structures where the oxygen of epoxide is placed above the plane of the larger ring in the bridged bicycle system:
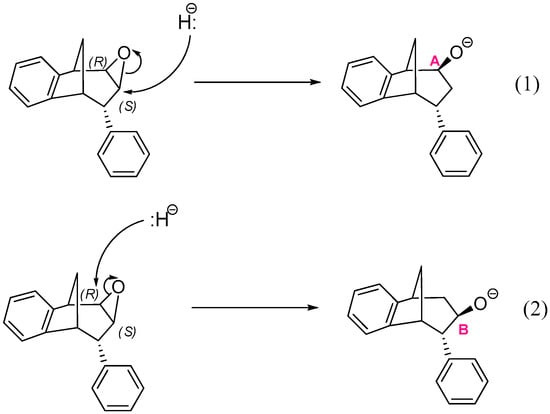
Scheme 4.
Two possible openings of epoxide, with oxygen placed above the plane of larger ring in the bridged bicyclic system.
Another two reactions, starting from the structure where an oxygen atom of epoxide is placed below the plane are also possible (Scheme 5):
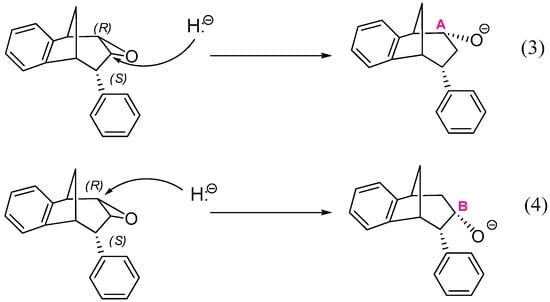
Scheme 5.
Two possible openings of epoxide, with oxygen positioned below the plane.
Reactions (1) and (3) lead to two stereoisomers of regioisomer where the hydroxyl group is placed at the CA atom of the larger ring in the bridged bicycle system, while (2) and (4) give two stereoisomers of an alternative regioisomer, with an OH group at the middle carbon atom of the larger ring (i.e., at CB).
The free energy profile for all four reactions (1)–(4) was calculated and is shown in Figure 6. The barrier for the ring opening was the highest and almost the same for reactions (1) and (2) (dotted lines, 93.0 and 92.7 kJ mol−1) where molecule endo-38 has an oxygen atom of the epoxide ring above the plane (structure (a) in Figure 7). Reaction (4) had the lowest activation energy of 73.0 kJ mol−1, meaning the fastest process among these four reactions was the ring opening which leads to the formation of alcohol with an OH group at the CB and placed below the plane.
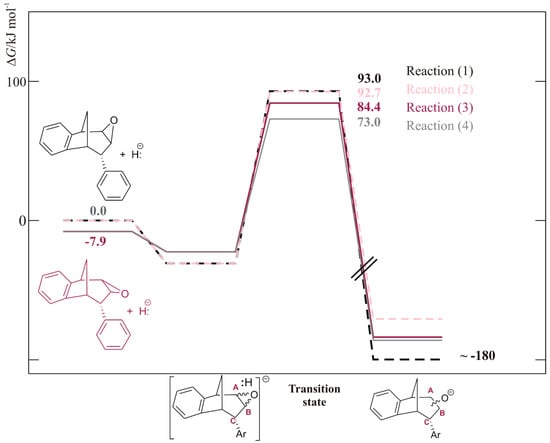
Figure 6.
The free energy profile for reactions (1)–(4), calculated at (SMD)/M06-2X/6-31G(d) level of theory.
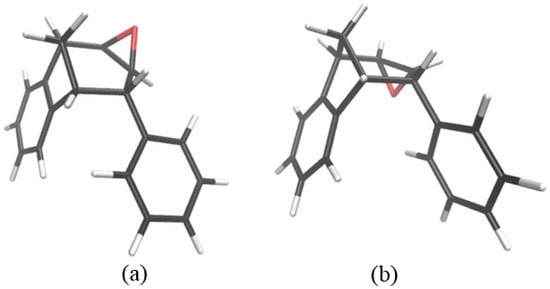
Figure 7.
Optimized structures of two stereoisomers of starting epoxide. Isomer (b) is thermodynamically more stable than isomer (a) by –7.9 kJ mol−1.
Thermodynamically, although the sum of free energies of reactants showed that an epoxide isomer with an oxygen above the plane was less stable compared to the other isomer (for 7.9 kJ mol−1), this ratio was inversed when an adduct between endo-38 and hydride formed: the adduct containing an isomer with an O above the plane becomes more stable (Figure 6). This makes the absolute barriers for the ring opening shown by reactions (1) and (2) even higher, being 123.9 and 124.2 kJ mol−1, respectively, leading to the conclusion that reactions (3) and (4), where the absolute barriers were 106.9 and 95.5 kJ mol−1, respectively, were favored. Due to the difference between activation energies for reactions (3) and (4) (Figure 6, full line), the product that was probably the most abundant in the mixture was a regioisomer of alcohol with an OH group at the CB atom and placed below the plane.
Optimized structures of alcohols derived from products of reactions (1) and (2) are shown in Figure 8, whereas the structures of alcohols corresponding to products of reactions (3) and (4) are depicted in Figure 9.
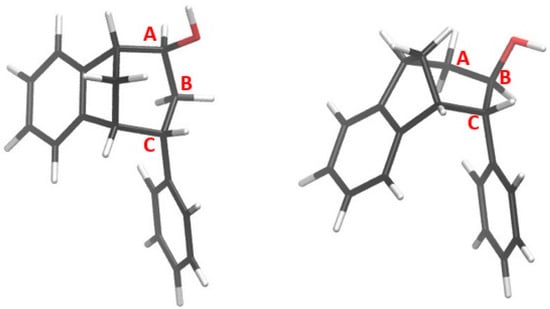
Figure 8.
Alcohols formed from products of reactions (1) and (2).
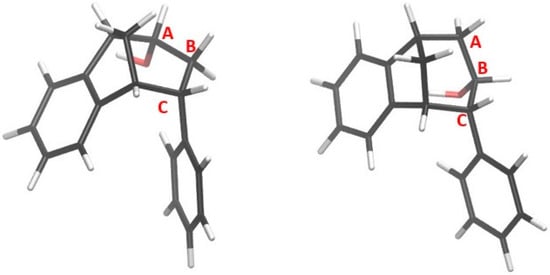
Figure 9.
Alcohols derived from products of reactions (3) and (4).
Figure 10 shows the aliphatic part of the 1H NMR spectrum of the isolated epoxy derivative endo-38 in comparison with the corresponding starting endo-36. It can be seen that there was no signal for protons on the sp2 carbon (between 5.00 and 6.50 ppm), which means that the double bond was broken and the epoxy ring has been formed. Instead of the signals in the mentioned region, new triplets can be seen at 3.45 and 3.39 ppm for protons A and B with coupling constants of 3.9 and 4.1 Hz, respectively.
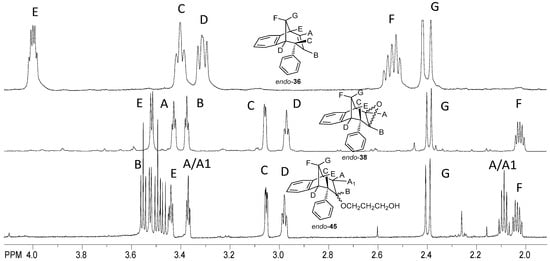
Figure 10.
Partial 1H NMR spectra of starting compound endo-36 and reaction products epoxyde endo-38 and ether endo-45.
The signal at 3.54 ppm was a doublet for the proton E with the coupling constant of 4.8 Hz. The triplet and doublet at 3.07 and 2.99 ppm belonged to the signals of protons C and D with coupling constants of 5.0 and 4.1 Hz, respectively. Proton G showed a doublet at 2.41 ppm with a coupling constant of 10.8 Hz. The signal at chemical shift 2.05 ppm was a multiplet belonging to proton F. The difference between the signals of endo-38 and the corresponding ether endo-45 is the presence of an expected number of new signals in the aliphatic region for the new aliphatic chain as a substituent following the substitution of the hydroxy proton. The same tendency can be seen for the methoxy derivatives starting with photoproduct endo-37, corresponding epoxide endo-39 and ether endo-48 (Figure 11).
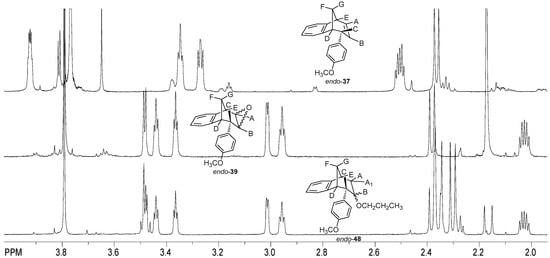
Figure 11.
Partial 1H NMR spectra of starting compound endo-37 and reaction products epoxyde endo-39 and ether endo-48.
The stereochemistry on the reactive carbon was determined for some ethers according to their NOESY spectra. From the NOESY spectrum of the ether endo-45 (Figure 12) the coupling of proton F with protons E, D and G, protons A/A1 with protons C and B, and proton G with protons E and D can be observed. The most significant coupling was of proton B with protons G and D within known stereochemistry.
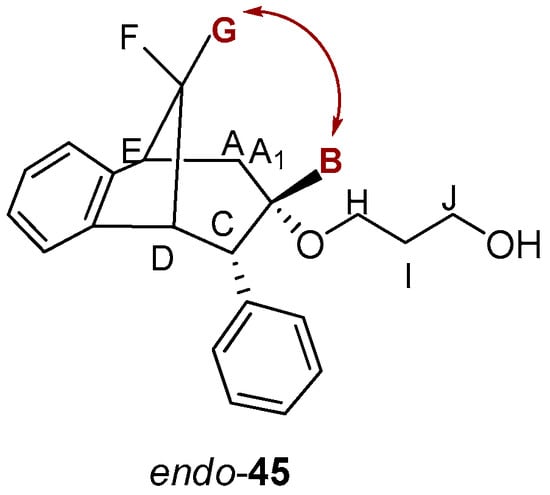
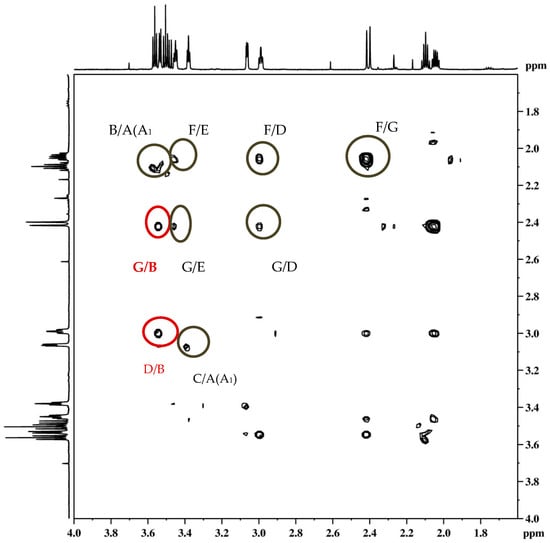
Figure 12.
NOESY spectrum of ether endo-45.
On the basis of the crucial interactions, it was concluded that the obtained stereoisomer was one in which the ether group was below the imaginary plane of the bicyclic skeleton. Accordingly, proton B was located above the plane of the bicyclic skeleton. These conclusions were also confirmed by calculations suggesting at the same time the stereochemistry of the corresponding alcohol endo-40 and epoxide endo-38 for the synthesis of the final ethers according to Scheme 3.
2.4. Friedel-Crafts Acylation of the Furo-Benzobicyclo[3.2.1]octadiene Photoproduct
Benzobicyclo[3.2.1]octadiene photoproduct 49 [41] with the annulated furan ring was the starting substrate (Scheme 6) for syntheses of the fourth group of compounds (Figure 2D). The Friedel–Crafts acylation reaction of 49 was performed with various carbonyl chlorides in the seal tube. During the reaction under applied conditions, in all cases the furan ring of 49 opened to give novel mono- or dihydroxy and mono- or dicarbonyl derivatives 50–57 (7–59%) with the characteristic benzobicyclo[3.2.1]-skeleton (Figure 13).
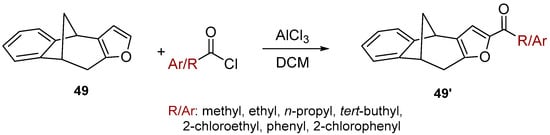
Scheme 6.
Acylation of the furo-benzobicyclo[3.2.1]octadiene photoproduct 49.
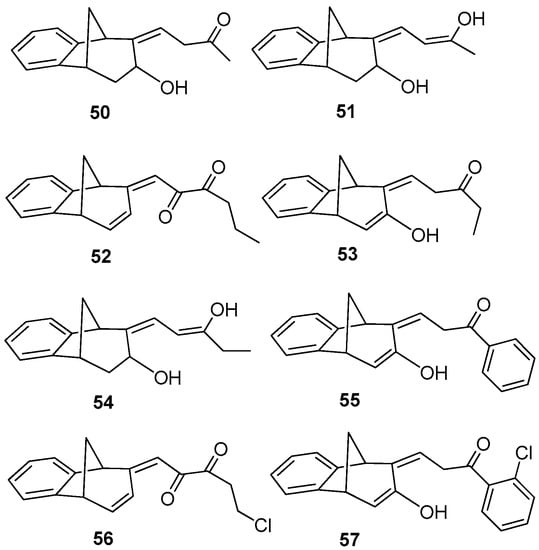
Figure 13.
Isolated products of acylation reaction.
The structure and purity of the synthesized products of acylation were confirmed by NMR techniques as well as High Resolution Mass Spectrometry (HRMS) analyses (see experimental and Figure 14). In comparison with the aliphatic part of the proton NMR spectrum of 49 and characteristic six signals, seven signals were present in the same region for compound 53. Together with the absence of the singlet for the furyl proton but two new doublets with allylic coupling constants, the opening of the heterocyclic ring as well as the structure were confirmed. However, as in the cases of several derivatives, the formed tautomers were in an equilibrium, and the products were isolated at lower amounts. Nevertheless, only two compounds from this group were tested as cholinesterase inhibitors.
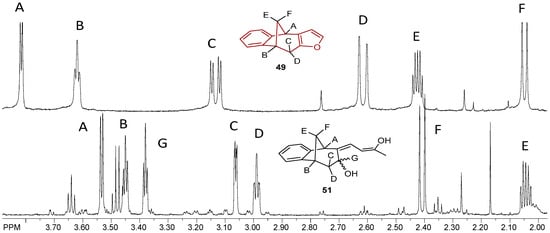
Figure 14.
Partial 1H NMR spectra of starting compound 49 and the acylation product 51 with the opened furan ring.
Acylated furo-benzobicyclo[3.2.1]octadiene 51 exhibited the maximum of anti-AChE activity of all of the tested compounds (Table 4). It seems that a smaller saturated molecule with two effective nucleophile groups can contribute to interactions with the AChE active site. Even though compound 51 was not a potent inhibitor of BChE, its structure is similar to opened oxime ether 31 (Figure 4), a potent inhibitor of BChE. However, none of tested compounds inhibited the enzymes in nanomolar range like etopropazine, a drug for Parkinson’s disease [42].
Table 4.
Inhibition of human AChE and BChE by acylated furo-benzobicyclo [3.2.1]octenes/octadienes expressed as IC50 ± SEM.
3. Materials and Methods
3.1. General
Petroleum ether (PE)(VWR Prolabo Chemicals, Fontenay-sous-Bois, France), bp 40–60 °C, was used. Solvents were purified by distillation. Column chromatography was carried out on columns with silica gel (Fluka 0.063–0.2 nm and Fluka 60 Å, technical grade, Merck, New York, NY, USA). Thin layer chromatography (TLC) was carried out using plates coated with silica gel (0.2 mm, 0.5 mm, 1.0 mm, Kiselgel 60 F254, Merck, New York, NY, USA). Organic layers were routinely dried with anhydrous MgSO4 and evaporated using a rotary evaporator (Heidolph, Schwabach, Germany). 1H and 13C NMR spectra were recorded on a spectrometer (Bruker AV-600 Spectrometer, Billerica, MA, USA) at 600 MHz. All NMR spectra were measured in CDCl3 using tetramethylsilane (Merck, New York, NY, USA) as reference. The following abbreviations were used: Sh—shoulder (in UV spectra), s—singlet, d—doublet, t—triplet, q—quartet, dd—doublet of doublets, m—multiplet, br—broad, PE—petroleum ether, E—diethyl ether. UV spectra were measured on a UV/VIS spectrophotometer (Varian Cary 50 UV/VIS Spectrophotometer, Palo Alto, CA, USA). Mass spectra were obtained on a UPLC-MS system (Acquity UPLC coupled with SQD mass spectrometer, Milford, MA, USA). Melting points were obtained using a microscope equipped apparatus (Original Kofler Mikroheitztisch apparatus Reichert, Vienna, Austria) and have not been corrected. HRMS analyses were carried out on a mass spectrometer (MALDI TOF/TOF analyzer (Merck, New York, NY, USA), equipped with Nd:YAG laser (Merck, New York, NY, USA) operating at 355 nm with a firing rate of 200 Hz in the positive (H+) or negative (-H) ion reflector mode. Compounds endo-1, endo-36, endo-37 and 49 were prepared in our laboratory by photochemical strategy and published before [31,41,43,44].
3.2. Photochemistry
Photochemical Synthesis of endo-1, endo-36 and endo-37
The solutions of a mixture of isomers of corresponding Wittig reaction products were purged with argon for 20 min and irradiated at 350 nm in petroleum ether (5 × 10−3 M) in a Rayonet reactor in a Pyrex tube for 4 h. The solvent was removed in vacuum and the oily residue chromatographed on silica gel column using petroleum ether as eluent.

After column chromatography, 77% of endo-1, 90% of endo-36 and 70% of endo-37 was isolated. High-molecular-weight products remained on the column.
11-(4-Chlorophenyl)tricyclo[6.3.1.0²,7]dodeca-2,4,6,9-tetraene (endo-1) [39]: colorless oil; Rf 0.66 (petroleum ether/dichloromethane = 9:1); UV (96% EtOH) λmax (log ε) 276 (3.19), 268 (3.22), 224 (4.20, sh); 1H NMR (CDCl3, 600 MHz) δ/ppm 7.14 (d, J = 8.4 Hz, 2H, Har2), 7.12 (d, J = 7.3 Hz, 1H), 7.03 (td, J = 7.4; 1.0 Hz, 1H), 6.84 (td, J = 7.4; 1.0 Hz, 1H), 6.65 (d, J = 8.3 Hz, 2H, Har1), 6.40–6.35 (m, 1H, HA), 6.23 (d, J = 7.3 Hz, 1H, Har), 5.25 (ddd, J = 9.6; 4.0; 1.9 Hz, 1H, HB), 3.96–3.92 (m, 1H), 3.34 (t, J = 9.4; 4.7 Hz, 1H), 3.29 (dd, J = 6.1; 4.7 Hz, 1H), 2.54–2.50 (m, 1H, HF), 2.37 (d, J = 10.0 Hz, 1H, HG); 13C NMR (CDCl3, 150 MHz) δ/ppm 140.6 (s), 137.3 (s), 132.9 (s), 130.9 (s), 134.7 (d), 129.2 (2d), 127.3 (d), 125.7 (d), 125.6 (d), 125.4 (2d), 124.7 (d), 119.7 (d), 48.0 (d), 45.3 (d), 43.7 (t), 39.9 (d); HRMS (m/z) for C18H15Cl: [M + H]+calcd = 265.0789, [M + H]+measured = 265.0786.
endo-6-Phenyl-6,9-dihydro-5H-5,9-methano-benzocycloheptene (endo-36) [38]: colorless crystals; mp 51 °C; Rf 0.44(petroleum ether); UV (EtOH) λmax (logε) 275 (2.88), 263 (2.90), 203 (4.46); 1H NMR (CDCl3, 600 MHz) δ 7.17–7.18 (m, 3H, Hf), 7.12 (d, J = 7.3 Hz, 1H, HAr4), 7.03 (t, J = 7.4 Hz, 1H, HAr3), 6.81 (t, J = 7.4 Hz, 1H, HAr2), 6.73–6.76 (m, 2H, Hf1), 6.37 (ddd, JAB = 9.5 Hz; JAE = 6.0 Hz; JAC = 2.5 Hz, 1H, HA), 6.18 (d, J = 7.3 Hz, 1H, HAr1), 5.33 (dt, JAB = 9.5 Hz; JBC = 2.5 Hz, 1H, HB), 3.98 (m, 1H, HC), 3.38 (t, JCD = JDF = 4.7 Hz, 1H, HD), 3.29 (dd, JAE = 6.0 Hz; JEF = 4.7 Hz, 1H, HE), 2.52 (dt, JFG = 9.9 Hz; JEF = JDF = 4.7 Hz, 1H, HF), 2.38 (d, JFG = 9.9 Hz, 1H, HG); 13C NMR (CDCl3, 150 MHz) δ 152.27 (s), 142.44 (s), 141.93 (s), 132.71 (d), 128.21 (d), 127.60 (d), 126.34 (d), 126.10 (d), 126.05 (d), 125.97 (d), 124.89 (d), 120.03 (d), 48.59 (d), 46.29 (d), 44.13 (t, CFG), 40.37 (d); MS m/z (EI) 232 (M+, 100%), 117 (25), 115 (10); HRMS (m/z) for C18H16: [M + H]+calcd = 232.1634, [M + H]+measured = 232.1655.
11-(4-Methoxyphenyl)tricyclo[6.3.1.0²,7]dodeca-2,4,6,9-tetraene (endo-37) [40]: colorless crystals; mp 45–47 °C; Rf (petroleum ether/dichloromethane = 8:2) = 0.27; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.11 (d, 1H, J = 7.3 Hz), 7.03 (dt, 1H, J = 7.3; 1.0 Hz), 6.82 (dt, 1H, J = 7.3; 1.0 Hz), 6.72 (d, 2H, J = 8.6 Hz), 6.64 (d, 2H, J = 8.6 Hz), 6.32-6.36 (m, 1H), 6.25 (d, 1H, J = 7.3 Hz), 5.28 (dt, 1H, J = 9.6; 2.6 Hz), 3.91-3.94 (m, 1H), 3.77 (s, 3H), 3.35 (t, 1H, J = 4.5 Hz), 3.27 (dd, 1H, J = 6.3; 4.7 Hz), 2.49-2.53 (m, 1H), 2.37 (d, 1H, J = 9.9 Hz); 13C NMR (CDCl3, 75 MHz) δ/ppm: 157.6 (s), 152.0 (s), 141.7 (s), 134.2 (s), 134.1 (d), 128.8 (2d), 126.3 (d), 125.8 (d), 125.5 (d), 124.5 (d), 119.6 (d), 112.6 (2d), 52.4 (q), 48.2 (d), 45.0 (d), 43.7 (t), 40.3 (d); MS m/z (%, fragment): 262 (100, M+), 154 (75), 115 (50); HRMS (m/z) for C19H18O: [M + H]+calcd = 262.1352, [M + H]+measured = 262.1351.
3.3. Synthesis of Benzylamine Derivatives
Amination Reactions of endo-1 with Primary Benzylic Amines
To a solution of BrettPhos (0.1 eq) and Pd(OAc)2 (0.05 eq) in dioxane, 4% of water was added. After the addition of water, the solution was heated to 120 °C for 2 min. The reaction mixture changed color from light yellow, over dark red to dark green. Compound endo-1, different amine (2 eq) and KOtBu (1.4 eq) were added into a sealed tube. The reaction mixture was stirred for 20 h on 180 °C. After the removal of the solvent, the crude product residue was purified by repeated column chromatography using petroleum ether/dichlorometane mixture as an eluent. All of the obtained compounds endo-4–20 were isolated in the first fractions as yellow oils, while BrettPhos remained on the chromatographic column. In all cases, the conversion of the amination reaction was complete. The spectroscopic data for some of the previously synthesized amines (endo-4, endo-6, endo-7 and endo-8) are given below.

N-Benzyl-4-((5R,6S,9S)-6,9-dihydro-5H-5,9-methanobenzo[7]annulen-6-yl)aniline (endo-4): Column chromatography on silica gel using petroleum ether/dichloromethane (0%–60%) as eluent afforded 0.080 g (48.3%) of endo-4: Rf (PE / DCM = 5:1) = 0.43; UV (EtOH) λmax/nm (ε/dm3mol−1cm−1): 251 (11,312), 306 (Sh, 2527); IR νmax/cm−1 (diamond ATR): 3408.78, 1613.45, 1517.61, 1468.98; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.38–7.25 (m, 5H), 7.10 (d, 1H, J = 7.4 Hz), 7.02 (dt, 1H, J = 7.4; 1.1 Hz), 6.83 (dt, 1H, J = 7.4; 1.1 Hz), 6.54 (d, 2H, J = 8.6 Hz), 6.47 (d, 2H, J = 8.6 Hz), 6.32 (d, 2H, J = 7.4 Hz, Har, HA), 5.27 (td, 1H, J = 9.5; 1.9 Hz, HB), 4.28 (s, 2H), 3.92–3.85 (m, 2H, HC, NH), 3.35 (t, 1H, J = 4.2 Hz, HD), 3.27 (dd, 1H, J = 6.3; 4.2 Hz, HE), 2.55–2.47 (m, 1H, HF), 2.34 (d, 1H, J = 10.1 Hz, HG); 13C NMR (CDCl3, 150 MHz) δ/ppm: 152.1 (s), 146.1 (s), 141.9 (s), 139.1 (s), 133.8 (d), 130.6 (s), 128.6 (2d), 128.1 (d), 128.1 (d), 127.1 (d), 126.7 (2d), 126.0 (d), 125.9 (d), 125.4 (d), 124.4 (d), 119.5 (d), 111.9 (2d), 48.4 (d), 48.2 (t), 45.2 (d), 43.7 (t), 40.0 (d); MS m/z (%, fragment): 337 (100, M+); HRMS (m/z) for C25H23N: [M + H]+calcd = 338.1822, [M + H]+measured = 338.1902.
4-((5R,6S,9S)-6,9-Dihydro-5H-5,9-methanobenzo[7]annulen-6-yl)-N-(pyridin-3-ylmethyl)-aniline (endo-6): Column chromatography on silica gel using dichloromethane/ethanol (variable ratio) as eluent afforded 0.062 g (21.2%) of endo-6 in the first fractions: Rf (DCM) = 0.1; UV (EtOH) λmax/nm (ε/dm3mol−1cm−1): 254 (15991); IR νmax/cm−1 (diamond ATR): 3370.11, 1609.55, 1531.76, 1444.89; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.62 (s, 1H), 8.52 (dd, 1H, J = 5.4; 1.3 Hz), 7.67 (d, 1H, J = 7.4 Hz), 7.30–7.23 (m, 3H), 7.13 (d, 1H, J = 7.4 Hz), 7.09 (dt, 1H, J = 7.7; 1.2 Hz), 7.03 (dt, 1H, J = 7.7; 1.2 Hz), 6.85 (d, 1H, J = 8.5 Hz), 6.83 (d, 2H, J = 8.5 Hz), 6.60-6.58 (m, 2H, Har, HA), 5.41 (t, 1H, J = 3.7 Hz, HB), 4.36 (s, 2H), 3.79–3.76 (m, 2H, HC, NH), 3.32 (t, 1H, J = 5.1 Hz, HD), 2.69 (td, 1H, J = 8.3; 1.2 Hz, HE), (signal for HF and HG can’t be assigned because of the presence of the catalyst BrettPhos); MS m/z (%, fragment): 338 (100, M+).
Column chromatography on silica gel using dichloromethane/ethanol (variable ratio) as eluent afforded 0.051 g (43.8%) of endo-7 and 0.064 g (51.5%) of endo-8.
4-((5R,6S,9S)-6,9-Dihydro-5H-5,9-methanobenzo[7]annulen-6-yl)-N-(fur-2-ylmethyl)aniline (endo-7): Rf (PE / DCM = 5:1) = 0.62; UV (EtOH) λmax/nm (ε/dm3mol−1cm−1): 255 (28024), 358 (12474); IR νmax/cm−1 (diamond ATR): 3412.68, 1603.44, 1517.61, 1468.98; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.34 (d, 1H, J = 1.6 Hz, H5f), 7.09 (d, 1H, J = 7.2 Hz), 7.00 (t, 1H, J = 7.4 Hz), 6.80 (t, 1H, J = 7.2 Hz), 6.53 (d, 2H, J = 8.5 Hz), 6.49 (d, 2H, J = 8.5 Hz), 6.33–6.24 (m, 3H, Har, HA), 6.20 (d, 1H, J= 3.2 Hz, H3f), 5.26 (d, 1H, J = 9.4 Hz, HB), 4.26 (s, 2H), 3.88–3.83 (m, 1H, HC), 3.31 (t, 1H, J = 4.8 Hz, HD), 3.24 (t, 1H, J = 4.8 Hz, HE), 2.51–2.43 (m, 1H, HF), 2.33 (d, 1H, J = 9.7 Hz, HG); 13C NMR (CDCl3, 150 MHz) δ/ppm: 152.9 (s), 152.5 (s), 146.0 (s), 142.3 (s), 141.8 (d), 134.3 (d), 132.2 (s), 129.1 (2d), 121.1 (d), 126.4 (d), 125.9 (d), 124.9 (d), 119.9 (d), 112.8 (2d), 110.2 (d), 106.9 (d), 48.8 (d), 45.7 (d), 44.1 (t), 40.5 (d); MS m/z (%, fragment): 327 (100, M+); HRMS (m/z) for C23H21NO: [M + H]+calcd = 328.1609, [M + H]+measured = 328.1689.
4-((5R,6S,9S)-6,9-dihydro-5H-5,9-methanobenzo[7]annulen-6-yl)-N-(thien-2-ylmethyl)aniline (endo-8): Rf (PE / DCM = 5:1) = 0.70; UV (EtOH) λmax/nm (ε/dm3mol−1cm−1): 247 (18373); IR νmax/cm−1 (diamond ATR): 3413.17, 1613.45, 1516.18, 1468.98; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.48 (d, 1H, J = 4.9 Hz) 7.38 (d, 1H, J = 7.2 Hz), 7.30 (t, 1H, J = 7.1 Hz), 7.27 (broad s), 7.25–7.22(m, 2H), 7.11 (t, 1H, J = 7.2 Hz), 6.83 (d, 2H, J = 8.6 Hz), 6.78 (d, 2H, J = 8.6 Hz), 6.62-6.56 (m, 2H, Har, HA), 5.55 (d, 1H, J = 9.5 Hz, HB), 4.75 (s, 2H), 4.20–4.13 (m, 2H, HC, NH), 3.61 (t, 1H, J = 4.7 Hz, HD), 3.53 (t, 1H, J = 4.7 Hz, HE), 2.79–2.74 (m, 1H, HF), 2.62 (d, 1H, J = 9.7 Hz, HG);13C NMR (CDCl3, 150 MHz) δ/ppm: 152.5 (s), 145.9 (s), 143.1 (s), 142.3 (s), 134.4 (d), 132.3 (s), 129.1 (2d), 127.1 (d), 126.7 (d), 126.4 (d), 125.8 (d),125.0 (d), 124.9(d), 124.5 (d), 119.9 (d), 112.7 (2d), 48.8 (d), 45.7 (d), 44.1 (t), 43.7 (t), 40.4 (d); MS m/z (%, fragment): 343 (100, M+); HRMS (m/z) for C23H21NS: [M + H]+calcd = 344.1394, [M + H]+measured = 344.1474.
3.4. Synthesis of Oxime Derivatives
3.4.1. Synthesis of Oxime
A mixture of aldehyde 21 (0.223 mmol), hydroxylamine hydrochloride (0.112 mmol, 0.5 eq), and pyridine (0.05 mL) was refluxed in ethanol (1 mL) for 1 h on a water bath. Completion of the reaction was checked by TLC (DCM/PE, 1:3). At the end of the reaction, ethanol was evaporated to dryness. Crude material was purified on column chromatography using different polarity of solvents. The product was additionally purified using preparative TLC (DCM/PE).
3.4.2. Synthesis of Oxime Ethers
A solution of oxime 22 (0.251 mmol) and alkyl bromide (0.276 mmol, 1.1 eq) in dry acetone (2 mL) was refluxed in the presence of dry potassium carbonate (0.376 mmol, 1.5 eq) for 48 h on a water bath. Completion of the reaction was checked by TLC (DCM/PE). At the end of the reaction, acetone was evaporated to dryness. Crude material was purified on column chromatography using different polarity of solvents (DCM/PE) and the product was also additionally purified using preparative TLC.

(4R,9R)-9,10-Dihydro-4H-4,9-methanobenzo[4,5]cyclohepta[1,2-b]furan-2-carbaldehyde (21) [33]: 180 mg (50.0%); Rf (petroleum ether/dichloromethane = 20:1) = 0.23; UV (EtOH) λmax (ε/dm3 mol−1 cm−1): 308 (16794) nm;IR νmax/cm−1 (NaCl): 1666, 1504; 1H NMR (CDCl3, 300 MHz) δ/ppm: 9.41 (s, 1H), 7.32 (dd, 1H, J = 7.2; 1.4 Hz), 7.16–7.08 (m, 4H, 3H), 3.91 (d, 1H, J = 4,4 Hz, HA), 3.66 (dt, 1H, J = 4.9; 0.5 Hz, HB), 3.20 (dd, 1H, J = 17.8; 4.9 Hz, HC), 2.72 (dd, 1H, J = 17.8; 0.5 Hz, HD), 2.48 (ddd, 1H, J = 10.7; 4.9; 4.4 Hz, HE), 2.4 (d, 1H, J = 10.7 Hz, HF); 13C NMR (CDCl3, 75 MHz) δ /ppm: 176.6 (d), 156.0 (s), 151.3 (s), 150.6 (s), 144.1 (s), 128.5 (s), 126.9 (d), 126.9 (d), 124.1 (d), 121.0 (2d), 42.5 (t), 39.6 (d), 39.1 (d), 31.4 (t); MS m/z (%, fragment): 224 (100, M+), 167 (75), 115 (50); HRMS (m/z) for C19H18O: [M + H]+calcd = 224.3355, [M + H]+measured = 224.3347.
(E)-9,10-Dihydro-4H-4,9-methanobenzo[4,5]cyclohepta[1,2-b]furan-2-carbaldehyde oxime (22): 16.3 mg (40%); Rf (PE / DCM = 1:1) = 0.33; IR νmax/cm−1 (NaCl): 3400, 2943, 1668, 1613, 1572, 1503, 1460; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.36 (d, 1H, J = 6.6 Hz), 7.18-7.05 (m, 4H), 6.06 (s, 1H), 4.62 (d, 1H, J = 2.5 Hz), 3.81 (d, 1H, J = 4.0 Hz), 3.59-3.54 (m, 2H), 2.40-2.37 (m, 2H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 152.1 (s), 146.6 (s), 141.9 (s), 135.9 (s), 131.6 (s), 126.9 (d), 126.4 (d), 125.1 (d), 121.5 (d), 104.6 (2d), 67.4 (t), 48.9 (d), 39.7 (t), 39.6 (d); MS m/z (%, fragment) (EI): 239 (100); HRMS (m/z) for C19H19ON: [M + H]+ calcd = 240.0946; [M + H]+measured = 240.0941.
(Z)-9,10-Dihydro-4H-4,9-methanobenzo[4,5]cyclohepta[1,2-b]furan-2-carbaldehyde O-propyl oxime (27): 13.6 mg (32%); Rf (PE / DCM = 1:5) = 0.55; IR νmax/cm−1 (NaCl): 3434, 2918, 1731, 1638, 1234; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.32 (d, 1H, J = 7.4 Hz), 7.19–7.07 (m, 4H), 6.94 (s, 1H), 3.86 (d, 1H, J = 4.5 Hz), 3.68 (d, 1H, J = 5.0 Hz), 3.48 (q, 2H, J = 7.0 Hz), 3.15 (dd, 1H, J = 17.4; 5.0 Hz), 2.67 (dd, 1H, J = 17.4; 1.3 Hz), 2.51–2.47 (m, 1H), 2.03 (d, 1H, J = 11.2 Hz), 1.34–1.29 (m, 2H), 0.88 (t, 3H, J = 6.9 Hz); 13C NMR (CDCl3, 150 MHz) δ/ppm: 152.0 (s),150.5 (s), 143.8 (s), 135.6 (s), 128.1 (s), 126.9 (d), 126.9 (d), 124.1 (d), 124.1 (d), 120.2 (d), 67.0 (t), 42.5 (t), 39.6 (d), 38.9 (d), 31.2 (t), 29.6 (t), 22.7 (q); MS m/z (%, fragment) (EI): 281 (100); HRMS (m/z): [M + H]+ calcd = 282.1416; [M + H]+measured = 282.1419.
3.5. Synthesis of Epoxide Derivatives
Synthesis of (2R,7R,8S,8aS)-8-Phenyl-1a,7,8,8a-tetrahydro-2H-2,7-methanobenzo[4,5]cyclohepta[1,2-b]oxirene (endo-38) and (2R,7R,8S,8aS)-8-(4-Methoxyphenyl)-1a,7,8,8a-tetrahydro-2H-2,7-methanobenzo[4,5]cyclohepta[1,2-b]oxirene (endo-39)
Epoxide derivatives endo-38 and endo-39 were prepared by epoxidation reaction using meta-chlorperbenzoic acid (m-CPBA). Photoproducts endo-36 and endo-37 were dissolved in 20 mL dry dioxane, and m-CPBA was added. The reaction mixture was stirred at room temperature overnight. After the reaction was complete, the saturated water solution of NaHCO3 was added. The organic layer was washed twice with water and dried over MgSO4, filtered and evaporated to obtain crude material. The crude material was purified using column chromatography.

(2R,7R,8S)-8-Phenyl-1a,7,8,8a-tetrahydro-2H-2,7-methanobenzo[4,5]cyclohepta[1,2-b]-oxirene (endo-38): Yield 61%; Rf (petroleum ether/dichloromethane): 0.27; 1H NMR (cdcl3, 600 mhz) δ/ppm: 7.53 (d, 2H, J = 8.8 Hz, Ar), 7.30 (d, 1H, J = 7.4 Hz, Ar), 7.16 (t, 2H, J = 7.3 Hz, Ar), 6.93 (t, 1H, J = 7.3 Hz, Ar), 6.70 (dd, 2H, J = 8.8; 7.3 Hz, Ar), 6.26 (d, 1H, J = 7.4 Hz, Ar), 3.54 (d, 1H, J = 4.8 Hz, HE), 3.45 (t, 1H, J = 4.1 Hz, HA), 3.39 (t, 1H, J = 3.9 Hz, HB), 3.07 (d, 1H, J = 4.1 Hz, HC), 2.99 (t, 1H, J = 5.0 Hz, HD), 2.41 (d, 1H, J = 10.8 Hz, HG), 2.0–2.02 (m, 1H, HF);13C NMR (cdcl3, 75 mhz) δ/ppm: 144.6 (s), 143.2 (s), 141.2 (s), 133.2 (d), 129.3 (d), 128.2 (2d), 127.3 (2d), 126.4 (d), 125.3 (d), 122.1 (d), 54.1 (d), 52.5 (d), 45.8 (d), 43.2 (t), 40.1 (d), 34.9 (t); MS m/z (%, fragment) (EI): 248; HRMS (m/z) for C18H16O: [M + H]+calcd = 249.1201, [M + H]+measured = 249.1190.
(2R,7R,8S,8as)-8-(4-Methoxyphenyl)-1a,7,8,8a-tetrahydro-2H-2,7-methanobenzo[4,5]cyclohepta[1,2-b]oxirene (endo-39): Yield 50%; Rf (petroleum ether): 0.13; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.29 (d, 1H, J = 7.3 Hz, Ar), 7.16 (t, 1H, J = 7.2 Hz, Ar), 6.95 (t, 1H, J = 7.2 Hz, Ar), 6.75 (d, 2H, J = 8.3 Hz, Ar), 6.61 (d, 2H, J = 8.3 Hz, Ar), 6.32 (d, 1H, J = 7.3 Hz, Ar), 3.79 (s, 3H, OCH3), 3.48 (d, 1H, J = 5.0 Hz, HE), 3.44 (t, 1H, J = 4.0 Hz, HA), 3.36 (t, 1H, J = 3.8 Hz, HB), 3.00 (d, 1H, J = 4.0 Hz, HC), 2.95 (t, 1H, J = 5.0 Hz, HD), 2.38 (d, 1H, J = 10.8 Hz, HG), 2.05–2.00 (m, 1H, HF); 13C NMR (CDCl3, 150 MHz) δ/ppm: 152.5 (s), 142.9 (s), 135.9 (s), 133.8 (s), 129.6 (2d), 126.8 (d), 126.7 (d), 125.8 (d), 122.5 (d), 113.8 (2d), 55.2 (d), 54.2 (d), 53.2 (q), 44.8 (d), 42.9 (t), 40.6 (d), 35.3 (d);MS m/z (%, fragment) (EI): 278; HRMS (m/z) for C19H16O2: [M + H]+calcd = 279.1306, [M + H]+measured = 279.1325.
3.6. Epoxide Ring Opening
To a solution of starting epoxide (0.8g) in THF, lithium aluminum hydride (2.5 eq) was added. The reaction mixture was stirred at 76 °C during 24 h. After the reaction was complete, NaHCO3 was added and pH was adjusted to 7. The water layer was washed twice with diethyl ether. Organic layers were dried over MgSO4, filtered and evaporated to obtain crude material. Crude material was purified on column chromatography using petroleum ether/dichloromethane as eluent.

(6S)-6-Phenyl-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]anulen-7-ol (endo-40): Yield 40%; Rf (petroleum ether/ dichloromethane 40%): 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm 8.00–7.30 (m, 4H, Ar), 6.93–6.20 (m, 5H, Ar), 4.13 (m, 1H, HB) 3.40 (dd, 1H, J = 12.7, 4.7 Hz, HA/A1/E) 3.20 (t, 1H, J = 4.5 Hz, HA/A1/E), 3.14 (d, 1H, J = 4.5 Hz, HA/A1/E), 2.56 (d, 1H, J = 10.8 Hz, HG), 2.35 (s, 1H, OH), 2.25–2.20 (m, 1H, HF) 1.63 (d, 1H, J = 4.8 Hz, HC/D), 1.60 (d, 1H, J = 4.8 Hz, HC/D); 13C NMR (CDCl3, 150 MHz) δ/ppm: 143.8 (s), 142.3 (s), 133.9 (s), 129.3 (d), 128.9 (d), 127.6 (d), 127.2 (2d), 126.5 (d), 124.3 (2d), 122.1 (d), 68.1 (d), 63.9 (t), 47.9 (d), 47.0 (d), 41.5 (d), 38.7 (t);MS m/z (%, fragment) (EI): 250; HRMS (m/z) for C18H18O: [M + H]+calcd = 251.1357, [M + H]+measured = 251.1349.
(5R,6S,9S)-6-(4-methoxyphenyl)-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]anulen-7-ol (endo-41): Yield 35%; Rf (petroleum ether/ dichloromethane): 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm 7.85–7.32 (m, 3H, Ar), 6.98–6.24 (m, 5H, Ar), 4.31–4.28 (m, 1H, HB) 3.79 (s, 1H, OCH3) 3.65–3.63 (m, 1H, HA/A1/E) 3.44 (t, 1H, J = 4.4 Hz, HA/A1/E), 3.36 (t, 1H, J = 3.8 Hz, HA/A1/E), 3.01 (dd, 1H, J = 4.0, 1.4 Hz, HC/D), 2,96 (t, 1H, J = 5,2 Hz, HC/D) 2,38 (d, 1H, J = 11,0 Hz, HG), 2,27 (s, 1H, OH), 2,05–2,00 (m, 1H, HF); MS m/z (%, fragment) (EI): 280; HRMS (m/z) for C19H20O2: [M + H]+calcd = 281.1463, [M + H]+measured = 281.1609.
3.7. Synthesis of Ethers from Alcohols
Alcohol obtained after epoxide cleavage (0.05 g) was dissolved in acetone (2 mL), K2CO3 (1.5 eq) and corresponding alkyl bromides (1.1 eq) were added. The reaction mixture was stirred over 24 h at 56 °C. After the reaction was complete, water and diethylether were added. The organic layer was dried over MgSO4, filtered and evaporated to obtain a crude product. The crude material was purified on column chromatography using petroleum ether and dichloromethane as eluents.

(5R,6S,9S)-7-Ethoxy-6-phenyl-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]annulene (endo-42): Yield 20%; Rf (petroleum ether/dichloromethane): 0.52; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.72 (d, 1H, J = 7.3 Hz), 7.33–7.19 (m, 4H), 7.05–7.01(m, 2H), 7.14 (t, 1H, J = 7.4 Hz), 6.74 (t, 1H, J = 7.3 Hz), 4.16–4.11 (m, 2H), 3.35–3.30 (m, 2H), 3.01 (d, 1H, J = 11.0 Hz), 2.18–2.13 (m, 1H), 2.09–2.07 (m, 1H), 1.89 (dd, 2H, J = 15.2, 4.5 Hz), 1.76–1.73 (t, 3H, J = 6.9 Hz, CH3); MS m/z (%, fragment) (EI): 280 (100).
(5R,6S,9S)-6-Phenyl-7-propoxy-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]annulene (endo-43): Yield 40%; Rf (petroleum ether/dichloromethane): 0.57; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.42 (t, 1H, J = 7.5 Hz), 7.33 (d,1H, J = 7.1 Hz), 7.28 (d, 2H, J = 7.9 Hz), 7.23 (t, 2H, J = 7.5 Hz), 7.14 (t, 2H, J = 7.9 Hz), 6.48 (d, 1H, J = 7.1 Hz), 4.68–4.63 (m, 2H), 4.19–4.14 (m, 2H), 3.31 (m, 2H), 3.03 (d, 1H, J = 11.0 Hz), 2.18–2.13 (m, 1H), 2.09–2.07 (m, 1H), 1.89 (dd, 2H, J = 15.2; 4.5 Hz), 1.71 (d, 1H, J = 10.5 Hz), 1.28 (t, 3H, J = 6.9 Hz, CH3); 13C NMR (CDCl3, 150 MHz) δ/ppm: 145.3 (s), 145.1 (s), 144.3 (s), 127.6 (d), 127.5 (2d), 126.9 (d), 126.6 (2d), 125.7 (d), 124.5 (d), 122.5 (d), 68.8 (d), 53.3 (d), 47.5 (t), 46.8 (d), 39.7 (t), 36.9 (t), 33.8 (t), 32.4 (d), 25.5 (q);MS m/z (%, fragment) (EI): 292 (100); HRMS (m/z) for C21H24O: [M + H]+calcd = 293.1827, [M + H]+measured = 293.1846.
(5R,6S,9S)-7-Isopropoxy-6-phenyl-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]annulene (endo-44): Yield 50%; Rf (petroleum ether/dichloromethane) = 0.55; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.33 (t, 1H, J = 7.0 Hz), 7.30–7.28 (m, 1H), 7.23 (t, 3H, J = 7.7 Hz), 7.14 (d, 1H, J = 7.3 Hz), 6.98 (d, 1H, J = 7.3 Hz), 6.47 (d, 2H, J = 7.7 Hz), 4.19–4.14 (m, 1H), 3.50–3.46 (m, 1H, isopropyl-CH), 3.24–3.20 (m, 2H), 2.98 (d, 1H, J = 11.1 Hz), 2.17–2.12 (m, 1H), 2.06–2.01 (m, 1H), 1.81 (dd, 1H, J = 15.0; 4.3 Hz), 1.67 (d, 1H, J = 15.0 Hz), 1.29 (broad s, 6H);13C NMR (CDCl3, 150 MHz) δ /ppm: 145.4 (s), 145.2 (s), 144.3 (s), 127.5 (2d), 126.9 (d), 126.8 (d), 126.5 (2d), 125.7 (d), 124.8 (d), 122.5 (d), 68.8 (d), 53.3 (d), 46.8 (d), 43.2 (d), 39.7 (d), 36.9 (t), 34.1 (t), 27.4 (2q); MS m/z (%, fragment) (EI): 292 (100); HRMS (m/z) for C21H24O: [M + H]+calcd = 293.1827, [M + H]+measured = 293.1814.
3-(((5R,6S,9S)-6-Phenyl-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]annulen-7-il)oxy)propan-1-ol (endo-45): Yield 45%; Rf (petroleum ether/dichloromethane): 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm 7.28 (d, 1H, J = 7.3 Hz, Ar), 7.21–7.18 (m, 3H, Ar), 7.15 (t, 1H, J = 7.3 Hz, Ar), 6.92 (t, 1H, J = 7.3 Hz, Ar), 6.69 (dd, 2H, J = 6.6; 3.3 Hz, Ar), 6.24 (d, 1H, J = 7.3 Hz, Ar), 3.57–3.44 (m, 4H), 3.52 (d, 1H, J = 4.8 Hz, HB), 3.44 (t, 1H, J = 4.2 Hz, HE), 3.37 (t, 1H, J = 3.8 Hz, HA/A1), 3.05 (dd, 1H, J = 4.2 Hz, HC), 2.98 (t, 1H, J = 5.1 Hz, HD), 2.40 (d, 1H, J = 10.7 Hz, HG), 2.11–2.06 (m, 1H, HA/A1), 2.06–2.01 (m, 1H, HF), 1.42 (s, 1H, OH), 1.24 (broad s, 2H); 13C NMR (CDCl3, 150 MHz) δ /ppm: 145.2 (s), 143.5 (s), 141.7 (s), 128.6 (2d), 127.7 (2d), 126.8 (d), 126.3 (d), 126.0 (d), 125.7 (d), 122.5 (d), 68.2 (d), 54.5 (t), 52.9 (d), 50.4 (t), 45.9 (d), 43.7 (d), 40.6 (t), 39.9 (t), 35.5 (t); MS m/z (%, fragment) (EI): 308; HRMS (m/z) for C21H24O2: [M + H]+calcd = 309.1776, [M + H]+measured = 309.4140.

(5R,6S,9S)-7-Ethoxy-6-(4-methoxyphenyl)-6,7,8,9-tetrahydro-5H-5,9-methanobenzo-[7]annulene (endo–46): Yield 20%; Rf (petroleum ether/dichloromethane): 0.67; 1H NMR (CDCl3, 600 MHz) δ/ppm 7.30 (d, 1H, J = 7.5 Hz), 7.17 (t, 1H, J = 7.5 Hz), 6.96 (t, 1H, J = 7.5 Hz), 6.76 (d, 2H, J = 8.6 Hz), 6.62 (d, 2H, J = 8.6 Hz), 6.33 (d, 1H, J = 7.5 Hz), 3.79 (s, 3H), 3.51–3.48 (m, 2H), 3.45 (t, 1H, J = 4.8 Hz), 3.38 (t, 1H, J = 4.2 Hz), 3.03 (d, 1H, J = 4.2 Hz), 2.97 (t, 1H, J = 4.8 Hz), 2.38-2.33 (m, 2H), 2.39 (d, 1H, J = 10.8 Hz), 2.06–2.01 (m, 1H), 1.27 (s, 3H); MS m/z (%, fragment) (EI): 308 (100).
(5R,6S,9S)-6-(4-Methoxyphenyl)-7-propoxy-6,7,8,9-tetrahydro-5H-5,9-methanobenzo[7]annulene (endo-47): Yield 35%; Rf (petroleum ether/dichloromethane):0.57; 1H NMR (CDCl3, 600 MHz) δ/ppm 7.21 (d, 1H, J = 7.3 Hz), 7.08 (t, 1H, J = 7.4 Hz), 6.87 (t, 1H, J = 7.4 Hz), 6.67 (d, 2H, J = 8.6 Hz), 6.53 (d, 2H, J = 8.6 Hz), 6.24 (d, 1H, J = 7.3 Hz), 3.70 (s, 3H, OCH3), 3.51 (q, 2H, J = 6.6 Hz), 3.40 (t, 1H, J = 4.8 Hz), 3.36 (t, 1H, J = 4.3 Hz), 2.93 (d, 1H, J = 3.7 Hz), 2.87 (t, 1H, J = 4.9 Hz), 2.39–2.35 (m, 4H), 2.30 (d, 1H, J = 10.8 Hz), 1.99–1.96 (m, 1H), 1.18 (broad s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 157.5 (s), 144.5 (s), 143.2 (s), 133.5 (s), 129.2 (d), 127.1 (d), 126.4 (2d), 126.2 (d), 122.1 (2d), 112.6 (d), 54.7 (d), 54.0 (d), 52.7 (d), 45.5 (d), 42.4 (d), 40.1 (d), 34.7 (t), 30.4 (q); MS m/z (%, fragment) (EI): 322; HRMS (m/z) for C22H26O2: [M + H]+calcd = 323.1932, [M + H]+measured = 323.4406.
3-(((5R,6S,9S)-6-(4-Methoxyphenyl)-6,7,8,9-tetrahydro-5H-5,9-methanobenzo-[7]annulen-7-il)oxy)propan-1-ol (endo-48): Yield 15%; Rf (petroleum ether/dichloromethane): 0.64; 1H NMR (CDCl3, 600 MHz) δ/ppm 6.95 (d, 1H, J = 7.4 Hz), 6.82 (t, 1H, J = 7.3 Hz), 6.75 (t, 1H, J = 7.3 Hz), 6,71 (d, 2H, J = 8,5 Hz, Ar), 6,63 (d, 1H, J = 7,4 Hz, Ar), 6,46 (d, 2H, J = 8,6 Hz, Ar), 4,20–4,16 (m, 1H), 3,72–3,66 (m, 4H, CH2CH2), 3,64 (s, 3H, OCH3), 3,53 (t, 1H, J = 4,8 Hz), 3,31–3,25 (m, 1H), 2,96 (s, 1H, OH), 2,57 (dt, 1H, J = 14,3; 3,4 Hz), 2,44–2,39 (m, 1H), 2,28–2,23 (m, 1H), 2,04–1,97 (m, 2H), 1,17 (broad s, 2H); MS m/z (%, fragment) (EI): 338; HRMS (m/z) for C22H26O3: [M + H]+calcd = 339.1881, [M + H]+measured = 339.4404.
3.8. Friedel-Crafts Acylation
To the solution of compound 49 [41] (100 mg, 0.510 mol) in dichloromethane (2 mL), AlCl3 (68 mg, 0.510 mol) and different carbonyl chlorides (1.1 eq) were added. The reaction was performed in a sealed tube and left overnight at room temperature. After removal of the solvent, the residue crude product was purified by repeated column chromatography using petroleum ether/dichloromethane mixture as eluent. All of the obtained compounds 50–57 were isolated in the first fractions.

(Z)-4-((5R,9R)-7-Hydroxy-5,7,8,9-tetrahydro-6H-5,9-methanobenzo[7]annulen-6-ilydene)butan-2-one (50): 16 mg (13%); yellow oil; Rf (dichloromethane): 0.5;1H NMR (CDCl3, 600 MHz) δ/ppm: 7.54 (d, 1H, J = 7.7 Hz, HAr), 7.29 (t, 1H, J = 7.7 Hz, HAr), 7.14 (t, 1H, J = 7.7 Hz, HAr), 7.10 (d, 1H, J = 7.7 Hz, HAr), 6.79 (m, 1H), 3.83 (m, 1H), 3.62 (d, 1H, J = 3.6 Hz), 3.24 (t, 1H, J = 6.6 Hz), 3.01 (d, 1H, J = 4.6 Hz), 2.45 (m, 1H), 2.28 (s, 1H, -OH), 2.31 (d, 1H, J = 10.7 Hz,), 2.29 (m, 2H), 2.06–2.02 (m, 1H), 1.95 (s, 3H); MS m/z (%, fragment) (EI): 242 (100); HRMS (m/z) for C16H18O2: [M + H]+calcd = 243.1306, [M + H]+measured = 243.3129.
(5R,9R,Z)-6-((Z)-3-Hydroxybut-2-ene-1-ilydene)-6,7,8,9-tetrahydro-5H-5,9-methano-benzo[7]annulen-7-ol (51): 30 mg (30%); yellow oil; Rf (dichloromethane/petroleum ether 70%): 0.10; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.30 (d, 1H, J = 7.5 Hz, HAr), 7.16 (t, 1H, J = 7.5 Hz, HAr), 6.93 (t, 1H, J = 7.5 Hz, HAr), 6.70 (m, 2H), 6.26 (d, J = 7.3 Hz, 1H), 3.53 (d, 1H, J = 5.1 Hz), 3.45 (t, 1H, J = 4.4 Hz), 3.38 (t, 1H, J = 3.9 Hz), 3.06 (dd, 1H, J = 4.0; 1.5 Hz), 2.99 (t, 1H, J = 5.1 Hz), 2.41 (d, 1H, J = 10.9 Hz), 2.10 (s, 1H, OH), 2.06–2.02 (m, 1H), 1.53 (s, 3H); MS m/z (%, fragment) (EI): 242 (100); HRMS (m/z) for C16H18O2: [M + H]+calcd = 243.1306, [M + H]+measured = 243.3131.
(E)-1-((5S,9R)-5,9-Dihydro-6H-5,9-methanobenzo[7]annulen-6-ilydene)hexa-2,3-dione (52): 21 mg (15%); yellow oil; Rf (dichloromethane): 0.40; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.07 (d, 1H, J= 7.9 Hz, HAr), 7.83 (d, 1H, J = 7.9 Hz, HAr), 7.13–7.09 (m, 2H), 6.26 (dd, 1H, J = 6.8; 1.8 Hz), 6.02 (d, 1H, J = 9.6 Hz), 5.91 (d, 1H, J = 9.6 Hz), 4.25–4.19 (m, 2H), 3.55–3.51 (m, 2H), 2.71 (d, 1H, J = 11.2 Hz), 2.35–2.31 (m, 2H), 2.04–2.01 (m, 1H), 1.63 (s, 3H); MS m/z (%, fragment) (EI): 266 (100); HRMS (m/z) for C18H18O2: [M + H]+calcd = 267.1306, [M + H]+measured = 267.3343.
(Z)-1-((5R,9R)-7-Hydroxy-5,9-dihydro-6H-5,9-methanobenzo[7]annulen-6-ilydene)-pentan-3-one (53): 76 mg (59%); yellow oil; Rf (dichloromethane): 0.29; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.04 (d, 1H, J = 8.5 Hz, HAr), 7.93 (d, 1H, J = 9.1 Hz, HAr), 7.50 (t, 1H, J = 7.7 Hz, HAr), 7.16 (m, 1H, HAr), 6.30 (dd, 1H, J = 3.1; 1.9 Hz), 6.10 (d, 1H, J = 3.1 Hz), 4.59 (d, 1H, J = 6.3 Hz), 4.42 (d, 1H, J = 5.6 Hz), 4.33 (s, 1H, OH), 4.10–4.07 (m, 2H), 2.28 (d, 1H, J = 11.5 Hz), 1.93–1.83 (m, 1H) 1.80–1.69 (m, 2H), 1.00 (t, 3H, J = 5.4 Hz); MS m/z (%, fragment) (EI): 254 (100); HRMS (m/z) for C17H18O2: [M + H]+calcd = 255.1306, [M + H]+measured = 255.3236.

(9Z)-9-[(Z)-3-Hydroxypent-2-enilydene]tricyclo[6.3.1.02,7]dodeca-2(7),3,5-trien-10-ol (54): 8.9 mg (12%); yellow oil; Rf (dichloromethane/petroleum ether 20%): 0.73; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.44–7.02 (m, 4H), 6,64 (m, 1H), 6.27 (d, 1H, J = 7.8 Hz), 3.63 (q, 2H, J = 6.8 Hz), 3.51 (d, 1H, J = 4.5 Hz), 3.44 (t, 1H, J = 4.5 Hz), 3.37 (t, 1H, J = 4.5 Hz), 2.99 (dd, 1H, J = 3.0; 1.5 Hz), 2.95 (t, 1H, J = 4.5 Hz), 2.38 (d, 1H, J = 10.6 Hz), 2.05-2.02 (m, 1H), 1.56 (t, 3H, J = 7.2 Hz); MS m/z (%, fragment) (EI): 256 (100).
(Z)-3-((5R,9R)-7-Hydroxy-5,9-dyhidro-6H-5,9-methanobenzo[7]annulen-6-ilydene)-1-phenylpropan-1-one (55): 29.4 mg (19%); colourless oil; Rf (dichloromethane): 0.38; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.01 (d, 2H, J = 8.7 Hz, HAr), 7.90 (t, 2H, J = 8.9 Hz, HAr), 7.71 (d, 1H, J = 8.3 Hz, HAr), 7.68 (d, 1H, J = 9.2 Hz, HAr), 7.50–7.43 (m, 3H, HAr), 6.30 (m, 1H), 6.10 (d, 1H, J = 3.3 Hz), 4.71 (s, 1H, OH), 4.58 (d, 1H, J = 4.3 Hz), 4.39 (d, 1H, J = 4.9 Hz), 4.12 (t, 2H, J = 4.9 Hz), 2.40 (d, 1H, J = 10.5 Hz), 2.33–2.28 (m, 1H); MS m/z (%, fragment) (EI): 302 (100).
(E)-5-Chloro-1-((5S,9R)-5,9-dyhdro-6H-5,9-methanobenzo[7]annulen-6-ilydene)penta-2,3-dione (56): 7.5 mg (7.3%); colorless oil; Rf (dichloromethane): 0.42; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.01 (d, 1H, J = 7.6 Hz, HAr), 7.13 (d, 1H, J = 7.6 Hz, HAr), 7.10–7.05 (m, 2H, HAr), 6.26 (dd, 1H, J = 6.5; 1.6 Hz), 6.14 (d, 1H, J = 9.3 Hz), 5.98 (d, 1H, J = 9.3 Hz), 4.25–4.19 (m, 2H), 3.55 (m, 2H), 2.71 (d, 1H, J = 11.1 Hz), 2.33 (m, 2H), 2.07–2.03 (m, 1H); MS m/z (%, fragment) (EI): 286 (100), 288 (32).
(Z)-1-(2-Chlorophenyl)-3-((5R,9R)-7-hydroxy-5,9-dihydro-6H-5,9-methanobenzo-[7]annulen-6-ilydene)propan-1-one (57): 9.5 mg (8%); colorless oil; Rf (dichloromethane): 0.40; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.02 (d, 1H, J = 8.9 Hz, HAr), 7.90 (t, 2H, J = 9.2 Hz, HAr), 7.81 (d, 1H, J = 8.3 Hz, HAr), 7.70 (d, 1H, J = 9.2 Hz, HAr), 7.50–7.43 (m, 3H, HAr), 6.28 (dd, 1H, J = 3.2; 1.8 Hz), 6.08 (d, 1H, J = 3.1 Hz), 4.55 (d, 1H, J = 6.1 Hz), 4.21 (d, 1H, J = 5.7 Hz), 3.98 (t, 2H, J = 6.1 Hz), 3.18 (s, 1H, -OH), 2.03–1.99 (m, 2H); MS m/z (%, fragment) (EI): 336 (100), 338 (32).
3.9. Reversible Inhibition of Cholinesterases by Novel Compounds
Inhibition of novel compounds was evaluated for recombinant human AChE and BChE derived from purified human plasma (a generous gift from Dr Florian Nachon, Département de Toxicologie et Risques Chimiques, Institut de Recherche Biomédicale des Armées, Bretigny-sur-Orge, France). The inhibition mixture contained a 0.1 M phosphate buffer, pH 7.4, enzyme (4.5 nM BChE or 0.2 nM AChE) tested compound, and DTNB (0.3 mM; Sigma Chemical Co., St. Louis, MO, USA). Enzyme activity was measured upon addition of ATCh (0.2 or 0.1 mM; Sigma Chemical Co., St. Louis, MO, USA) by the Ellman method [45] at 25 °C and 412 nm, on a Tecan Infinite M200PRO plate reader (Tecan Austria, GmbH, Salzburg, Austria). Due to the low solubility, a 100 mM stock solution of the tested compounds was prepared in DMSO, and the same solvent was in controls as well. The IC50 values were determined from at least three experiments by a nonlinear fit of the compound concentration logarithm values vs.% of enzyme activity using Prism6 software (GraphPad Prism 6 Software, San Diego, USA).
4. Conclusions
The photochemical synthesis resulted in four groups of molecules with different functionalities on the methano-bridged benzobicyclo[3.2.1]octadiene skeleton. The majority of the compounds were more selective for BChE than for AChE, although their inhibitory potency varied with compound functionalities. The structure-activity relationship exhibited an increase in inhibition potency for both enzymes by elimination of the endo-benzylamine substituent. Although none of the tested compounds inhibited in nanomolar range, benzobicyclo[3.2.1]octene compounds 51 and 31 arose as lead compounds for the future development of new AChE and BChE inhibitors, respectively. Therefore, for future design of cholinesterase inhibitors and search for therapeutics of neurological disorders, compounds with a benzobicyclo[3.2.1]octene skeleton, a hydroxyl group on it, and an opened furan ring, should be taken into consideration.
Supplementary Materials
The following are available online. Figures S1–S31 with 1H NMR, 13C NMR, COSY, LR COSY, and NOESY spectrum of prepared compounds, and Cartesian coordinates of optimized geometries of reactants, transition states, and products of reactions 1–4.
Author Contributions
Conceptualization, I.Š. and Z.K.; syntheses and characterization, A.G., R.A.; modelling, D.B.; biological activity measurements, T.Č. and N.M.H.; writing—original draft preparation, I.Š., D.B. and Z.K.; writing—review and editing, T.Č., A.G., D.B., Z.K. and I.Š.; funding acquisition, Z.K. and I.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Croatian Science Foundation (project IP-2018-01-7683) and the University of Zagreb short term scientific support (2019) under the title Synthesis and functionalization of novel (hetero)polycyclic photoproducts as potential cholinesterase inhibitors.
Acknowledgments
The competent help by Željko Marinić in the NMR measurements and computational resources provided by the University of Zagreb Computing Centre (SRCE) is acknowledged. A generous gift of enzymes (AChE; BChE) from Dr Florian Nachon, Département de Toxicologie et Risques Chimiques, Institut de Recherche Biomédicale des Armées, Bretigny-sur-Orge, France is also gratefully acknowledged. The authors thank Makso Herman for language editing.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Darvesh, S.; Walsh, R.; Kumar, R.; Caines, A.; Roberts, S.; Magee, D.; Rockwood, K.; Martin, E. Inhibition of human cholinesterases by drugs used to treat Alzheimer disease. Alzheimer Dis. Assoc. 2003, 17, 117–126. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinergic function and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 1403. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Alvarez, A.; Opazo, C.; Alarcón, R.; Garrido, J.; Inestrosa, N.C. Acetylcholinesterase promotes the aggregation of amyloid-β-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol. 1997, 272, 348–361. [Google Scholar] [CrossRef]
- Calderón, F.H.; Von Bernhardi, R.; De Ferrari, G.; Luza, S.; Aldunate, R.; Inestrosa, N.C. Toxic effects of acetylcholinesterase on neuronal and glial-like cells in vitro. Mol. Psychiatry 1998, 3, 247–255. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alarcón, R. Molecular interactions of acetylcholinesterase with senile plaques. J. Physiol. Paris 1998, 92, 341–344. [Google Scholar] [CrossRef]
- Reid, G.A.; Darvesh, S. Butyrylcholinesterase-knockout reduces brain deposition of fibrillar β-amyloid in an Alzheimer mouse model. Neuroscience 2015, 298, 424–435. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterases in human brain: The effect of cholinesterase inhibitors on Alzheimer’s disease and related disorders. In The Brain Cholinergic System in Health and Disease; Giacobini, E., Pepeu, E., Eds.; Informa Healthcare: Abingdon, UK, 2006; pp. 235–264. [Google Scholar]
- Katz, N.K.; Barohn, R.J. The history of acetylcholinesterase inhibitors in the treatment of myasthenia gravis. Neuropharmacology 2020, 108303. [Google Scholar] [CrossRef] [PubMed]
- Shaydenfish, D.; Wongtangman, K.; Eikermann, M.; Schaefer, M.S. The effects of acetylcholinesterase inhibitors on morbidity after general anesthesia and surgery. Neuropharmacology 2020, 173, 108134. [Google Scholar] [CrossRef]
- Mohammad, D.; Chan, P.; Bradley, P.J.; Lanctôt, K.; Herrmann, N. Acetylcholinesterase inhibitors for treating dementia symptoms—A safety evaluation. Expert Opin. Drug Saf. 2017, 16, 1009–1019. [Google Scholar] [CrossRef]
- Semenov, V.E.; Zueva, I.V.; Mukhamedyarov, M.A.; Lushchekina, S.V.; Petukhova, E.O.; Gubaidullina, L.M.; Krylova, E.S.; Saifina, L.F.; Lenina, O.A.; Petrov, K.A. Novel acetylcholinesterase inhibitors based on uracil moiety for possible treatment of Alzheimer disease. Molecules 2020, 25, 4191. [Google Scholar] [CrossRef] [PubMed]
- Radić, Z.; Pickering, N.A.; Vellom, D.C.; Camp, S.; Taylor, P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993, 32, 12074–12084. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Radić, Z.; Hosea, N.A.; Camp, S.; Marchot, P.; Berman, H.A. Structural bases for the specificity of cholinesterase catalysis and inhibition. Toxicol. Lett. 1995, 82–83, 453–458. [Google Scholar] [CrossRef]
- Saxena, A.; Redman, A.M.G.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in active site gorge dimensions of cholinesterase revealed by binding of inhibitors to human butyrylcholinesterase. Chem. Biol. Interact. 1999, 119–120, 61–69. [Google Scholar] [CrossRef]
- Bosak, A.; Gazić Smilović, I.; Šinko, G.; Vinković, V.; Kovarik, Z. Metaproterenol, isoproterenol, and their bisdimethylcarbamate derivatives as human cholinesterase inhibitors. J. Med. Chem. 2012, 55, 6716–6723. [Google Scholar] [CrossRef]
- Katalinić, M.; Bosak, A.; Kovarik, Z. Flavonoids as inhibitors of human butyrylcholinesterase variants. Food Technol. Biotechnol. 2014, 52, 64–67. [Google Scholar] [CrossRef]
- Bosak, A.; Knežević, A.; Gazić Smilović, I.; Šinko, G.; Kovarik, Z. Resorcinol-, catechol- and saligenin-based bronchodilating β2-agonists as inhibitors of human cholinesterase activity. J. Enzyme Inhib. Med. Chem. 2017, 32, 789–797. [Google Scholar] [CrossRef]
- Bosak, A.; Ramić, A.; Šmidlehner, T.; Hrenar, T.; Primožič, I.; Kovarik, Z. Design and evaluation of selective butyrylcholinesterase inhibitors based on Cinchona alkaloid scaffold. PLoS ONE 2018, 13, e0205193. [Google Scholar] [CrossRef] [PubMed]
- Bosak, A.; Opsenica, D.M.; Šinko, G.; Zlatar, M.; Kovarik, Z. Structural aspects of 4-aminoquinolines as reversible inhibitors of human acetylcholinesterase and butyrylcholinesterase. Chem. Biol. Interact. 2019, 308, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Šagud, I.; Škorić, I.; Burčul, F. Naphthoxazoles and heterobenzoxazoles: Cholinesterase inhibiting and antioxidant activity. Turk. J. Chem. 2019, 43, 118–124. Available online: https://dergipark.org.tr/en/pub/tbtkchem/issue/45572/572769 (accessed on 5 October 2020). [CrossRef]
- Šagud, I.; Maček Hrvat, N.; Grgičević, A.; Čadež, T.; Hodak, J.; Dragojević, M.; Lasić, K.; Kovarik, Z.; Škorić, I. Design, synthesis and cholinesterase inhibitory properties of new oxazole benzylamine derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Maraković, N.; Knežević, A.; Rončević, I.; Brazzolotto, X.; Kovarik, Z.; Šinko, G. Enantioseparation, in vitro testing, and structural characterization of triple-binding reactivators of organophosphate-inhibited cholinesterases. Biochem. J. 2020, 15, 2771–2790. [Google Scholar] [CrossRef]
- Ratković, A.; Marinić, Ž.; Škorić, I. Flow-photochemical synthesis of the functionalized benzobicyclo[3.2.1]octadiene skeleton. J. Mol. Struct. 2018, 1168, 165–174. [Google Scholar] [CrossRef]
- Šagud, I.; Škorić, I.; Vuk, D.; Ratković, A.; Burčul, F. Acetyl- and butyrylcholinesterase inhibitory activity of selected photochemically synthesized polycycles. Turk. J. Chem. 2019, 43, 1170–1182. [Google Scholar] [CrossRef]
- Su, X.; Sun, Y.; Yao, J.; Chen, H.; Chen, C. Acid-promoted bicyclization of arylacetylenes to benzobicyclo[3.2.1]octanes through cationic rearrangements. Chem. Commun. 2016, 52, 4537–4540. [Google Scholar] [CrossRef]
- Thomson, C.G.; Carlson, E.; Chicchi, G.G.; Kulagowski, J.J.; Kurtz, M.M.; Swain, C.J.; Tsao, K.L.C.; Wheeldon, A. Synthesis and structure-activity relationships of 8-azabicyclo[3.2.1]octane benzylamine NK1 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 811–814. [Google Scholar] [CrossRef]
- Škorić, I.; Šmehil, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Photochemistry of ω-(o-vinylphenyl)-ω’-(phenyl/2-furyl)butadienes: New approach to 4-substituted benzobicyclo[3.2.1]octadienes. J. Photochem. Photobiol. A Chem. 2009, 207, 190–196. [Google Scholar]
- Ratković, A.; Kelava, V.; Marinić, Ž.; Škorić, I. Buchwald-Hartwig amination of the chloro substituted benzobicyclo[3.2.1]octadiene skeleton using primary benzylic amines. J. Mol. Struct. 2019, 1179, 597–607. [Google Scholar] [CrossRef]
- Kikaš, I.; Škorić, I.; Marinić, Ž.; Šindler-Kulyk, M. Synthesis and phototransformations of novel styryl-substituted furo-benzobicyclo[3.2.1]octadiene derivatives. Tetrahedron 2010, 66, 9405–9414. [Google Scholar] [CrossRef]
- Ratković, A.; Pavlović, K.; Barić, D.; Marinić, Ž.; Grgičević, I.; Škorić, I. Modeling and synthesis of novel oxime derivatives as potential cholinesterase inhibitors. J. Mol. Struct. 2020, 1200, 127149. [Google Scholar] [CrossRef]
- Srinivasan, R.; Chandrasekharam, M.; Vani, P.V.S.N.; Seema Chida, A.; Singh, A.K. Epoxidation of olefins at low temperature using m-chloroperbenzoic acid. Synth. Commun. 2002, 32, 1853–1858. [Google Scholar] [CrossRef]
- Fringuelli, F.; Germani, R.; Pizzo, F.; Savelli, G. Epoxidation reaction with m-chloroperoxybenzoic acid in water. Tetrahedron Lett. 1989, 30, 1427–1428. [Google Scholar] [CrossRef]
- Nirmal, K.J.; Verkade, J.G. Phase-Vanishing Methodology for Efficient Bromination, Alkylation, Epoxidation, and Oxidation Reactions of Organic Substrates. Org. Lett. 2003, 5, 3787–3790. [Google Scholar] [CrossRef]
- Mattay, J.; Griesbeck, A. Photochemical Key Steps in Organic Synthesis. An Experimental Course Book, 1st ed.; VCH: Veinheim, Germany, 1994; pp. 214–215. [Google Scholar]
- Mohammadi-Farani, A.; Ahmadi, A.; Nadri, H.; Aliabadi, A. Synthesis, docking and acetylcholinesterase inhibitory assessment of 2-(2-(4-Benzylpiperazin-1-yl)ethyl)isoindoline-1,3-dione derivatives with potential anti-Alzheimer effects. DARU J. Pharm. Sci. 2013, 21, 1–9. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Šindler-Kulyk, M.; Kragol, G.; Piantanida, I.; Tomšić, S.; Vujković-Cvijin, I.; Marinić, Ž.; Metelko, B. Photochemistry of β-(5-Substituted-2-furyl)-o-divinylbenzenes: Substituent effects on the reaction course. Croat. Chem. Acta 1996, 69, 1593–1602. [Google Scholar] [CrossRef]
- Šinko, G.; Kovarik, Z.; Reiner, E.; Simeon-Rudolf, V.; Stojan, J. Mechanism of stereoselective interaction between butyrylcholinesterase and ethopropazine enantiomers. Biochimie 2011, 93, 1797–1807. [Google Scholar] [CrossRef]
- Vuk, D.; Potroško, D.; Šindler-Kulyk, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Škorić, I. Synthesis and photochemical transformations of new butadiene chromophores: The influence of the nature and position of chlorine substituent on the photoinduced behavior. J. Mol. Struct. 2013, 1051, 1–14. [Google Scholar] [CrossRef]
- Vuk, D.; Molčanov, K.; Škorić, I. Novel arylated chloro- and methoxy-1,3-dibutadienes: Influence of substituents on molecular conformation and crystal packing. J. Mol. Struct. 2014, 1068, 124–129. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. New and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]





















Sample Availability: Samples of the compounds endo-1, endo-36, endo-37–39, endo-42, endo-43, endo-45–48 are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).