
Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods



Abstract
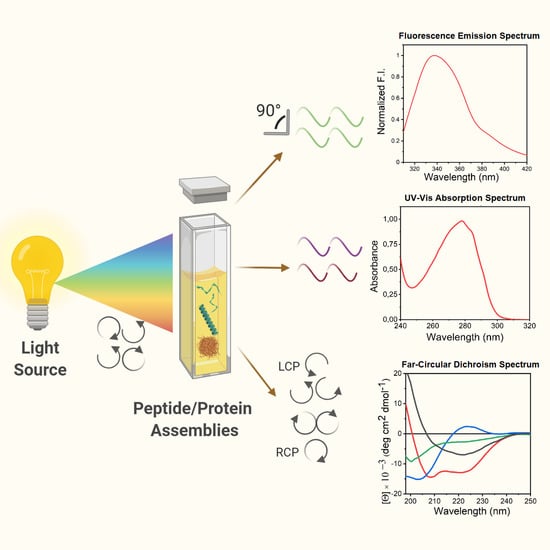
1. Setting the Frame and Initial Considerations
An Overview of Protein Structure and Self-Assembly
2. UV-Vis Absorption Spectroscopy and Turbidity Are Initial Approaches for Detecting the Self-Assembly/Aggregation-Prone Tendency of Proteins
2.1. Principles and General Considerations
2.2. Indirect Methods: Evaluating Protein Absorption Spectra
2.2.1. Intrinsic Chromophores
2.2.2. Extrinsic Chromophores
2.3. Direct Methods: Analyzing Protein Aggregation in Solution
3. Fluorescence Spectroscopy Is a Versatile Tool for Evaluating Peptide and Protein Self-Assembly
3.1. Principles and General Considerations
3.2. Intrinsic Fluorophore
3.3. Extrinsic Fluorophore
3.4. Assessment of Protein and Peptide Self-Assembly Processes by Fluorescence Spectroscopy: Methods and Selected Examples
3.4.1. Steady-State Fluorescence Methods
3.4.2. Time-Resolved Methods
3.4.3. Fluorescence Correlation Spectroscopy (FCS)
4. Circular Dichroism Makes It Possible to Follow Secondary and Tertiary Structure Changes Due to Self-Assembly
4.1. Main Principles and Structural Protein Features
4.2. Experimental Considerations of CD Spectroscopy
4.3. Uses of Far-UV CD Spectroscopy on the Study of Self-Assembled Systems
4.3.1. Analysis of Amyloidogenic Proteins
4.3.2. Evaluation of Non-Amyloid Systems
4.3.3. The Use of CD to Assess Conformational Equilibriums during Self-Assembly by Changes on the Microenvironment
4.4. Near-UV CD Analysis of Protein Aggregation
5. Some Considerations in Order to Avoid Protein Aggregation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiesa, G.; Kiriakov, S.; Khalil, A.S. Protein assembly systems in natural and synthetic biology. BMC Biol. 2020, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Engelberth, S.A.; Bacino, M.S.; Sandhu, S.; Li, W.; Bonde, J.; Habelitz, S. Progression of Self-Assembly of Amelogenin Protein Supramolecular Structures in Simulated Enamel Fluid. Biomacromolecules 2018, 19, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef]
- Wolz, M.; Mersch, E.; Kulozik, U. Thermal aggregation of whey proteins under shear stress. Food Hydrocoll. 2016, 56, 396–404. [Google Scholar] [CrossRef]
- Ma, S.; Han, W.; Li, L.; Zheng, X.; Wang, X. The thermal stability, structural changeability, and aggregability of glutenin and gliadin proteins induced by wheat bran dietary fiber. Food Funct. 2019, 10, 172–179. [Google Scholar] [CrossRef]
- Sun, X.; Jin, H.; Li, Y.; Feng, H.; Liu, C.; Xu, J. The Molecular Properties of Peanut Protein: Impact of Temperature, Relative Humidity and Vacuum Packaging during Storage. Molecules 2018, 23, 2618. [Google Scholar] [CrossRef]
- Singh, T.K.; Oiseth, S.K.; Lundin, L.; Day, L. Influence of heat and shear induced protein aggregation on the in vitro digestion rate of whey proteins. Food Funct. 2014, 5, 2686–2698. [Google Scholar] [CrossRef]
- Satitsuksanoa, P.; Jansen, K.; Globinska, A.; van de Veen, W.; Akdis, M. Regulatory Immune Mechanisms in Tolerance to Food Allergy. Front. Immunol. 2018, 9, 2939. [Google Scholar] [CrossRef]
- Islam, M.S.; Reineke, J.; Kaushik, R.; Woyengo, T.; Baride, A.; Alqahtani, M.S.; Perumal, O. Bioadhesive Food Protein Nanoparticles as Pediatric Oral Drug Delivery System. ACS Appl. Mater. Interfaces 2019, 11, 18062–18073. [Google Scholar] [CrossRef] [PubMed]
- Gulfam, M.; Kim, J.E.; Lee, J.M.; Ku, B.; Chung, B.H.; Chung, B.G. Anticancer drug-loaded gliadin nanoparticles induce apoptosis in breast cancer cells. Langmuir 2012, 28, 8216–8223. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.K.; Rankin, S.A.; Lee, M.R.; Lee, W.J. Development and Characterization of Whey Protein-Based Nano-Delivery Systems: A Review. Molecules 2019, 24, 3254. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, R.; Velyvis, A.; Kay, L.E. Solution NMR of supramolecular complexes: Providing new insights into function. Nat. Methods 2007, 4, 697–703. [Google Scholar] [CrossRef]
- Riek, R.; Pervushin, K.; Wüthrich, K. TROSY and CRINEPT: NMR with large molecular and supramolecular structures in solution. Trends Biochem. Sci. 2000, 25, 462–468. [Google Scholar] [CrossRef]
- Naito, A.; Kawamura, I. Solid-state NMR as a method to reveal structure and membrane-interaction of amyloidogenic proteins and peptides. Biochim. Biophys. Acta 2007, 1768, 1900–1912. [Google Scholar] [CrossRef] [PubMed]
- Weingarth, M.; Baldus, M. Solid-state NMR-based approaches for supramolecular structure elucidation. Acc. Chem. Res. 2013, 46, 2037–2046. [Google Scholar] [CrossRef]
- Linser, R. Solid-state NMR spectroscopic trends for supramolecular assemblies and protein aggregates. Solid State Nucl. Magn. Reson. 2017, 87, 45–53. [Google Scholar] [CrossRef]
- Duer, M.J. Solid state NMR Spectroscopy: Principles and Applications; Blackwell Science Ltd.; John Wiley & Sons: Oxford, UK, 2008. [Google Scholar]
- Cavanagh, J.; Fairbrother, W.J.; Palmer III, A.G.; Skelton, N.J. Protein NMR Spectroscopy: Principles and Practice; Academic Press; Elsevier: Burllington, NJ, USA, 2007. [Google Scholar]
- Uversky, V.N.; Lyubchenko, Y. Bio-nanoimaging: Protein Misfolding and Aggregation; Academic Press; Elsevier: London, UK, 2013. [Google Scholar]
- Lu, M.; Kaminski, C.F.; Schierle, G.S.K. Advanced fluorescence imaging of in situ protein aggregation. Phys. Biol. 2020, 17, 021001. [Google Scholar] [CrossRef]
- Creasey, R.G.; Gibson, C.T.; Voelcker, N.H. Characterization of fiber-forming peptides and proteins by means of atomic force microscopy. Curr. Protein Pept. Sci. 2012, 13, 232–257. [Google Scholar] [CrossRef]
- Ognjenovic, J.; Grisshammer, R.; Subramaniam, S. Frontiers in Cryo Electron Microscopy of Complex Macromolecular Assemblies. Annu. Rev. Biomed. Eng. 2019, 21, 395–415. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Saibil, H.R. Cryo-EM of amyloid fibrils and cellular aggregates. Curr. Opin. Struct. Biol. 2019, 58, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. A review on protein oligomerization process. Int. J. Precis. Eng. Manuf. 2015, 16, 2731–2760. [Google Scholar] [CrossRef]
- Koehl, P. Protein Structure Classification. In Reviews in Computational Chemistry; Lipkowitz, K.B., Cundari, T.R., Gillet, V.J., Boyd, D.B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2006; Chapter 1; pp. 1–55. [Google Scholar] [CrossRef]
- Dhotel, A.; Chen, Z.; Delbreilh, L.; Youssef, B.; Saiter, J.M.; Tan, L. Molecular motions in functional self-assembled nanostructures. Int. J. Mol. Sci. 2013, 14, 2303–2333. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, E.; Venema, P. Self-assembly and aggregation of proteins. Curr. Opin. Coll. Interface Sci. 2007, 12, 158–165. [Google Scholar] [CrossRef]
- Herrera, M.G.; Benedini, L.A.; Lonez, C.; Schilardi, P.L.; Hellweg, T.; Ruysschaert, J.M.; Dodero, V.I. Self-assembly of 33-mer gliadin peptide oligomers. Soft Matter 2015, 11, 8648–8660. [Google Scholar] [CrossRef]
- Khire, T.S.; Kundu, J.; Kundu, S.C.; Yadavalli, V.K. The fractal self-assembly of the silk protein sericin. Soft Matter 2010, 6, 2066–2071. [Google Scholar] [CrossRef]
- van Raaij, M.J. and Mitraki, A. Natural Fibrous Proteins: Structural Analysis, Assembly, and Applications. In Proteins in Solution and at Interfaces; Ruso, J.M., Piñeiro, Á., Eds.; Jon Wiley & Sons: Hoboken, NJ, USA, 2013; Chapter 11; pp. 219–232. [Google Scholar] [CrossRef]
- Crick, S. and Pappu, R. Thermodynamic and Kinetic Models for Aggregation of Intrinsically Disordered Proteins. In Peptide Folding, Misfolding, and Nonfolding; Schweitzer-Stenner, R., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 413–440. [Google Scholar]
- Dine, E.; Gil, A.A.; Uribe, G.; Brangwynne, C.P.; Toettcher, J.E. Protein Phase Separation Provides Long-Term Memory of Transient Spatial Stimuli. Cell Syst. 2018, 6, 655–663. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- McManus, J.J.; Charbonneau, P.; Zaccarelli, E.; Asherie, N. The physics of protein self-assembly. Current Opin. Coll. Interface Sci. 2016, 22, 73–79. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils: Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.; Hsu, C.C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. alpha-Sheet secondary structure in amyloid beta-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Lu, T.; Li, F. Stabilization of Solvent to alpha-Sheet Structure and Conversion between alpha-Sheet and beta-Sheet in the Fibrillation Process of Amyloid Peptide. J. Phys. Chem. B 2019, 123, 9576–9583. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.C.; Daggett, V. Drivers of alpha-Sheet Formation in Transthyretin under Amyloidogenic Conditions. Biochemistry 2019, 58, 4408–4423. [Google Scholar] [CrossRef] [PubMed]
- Whittington, S.J.; Creamer, T.P. Salt bridges do not stabilize polyproline II helices. Biochemistry 2003, 42, 14690–14695. [Google Scholar] [CrossRef] [PubMed]
- Berisio, R.; Loguercio, S.; De Simone, A.; Zagari, A.; Vitagliano, L. Polyproline helices in protein structures: A statistical survey. Protein Pept. Lett. 2006, 13, 847–854. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Sternberg, M.J. Left-handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 1993, 229, 472–493. [Google Scholar] [CrossRef]
- Bochicchio, B.; Tamburro, A.M. Polyproline II structure in proteins: Identification by chiroptical spectroscopies, stability, and functions. Chirality 2002, 14, 782–792. [Google Scholar] [CrossRef]
- Cubellis, M.V.; Caillez, F.; Blundell, T.L.; Lovell, S.C. Properties of polyproline II, a secondary structure element implicated in protein-protein interactions. Proteins 2005, 58, 880–892. [Google Scholar] [CrossRef]
- Blanch, E.W.; Morozova-Roche, L.A.; Cochran, D.A.; Doig, A.J.; Hecht, L.; Barron, L.D. Is polyproline II helix the killer conformation? A Raman optical activity study of the amyloidogenic prefibrillar intermediate of human lysozyme. J. Mol. Biol. 2000, 301, 553–563. [Google Scholar] [CrossRef]
- Eker, F.; Griebenow, K.; Schweitzer-Stenner, R. Abeta (1-28) fragment of the amyloid peptide predominantly adopts a polyproline II conformation in an acidic solution. Biochemistry 2004, 43, 6893–6898. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.G.; Zamarreno, F.; Costabel, M.; Ritacco, H.; Hutten, A.; Sewald, N.; Dodero, V.I. Circular dichroism and electron microscopy studies in vitro of 33-mer gliadin peptide revealed secondary structure transition and supramolecular organization. Biopolymers 2014, 101, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.G.; Gomez Castro, M.F.; Prieto, E.; Barrera, E.; Dodero, V.I.; Pantano, S.; Chirdo, F. Structural conformation and self-assembly process of p31-43 gliadin peptide in aqueous solution. Implications for celiac disease. FEBS J. 2020, 287, 2134–2149. [Google Scholar] [CrossRef]
- Dodero, V.I.; Messina, P.V. Analyzing the Solution State of Protein Structure, Interactions, and Ligands by Spectroscopic Methods. In Proteins in Solution and at Interfaces; Ruso, J.M., Piñeiro, Á., Eds.; Jon Wiley & Sons: Hoboken, NJ, USA, 2013; Chapter 4; pp. 73–98. [Google Scholar] [CrossRef]
- Esfandiary, R.; Middaugh, C.R. Ultraviolet Absorption Spectroscopy. In Analysis of Aggregates and Particles in Protein Pharmaceuticals; Mahler, H.-C., Jiskoot, W., Eds.; Jon Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 169–200. [Google Scholar] [CrossRef]
- Berns, R.S. Billmeyer and Saltzman’s Principles of Color Technology, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Perkampus, H.-H. UV-VIS Spectroscopy and Its Applications; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press; Springer: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Schmid, F. Spectroscopic Techniques to Study Protein Folding and Stability. In Protein Folding Handbook; Buchner, J., Kiefhaber, T., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2005; Chapter 2; pp. 22–44. [Google Scholar]
- Correia, M.; Neves-Petersen, M.T.; Jeppesen, P.B.; Gregersen, S.; Petersen, S.B. UV-light exposure of insulin: Pharmaceutical implications upon covalent insulin dityrosine dimerization and disulphide bond photolysis. PLoS ONE 2012, 7, e50733. [Google Scholar] [CrossRef] [PubMed]
- Lavrinenko, I.A.; Holyavka, M.G.; Chernov, V.E.; Artyukhov, V.G. Second derivative analysis of synthesized spectra for resolution and identification of overlapped absorption bands of amino acid residues in proteins: Bromelain and ficin spectra in the 240-320nm range. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 227, 117722. [Google Scholar] [CrossRef] [PubMed]
- Lucas, L.H.; Ersoy, B.A.; Kueltzo, L.A.; Joshi, S.B.; Brandau, D.T.; Thyagarajapuram, N.; Peek, L.J.; Middaugh, C.R. Probing protein structure and dynamics by second-derivative ultraviolet absorption analysis of cation-{pi} interactions. Protein Sci. 2006, 15, 2228–2243. [Google Scholar] [CrossRef] [PubMed]
- Maciazek-Jurczyk, M.; Janas, K.; Pozycka, J.; Szkudlarek, A.; Rogoz, W.; Owczarzy, A.; Kulig, K. Human Serum Albumin Aggregation/Fibrillation and its Abilities to Drugs Binding. Molecules 2020, 25, 618. [Google Scholar] [CrossRef]
- Herrera, M.G.; Veuthey, T.V.; Dodero, V.I. Self-organization of gliadin in aqueous media under physiological digestive pHs. Coll. Surf. B Biointerfaces 2016, 141, 565–575. [Google Scholar] [CrossRef]
- Sequeira, M.A.; Herrera, M.G.; Dodero, V.I. Modulating amyloid fibrillation in a minimalist model peptide by intermolecular disulfide chemical reduction. Phys. Chem. Chem. Phys. 2019, 21, 11916–11923. [Google Scholar] [CrossRef]
- Karolczak, J.; Kowalska, D.; Lukaszewicz, A.; Maciejewski, A.; Steer, R.P. Photophysical studies of porphyrins and metalloporphyrins: Accurate measurements of fluorescence spectra and fluorescence quantum yields for Soret band excitation of zinc tetraphenylporphyrin. J. Phys. Chem. A 2004, 108, 4570–4575. [Google Scholar] [CrossRef]
- Karnaukhova, E.; Rutardottir, S.; Rajabi, M.; Wester Rosenlof, L.; Alayash, A.I.; Akerstrom, B. Characterization of heme binding to recombinant alpha1-microglobulin. Front. Physiol. 2014, 5, 465. [Google Scholar] [CrossRef]
- Hasan, S.; Naeem, A. Consequence of macromolecular crowding on aggregation propensity and structural stability of haemoglobin under glycating conditions. Int. J. Biol. Macromol. 2020, 162, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Iram, A.; Alam, T.; Khan, J.M.; Khan, T.A.; Khan, R.H.; Naeem, A. Molten globule of hemoglobin proceeds into aggregates and advanced glycated end products. PLoS ONE 2013, 8, e72075. [Google Scholar] [CrossRef] [PubMed]
- Inouye, H.; Kirschner, D.A. Alzheimer’s beta-amyloid: Insights into fibril formation and structure from Congo red binding. Subcell. Biochem. 2005, 38, 203–224. [Google Scholar] [CrossRef]
- Wu, C.; Scott, J.; Shea, J.E. Binding of Congo red to amyloid protofibrils of the Alzheimer Abeta (9-40) peptide probed by molecular dynamics simulations. Biophys. J. 2012, 103, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Inouye, H.; Kirschner, D.A. A beta fibrillogenesis: Kinetic parameters for fibril formation from congo red binding. J. Struct. Biol. 2000, 130, 123–129. [Google Scholar] [CrossRef]
- Wu, J.W.; Liu, K.N.; How, S.C.; Chen, W.A.; Lai, C.M.; Liu, H.S.; Hu, C.J.; Wang, S.S. Carnosine’s effect on amyloid fibril formation and induced cytotoxicity of lysozyme. PLoS ONE 2013, 8, e81982. [Google Scholar] [CrossRef]
- Coelho, F.R.; Iqbal, A.; Linares, E.; Silva, D.F.; Lima, F.S.; Cuccovia, I.M.; Augusto, O. Oxidation of the tryptophan 32 residue of human superoxide dismutase 1 caused by its bicarbonate-dependent peroxidase activity triggers the non-amyloid aggregation of the enzyme. J. Biol. Chem. 2014, 289, 30690–30701. [Google Scholar] [CrossRef]
- Rymer, D.L.; Good, T.A. The role of prion peptide structure and aggregation in toxicity and membrane binding. J. Neurochem. 2000, 75, 2536–2545. [Google Scholar] [CrossRef]
- Klunk, W.E.; Jacob, R.F.; Mason, R.P. Quantifying amyloid beta-peptide (A beta) aggregation using the Congo red-Abeta (CR-abeta) spectrophotometric assay. Anal. Biochem. 1999, 266, 66–76. [Google Scholar] [CrossRef]
- Krimm, S. The hydrophobic effect: Formation of micelles and biological membranes, Charles Tanford, Wiley-Interscience. J. Polym. Sci. Polym. Lett. 1980, 18, 687. [Google Scholar] [CrossRef]
- Siddiqui, G.A.; Naeem, A. Aggregation of globular protein as a consequences of macromolecular crowding: A time and concentration dependent study. Int. J. Biol. Macromol. 2018, 108, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Nonaka, T.; Terada, M.; Tamaoka, A.; Hisanaga, S.; Hasegawa, M. alpha-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J. Biol. Chem. 2016, 291, 15046–15056. [Google Scholar] [CrossRef]
- Mangione, P.P.; Verona, G.; Corazza, A.; Marcoux, J.; Canetti, D.; Giorgetti, S.; Raimondi, S.; Stoppini, M.; Esposito, M.; Relini, A.; et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018, 293, 14192–14199. [Google Scholar] [CrossRef]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11, 302. [Google Scholar] [CrossRef]
- Mahler, H.C.; Muller, R.; Friess, W.; Delille, A.; Matheus, S. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur. J. Pharm. Biopharm. 2005, 59, 407–417. [Google Scholar] [CrossRef]
- Gupta, A.; Mahalakshmi, R. Helix–strand interaction regulates stability and aggregation of the human mitochondrial membrane protein channel VDAC3. J. Gen. Physiol. 2019, 151, 489–504. [Google Scholar] [CrossRef]
- Gupta, A.; Mahalakshmi, R. Single-residue physicochemical characteristics kinetically partition membrane protein self-assembly and aggregation. J. Biol. Chem. 2020, 295, 1181–1194. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Poole, R.A.; Hawe, A.; Jiskoot, W.; Braeckmans, K. Fluorescence Spectroscopy to Characterize Protein Aggregates and Particles. In Analysis of Aggregates and Particles in Protein Pharmaceuticals; Mahler, H.-C., Jiskoot, W., Eds.; Jon Wiley & Sons: Hoboken, NJ, USA, 2012; Chapter 9; pp. 201–226. [Google Scholar] [CrossRef]
- Jameson, D. Introduction to Fluorescence; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Lawaetz, A.J.; Stedmon, C.A. Fluorescence intensity calibration using the Raman scatter peak of water. Appl. Spectrosc. 2009, 63, 936–940. [Google Scholar] [CrossRef]
- Lee, J.; Ross, R.T. Absorption and Fluorescence of Tyrosine Hydrogen-Bonded to Amide-like Ligands. J. Phys. Chem. B 1998, 102, 4612–4618. [Google Scholar] [CrossRef]
- Vivian, J.T.; Callis, P.R. Mechanisms of Tryptophan Fluorescence Shifts in Proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef]
- Fuentes, L.; Oyola, J.; Fernández, M.; Quiñones, E. Conformational Changes in Azurin from Pseudomona aeruginosa Induced through Chemical and Physical Protocols. Biophys. J. 2004, 87, 1873–1880. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burstein, E.A.; Abornev, S.M.; Reshetnyak, Y.K. Decomposition of Protein Tryptophan Fluorescence Spectra into Log-Normal Components. I. Decomposition Algorithms. Biophys. J. 2001, 81, 1699–1709. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1862–1885. [Google Scholar] [CrossRef]
- Laptenok, S.P.; Visser, N.V.; Engel, R.; Westphal, A.H.; van Hoek, A.; van Mierlo, C.P.; van Stokkum, I.H.; van Amerongen, H.; Visser, A.J. A general approach for detecting folding intermediates from steady-state and time-resolved fluorescence of single-tryptophan-containing proteins. Biochemistry 2011, 50, 3441–3450. [Google Scholar] [CrossRef]
- Bekard, I.B.; Dunstan, D.E. Tyrosine autofluorescence as a measure of bovine insulin fibrillation. Biophys. J. 2009, 97, 2521–2531. [Google Scholar] [CrossRef]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef]
- Bertoncini, W.C.; Celej, M.S.; Science, P. Small molecule fluorescent probes for the detection of amyloid self-assembly in vitro and in vivo. Curr. Protein Pept. Sci. 2011, 12, 206–220. [Google Scholar] [CrossRef]
- Demchenko, A.P. Ultraviolet Spectroscopy of Proteins; Springer: Berlin, Germany, 1986. [Google Scholar]
- Engelborghs, Y.; Visser, A. Fluorescence Spectroscopy and Microscopy; Springer: New York, NY, USA, 2016; pp. 1–536. [Google Scholar]
- LeVine III, H. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Giasson, B.I.; Crowe, A.; Kung, M.P.; Zhuang, Z.P.; Trojanowski, J.Q.; Lee, V.M. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J. Neurochem. 2003, 86, 1359–1368. [Google Scholar] [CrossRef]
- LeVine, H. Mechanism of A beta (1-40) fibril-induced fluorescence of (trans,trans)-1-bromo-2,5-bis(4-hydroxystyryl)benzene (K114). Biochemistry 2005, 44, 15937–15943. [Google Scholar] [CrossRef] [PubMed]
- Aslund, A.; Sigurdson, C.J.; Klingstedt, T.; Grathwohl, S.; Bolmont, T.; Dickstein, D.L.; Glimsdal, E.; Prokop, S.; Lindgren, M.; Konradsson, P.; et al. Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem. Biol. 2009, 4, 673–684. [Google Scholar] [CrossRef]
- Ran, C.; Xu, X.; Raymond, S.B.; Ferrara, B.J.; Neal, K.; Bacskai, B.J.; Medarova, Z.; Moore, A. Design, synthesis, and testing of difluoroboron-derivatized curcumins as near-infrared probes for in vivo detection of amyloid-beta deposits. J. Am. Chem. Soc. 2009, 131, 15257–15261. [Google Scholar] [CrossRef] [PubMed]
- Benzeid, H.; Mothes, E.; Essassi, E.M.; Faller, P.; Pratviel, G. A thienoquinoxaline and a styryl-quinoxaline as new fluorescent probes for amyloid-β fibrils. C. R. Chim. 2012, 15, 79–85. [Google Scholar] [CrossRef]
- Ono, M.; Watanabe, H.; Kimura, H.; Saji, H. BODIPY-based molecular probe for imaging of cerebral beta-amyloid plaques. ACS Chem. Neurosci. 2012, 3, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Tonali, N.; Dodero, V.I.; Kaffy, J.; Hericks, L.; Ongeri, S.; Sewald, N. Real-Time BODIPY-Binding Assay To Screen Inhibitors of the Early Oligomerization Process of Abeta1-42 Peptide. ChemBioChem 2020, 21, 1129–1135. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Rodina, N.P.; Sulatsky, M.I.; Povarova, O.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Investigation of alpha-Synuclein Amyloid Fibrils Using the Fluorescent Probe Thioflavin T. Int. J. Mol. Sci. 2018, 19, 2486. [Google Scholar] [CrossRef]
- Celej, M.S.; Jares-Erijman, E.A.; Jovin, T.M. Fluorescent N-arylaminonaphthalene sulfonate probes for amyloid aggregation of alpha-synuclein. Biophys. J. 2008, 94, 4867–4879. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, I.-H.; Choe, Y.-J.; Kang, S.; Kim, H.Y.; Gai, W.-P.; Hahn, J.-S.; Paik, S.R. Real-time analysis of amyloid fibril formation of α-synuclein using a fibrillation-state-specific fluorescent probe of JC-1. Biochem. J. 2009, 418, 311–323. [Google Scholar] [CrossRef]
- Paslawski, W.; Andreasen, M.; Nielsen, S.B.; Lorenzen, N.; Thomsen, K.; Kaspersen, J.D.; Pedersen, J.S.; Otzen, D.E. High stability and cooperative unfolding of alpha-synuclein oligomers. Biochemistry 2014, 53, 6252–6263. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Kasoju, N.; Bora, U. Fluorescence study of the curcumin-casein micelle complexation and its application as a drug nanocarrier to cancer cells. Biomacromolecules 2008, 9, 2905–2912. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.A.; Risso, P.H.; Zerpa, S.M. Study of the inhibitory effect of hydrophobic fluorescent markers on the enzymic coagulation of bovine casein micelles: Action of TNS. Food Hydrocoll. 1998, 12, 393–400. [Google Scholar] [CrossRef]
- Risso, P.H.; Gatti, C.A.; Zerpa, S.M.; Perez, G.R. Comparative study of the action of anionic and non-ionic hydrophobic fluorescent markers on the enzymic coagulation of heated bovine casein micelles. Food Hydrocoll. 2000, 14, 179–185. [Google Scholar] [CrossRef]
- Demchenko, A.P. The red-edge effects: 30 years of exploration. Luminescence 2002, 17, 19–42. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Keating-Nakamoto, S. Red-edge excitation of fluorescence and dynamic properties of proteins and membranes. Biochemistry 1984, 23, 3013–3021. [Google Scholar] [CrossRef]
- Vus, K.; Trusova, V.; Gorbenko, G.; Kirilova, E.; Kirilov, G.; Kalnina, I.; Kinnunen, P. Novel aminobenzanthrone dyes for amyloid fibril detection. Chem. Phys. Lett. 2012, 532, 110–115. [Google Scholar] [CrossRef]
- Sutter, M.; Oliveira, S.; Sanders, N.N.; Lucas, B.; van Hoek, A.; Hink, M.A.; Visser, A.J.; De Smedt, S.C.; Hennink, W.E.; Jiskoot, W. Sensitive spectroscopic detection of large and denatured protein aggregates in solution by use of the fluorescent dye Nile red. J. Fluoresc. 2007, 17, 181–192. [Google Scholar] [CrossRef]
- Matveeva, E.G.; Rudolph, A.; Moll, J.R.; Thompson, R.B. Structure-selective anisotropy assay for amyloid Beta oligomers. ACS Chem. Neurosci. 2012, 3, 982–987. [Google Scholar] [CrossRef][Green Version]
- Jiang, B.; Aliyan, A.; Cook, N.P.; Augustine, A.; Bhak, G.; Maldonado, R.; Smith McWilliams, A.D.; Flores, E.M.; Mendez, N.; Shahnawaz, M.; et al. Monitoring the Formation of Amyloid Oligomers Using Photoluminescence Anisotropy. J. Am. Chem. Soc. 2019, 141, 15605–15610. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Jain, N.; Mukhopadhyay, S. Insights into the mechanism of aggregation and fibril formation from bovine serum albumin. J. Phys. Chem. B 2011, 115, 4195–4205. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Sorgjerd, K.; Hammarstrom, P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 2005, 88, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Vaneyck, J.; Blum, C.; Segers-Nolten, I.; Subramaniam, V. Polymorph-specific distribution of binding sites determines thioflavin-T fluorescence intensity in alpha-synuclein fibrils. Amyloid 2018, 25, 189–196. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.S.; Das, S.; Ghosh, S.; Anoop, A.; Jha, N.N.; Khan, T.; Singru, P.; Kumar, A.; Maji, S.K. Amyloid formation of growth hormone in presence of zinc: Relevance to its storage in secretory granules. Sci. Rep. 2016, 6, 23370. [Google Scholar] [CrossRef]
- Amaro, M.; Birch, D.J.; Rolinski, O.J. Beta-amyloid oligomerisation monitored by intrinsic tyrosine fluorescence. Phys. Chem. Chem. Phys. 2011, 13, 6434–6441. [Google Scholar] [CrossRef]
- Saraiva, M.A.; Jorge, C.D.; Santos, H.; Macanita, A.L. Earliest events in alpha-synuclein fibrillation probed with the fluorescence of intrinsic tyrosines. J. Photochem. Photobiol. B 2016, 154, 16–23. [Google Scholar] [CrossRef]
- Dusa, A.; Kaylor, J.; Edridge, S.; Bodner, N.; Hong, D.P.; Fink, A.L. Characterization of oligomers during alpha-synuclein aggregation using intrinsic tryptophan fluorescence. Biochemistry 2006, 45, 2752–2760. [Google Scholar] [CrossRef]
- von Bergen, M.; Li, L.; Mandelkow, E. Intrinsic fluorescent detection of tau conformation and aggregation. Methods Mol. Biol. 2005, 299, 175–184. [Google Scholar] [CrossRef]
- Amundarain, M.J.; Herrera, M.G.; Zamarreno, F.; Viso, J.F.; Costabel, M.D.; Dodero, V.I. Molecular mechanisms of 33-mer gliadin peptide oligomerisation. Phys. Chem. Chem. Phys. 2019, 21, 22539–22552. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Biasetti, L.; Blakeman, B.J.F.; Pollack, S.J.; Zibaee, S.; Abdul-Sada, A.; Thorpe, J.R.; Xue, W.-F.; Serpell, L.C. The involvement of dityrosine crosslinking in α-synuclein assembly and deposition in Lewy Bodies in Parkinson’s disease. Sci. Rep. 2016, 6, 39171. [Google Scholar] [CrossRef]
- Gu, M.; Bode, D.C.; Viles, J.H. Copper Redox Cycling Inhibits Abeta Fibre Formation and Promotes Fibre Fragmentation, while Generating a Dityrosine Abeta Dimer. Sci. Rep. 2018, 8, 16190. [Google Scholar] [CrossRef]
- Brosey, C.A.; Tainer, J.A. Evolving SAXS versatility: Solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Curr. Opin. Struct. Biol. 2019, 58, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Boldon, L.; Laliberte, F.; Liu, L. Review of the fundamental theories behind small angle X-ray scattering, molecular dynamics simulations, and relevant integrated application. Nano Rev. 2015, 6, 25661. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Yip, S. Spectroscopy in Biology and Chemistry: Neutron, X-Ray, Laser; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics; Courier Corporation: Mineloa, NY, USA, 2000.
- Svergun, D.I.; Koch, M.H.; Timmins, P.A.; May, R.P. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Mittag, J.J.; Radler, J.O.; McManus, J.J. Peptide Self-Assembly Measured Using Fluorescence Correlation Spectroscopy. Methods Mol. Biol. 2018, 1777, 159–171. [Google Scholar] [CrossRef]
- Garai, K.; Sureka, R.; Maiti, S. Detecting amyloid-beta aggregation with fiber-based fluorescence correlation spectroscopy. Biophys. J. 2007, 92, L55–L57. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Meuvis, J.; Hendrix, J.; Carl, S.A.; Engelborghs, Y. Early aggregation steps in alpha-synuclein as measured by FCS and FRET: Evidence for a contagious conformational change. Biophys. J. 2010, 98, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Comas-Garcia, M.; Garmann, R.F.; Singaram, S.W.; Ben-Shaul, A.; Knobler, C.M.; Gelbart, W.M. Characterization of Viral Capsid Protein Self-Assembly around Short Single-Stranded RNA. J. Phys. Chem. B 2014, 118, 7510–7519. [Google Scholar] [CrossRef]
- Drzewiecki, K.E.; Grisham, D.R.; Parmar, A.S.; Nanda, V.; Shreiber, D.I. Circular Dichroism Spectroscopy of Collagen Fibrillogenesis: A New Use for an Old Technique. Biophys. J. 2016, 111, 2377–2386. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef]
- Rodger, A. Far UV Protein Circular Dichroism. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin, Germany, 2013; pp. 726–730. [Google Scholar] [CrossRef]
- Hopping, G.; Kellock, J.; Barnwal, R.P.; Law, P.; Bryers, J.; Varani, G.; Caughey, B.; Daggett, V. Designed alpha-sheet peptides inhibit amyloid formation by targeting toxic oligomers. eLife 2014, 3, e01681. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic. Acids Res. 2004, 32 (Suppl. 2), W668–W673. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. Evaluation of the information content in infrared spectra for protein secondary structure determination. Biophys. J. 2006, 90, 2946–2957. [Google Scholar] [CrossRef]
- Sarroukh, R.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. ATR-FTIR: A “rejuvenated” tool to investigate amyloid proteins. Biochim. Biophys. Acta 2013, 1828, 2328–2338. [Google Scholar] [CrossRef]
- Martin, I.; Goormaghtigh, E.; Ruysschaert, J.M. Attenuated total reflection IR spectroscopy as a tool to investigate the orientation and tertiary structure changes in fusion proteins. Biochim. Biophys. Acta (BBA) Biomembr. 2003, 1614, 97–103. [Google Scholar] [CrossRef]
- Ruysschaert, J.M.; Raussens, V. ATR-FTIR Analysis of Amyloid Proteins. Methods Mol. Biol. 2018, 1777, 69–81. [Google Scholar] [CrossRef]
- Waeytens, J.; Van Hemelryck, V.; Deniset-Besseau, A.; Ruysschaert, J.M.; Dazzi, A.; Raussens, V. Characterization by Nano-Infrared Spectroscopy of Individual Aggregated Species of Amyloid Proteins. Molecules 2020, 25, 2899. [Google Scholar] [CrossRef] [PubMed]
- Skamris, T.; Marasini, C.; Madsen, K.L.; Fodera, V.; Vestergaard, B. Early Stage Alpha-Synuclein Amyloid Fibrils are Reservoirs of Membrane-Binding Species. Sci. Rep. 2019, 9, 1733. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Noji, M.; So, M.; Sasahara, K.; Kardos, J.; Naiki, H.; Goto, Y. Aggregation-phase diagrams of beta2-microglobulin reveal temperature and salt effects on competitive formation of amyloids versus amorphous aggregates. J. Biol. Chem. 2018, 293, 14775–14785. [Google Scholar] [CrossRef] [PubMed]
- Aggeli, A.; Bell, M.; Boden, N.; Keen, J.N.; McLeish, T.C.B.; Nyrkova, I.; Radford, S.E.; Semenov, A. Engineering of peptide β-sheet nanotapes. J. Mater. Chem. 1997, 7, 1135–1145. [Google Scholar] [CrossRef]
- Tipping, K.W.; Karamanos, T.K.; Jakhria, T.; Iadanza, M.G.; Goodchild, S.C.; Tuma, R.; Ranson, N.A.; Hewitt, E.W.; Radford, S.E. pH-induced molecular shedding drives the formation of amyloid fibril-derived oligomers. Proc. Natl. Acad. Sci. USA 2015, 112, 5691–5696. [Google Scholar] [CrossRef]
- Brudar, S.; Hribar-Lee, B. The Role of Buffers in Wild-Type HEWL Amyloid Fibril Formation Mechanism. Biomolecules 2019, 9, 65. [Google Scholar] [CrossRef]
- Chan, S.W.; Yau, J.; Ing, C.; Liu, K.; Farber, P.; Won, A.; Bhandari, V.; Kara-Yacoubian, N.; Seraphim, T.V.; Chakrabarti, N.; et al. Mechanism of Amyloidogenesis of a Bacterial AAA+ Chaperone. Structure 2016, 24, 1095–1109. [Google Scholar] [CrossRef]
- Dasari, A.K.R.; Hughes, R.M.; Wi, S.; Hung, I.; Gan, Z.; Kelly, J.W.; Lim, K.H. Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers. Sci. Rep. 2019, 9, 33. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Gursky, O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. USA 2017, 114, E6507–E6515. [Google Scholar] [CrossRef]
- Mawhinney, M.T.; Williams, T.L.; Hart, J.L.; Taheri, M.L.; Urbanc, B. Elucidation of insulin assembly at acidic and neutral pH: Characterization of low molecular weight oligomers. Proteins 2017, 85, 2096–2110. [Google Scholar] [CrossRef]
- Gelenter, M.D.; Smith, K.J.; Liao, S.Y.; Mandala, V.S.; Dregni, A.J.; Lamm, M.S.; Tian, Y.; Xu, W.; Pochan, D.J.; Tucker, T.J.; et al. The peptide hormone glucagon forms amyloid fibrils with two coexisting beta-strand conformations. Nat. Struct. Mol. Biol. 2019, 26, 592–598. [Google Scholar] [CrossRef]
- Chao, Y.-J.; Wu, K.; Chang, H.-H.; Chien, M.-J.; Chan, J.C.C. Manifold of self-assembly of a de novo designed peptide: Amyloid fibrils, peptide bundles, and fractals. RSC Adv. 2020, 10, 29510–29515. [Google Scholar] [CrossRef]
- Doti, N.; Monti, A.; Bruckmann, C.; Calvanese, L.; Smaldone, G.; Caporale, A.; Falcigno, L.; D’Auria, G.; Blasi, F.; Ruvo, M.; et al. Identification and characterization of cytotoxic amyloid-like regions in human Pbx-regulating protein-1. Int. J. Biol. Macromol. 2020, 163, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, Z.; Nasiri Khalili, M.A.; Moradi, S.; Hassan Sajedi, R.; Zeinoddini, M. The Molecular Chaperone Artemin Efficiently Blocks Fibrillization of TAU Protein In Vitro. Cell J. 2018, 19, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.Y.; Zhang, F.; Balch, W.E.; Linhardt, R.J.; Kelly, J.W. Heparin accelerates gelsolin amyloidogenesis. Biochemistry 2006, 45, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.; Hughes, E.; Hussain, R.; Siligardi, G.; Baldock, S.; Madine, J.; Middleton, D.A. Heparin and Methionine Oxidation Promote the Formation of Apolipoprotein A-I Amyloid Comprising alpha-Helical and beta-Sheet Structures. Biochemistry 2017, 56, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Siddiqi, M.K.; Chaturvedi, S.K.; Zaman, M.; Khan, R.H. Vitamin B12 offers neuronal cell protection by inhibiting Abeta-42 amyloid fibrillation. Int. J. Biol. Macromol. 2017, 99, 477–482. [Google Scholar] [CrossRef]
- Batkulwar, K.B.; Jana, A.K.; Godbole, R.K.; Khandelwal, P.; Sengupta, N.; Kulkarni, M.J. Hydralazine inhibits amyloid beta (Aβ) aggregation and glycation and ameliorates Aβ 1–42 induced neurotoxicity. RSC Adv. 2016, 6, 108768–108776. [Google Scholar] [CrossRef]
- Sharma, S.; Nehru, B.; Saini, A. Inhibition of Alzheimer’s amyloid-beta aggregation in-vitro by carbenoxolone: Insight into mechanism of action. Neurochem. Int. 2017, 108, 481–493. [Google Scholar] [CrossRef]
- Mohammadi, F.; Mahmudian, A.; Moeeni, M.; Hassani, L. Inhibition of amyloid fibrillation of hen egg-white lysozyme by the natural and synthetic curcuminoids. RSC Adv. 2016, 6, 23148–23160. [Google Scholar] [CrossRef]
- Ioannou, J.; Donald, A.; Tromp, R. Characterising the secondary structure changes occurring in high density systems of BLG dissolved in aqueous pH 3 buffer. Food Hydrocoll. 2015, 46, 216–225. [Google Scholar] [CrossRef]
- Hu, Y.; He, C.; Woo, M.W.; Xiong, H.; Hu, J.; Zhao, Q. Formation of fibrils derived from whey protein isolate: Structural characteristics and protease resistance. Food Funct. 2019, 10, 8106–8115. [Google Scholar] [CrossRef]
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium ions promote superoxide dismutase 1 (SOD1) aggregation into non-fibrillar amyloid: A link to toxic effects of calcium overload in amyotrophic lateral sclerosis (ALS)? J. Biol. Chem. 2013, 288, 25219–25228. [Google Scholar] [CrossRef] [PubMed]
- Ruano, M.L.; Garcia-Verdugo, I.; Miguel, E.; Perez-Gil, J.; Casals, C. Self-aggregation of surfactant protein A. Biochemistry 2000, 39, 6529–6537. [Google Scholar] [CrossRef]
- Joshi, V.; Shivach, T.; Yadav, N.; Rathore, A.S. Circular dichroism spectroscopy as a tool for monitoring aggregation in monoclonal antibody therapeutics. Anal. Chem. 2014, 86, 11606–11613. [Google Scholar] [CrossRef]
- Benjwal, S.; Verma, S.; Rohm, K.H.; Gursky, O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006, 15, 635–639. [Google Scholar] [CrossRef]
- Waterhous, D.V.; Johnson, W.C., Jr. Importance of environment in determining secondary structure in proteins. Biochemistry 1994, 33, 2121–2128. [Google Scholar] [CrossRef]
- Sonnichsen, F.D.; Van Eyk, J.E.; Hodges, R.S.; Sykes, B.D. Effect of trifluoroethanol on protein secondary structure: A NMR and CD study using a synthetic actin peptide. Biochemistry 1992, 31, 8790–8798. [Google Scholar] [CrossRef]
- Storrs, R.W.; Truckses, D.; Wemmer, D.E. Helix propagation in trifluoroethanol solutions. Biopolymers 1992, 32, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- MacPhee, C.E.; Perugini, M.A.; Sawyer, W.H.; Howlett, G.J. Trifluoroethanol induces the self-association of specific amphipathic peptides. FEBS Lett. 1997, 416, 265–268. [Google Scholar] [CrossRef]
- Fezoui, Y.; Teplow, D.B. Kinetic studies of amyloid beta-protein fibril assembly. Differential effects of alpha-helix stabilization. J. Biol. Chem. 2002, 277, 36948–36954. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.L.; Ramlall, T.F.; Rospigliosi, C.C.; Webb, W.W.; Eliezer, D. Identification of a helical intermediate in trifluoroethanol-induced alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2010, 107, 18850–18855. [Google Scholar] [CrossRef] [PubMed]
- Muta, H.; Lee, Y.H.; Kardos, J.; Lin, Y.; Yagi, H.; Goto, Y. Supersaturation-limited amyloid fibrillation of insulin revealed by ultrasonication. J. Biol. Chem. 2014, 289, 18228–18238. [Google Scholar] [CrossRef] [PubMed]
- Calamai, M.; Chiti, F.; Dobson, C.M. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys. J. 2005, 89, 4201–4210. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.M.; Qadeer, A.; Chaturvedi, S.K.; Ahmad, E.; Rehman, S.A.A.; Gourinath, S.; Khan, R.H. SDS can be utilized as an amyloid inducer: A case study on diverse proteins. PLoS ONE 2012, 7, e29694. [Google Scholar] [CrossRef] [PubMed]
- Castro, I.H.; Bringas, M.; Doni, D.; Noguera, M.E.; Capece, L.; Aran, M.; Blaustein, M.; Costantini, P.; Santos, J. Relationship between activity and stability: Design and characterization of stable variants of human frataxin. Arch. Biochem. Biophys. 2020, 691, 108491. [Google Scholar] [CrossRef]
- Faraj, S.E.; Venturutti, L.; Roman, E.A.; Marino-Buslje, C.B.; Mignone, A.; Tosatto, S.C.; Delfino, J.M.; Santos, J. The role of the N-terminal tail for the oligomerization, folding and stability of human frataxin. FEBS Open Bio 2013, 3, 310–320. [Google Scholar] [CrossRef]
- Curto, L.M.; Angelani, C.R.; Caramelo, J.J.; Delfino, J.M. Truncation of a beta-barrel scaffold dissociates intrinsic stability from its propensity to aggregation. Biophys. J. 2012, 103, 1929–1939. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef]
- Valsecchi, W.M.; Cousido-Siah, A.; Defelipe, L.A.; Mitschler, A.; Podjarny, A.; Santos, J.; Delfino, J.M. The role of the C-terminal region on the oligomeric state and enzymatic activity of Trypanosoma cruzi hypoxanthine phosphoribosyl transferase. Biochim. Biophys. Acta 2016, 1864, 655–666. [Google Scholar] [CrossRef]
- Honisch, C.; Donadello, V.; Hussain, R.; Peterle, D.; De Filippis, V.; Arrigoni, G.; Gatto, C.; Giurgola, L.; Siligardi, G.; Ruzza, P. Application of Circular Dichroism and Fluorescence Spectroscopies To Assess Photostability of Water-Soluble Porcine Lens Proteins. ACS Omega 2020, 5, 4293–4301. [Google Scholar] [CrossRef]
- Visentin, C.; Navarro, S.; Grasso, G.; Regonesi, M.E.; Deriu, M.A.; Tortora, P.; Ventura, S. Protein Environment: A Crucial Triggering Factor in Josephin Domain Aggregation: The Role of 2,2,2-Trifluoroethanol. Int. J. Mol. Sci. 2018, 19, 2151. [Google Scholar] [CrossRef] [PubMed]
- Muzaffar, M.; Ahmad, A. The mechanism of enhanced insulin amyloid fibril formation by NaCl is better explained by a conformational change model. PLoS ONE 2011, 6, e27906. [Google Scholar] [CrossRef] [PubMed]
- Baynes, B.M.; Trout, B.L. Rational design of solution additives for the prevention of protein aggregation. Biophys. J. 2004, 87, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.G.; Pignataro, M.F.; Noguera, M.E.; Cruz, K.M.; Santos, J. Rescuing the Rescuer: On the Protein Complex between the Human Mitochondrial Acyl Carrier Protein and ISD11. ACS Chem. Biol. 2018, 13, 1455–1462. [Google Scholar] [CrossRef]
- Bondos, S.E.; Bicknell, A. Detection and prevention of protein aggregation before, during, and after purification. Anal. Biochem. 2003, 316, 223–231. [Google Scholar] [CrossRef]
- Kumat, T.; Samuel, D.; Jayaraman, G.; Srimathi, T.; Yu, C.J. The role of proline in the prevention of aggregation during protein folding in vitro. IUBMB Life 1998, 46, 509–517. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, H.; Wang, Q.; Yuan, H.; He, W.; Zhao, Z.; Li, Y. Purification, refolding of hybrid hIFNgamma-kringle 5 expressed in Escherichia coli. Curr. Microbiol. 2001, 42, 211–216. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorbance | Fluorescence | ||||
|---|---|---|---|---|---|
| Compound | λ max (nm) | ɛ max (M−1 cm−1) | Ɛ280 (M−1 cm−1) | λ max (nm) | Φi |
| Tryptophan | 280 | 5600 | 5500 | 355 | 0.14 |
| Tyrosine | 275 | 1400 | 1490 | 204 | 0.13 |
| Phenylalanine | 258 | 200 | 200 | 282 | 0.02 |
| Cysteine | 125 | ||||
| Method | Principle | Application |
|---|---|---|
| Steady-state fluorescence | Generally, the emission spectrum of the sample is acquired by exciting the sample at a specific wavelength. The emission maximum, the shape of the spectra and intensity could be analyzed to obtain information about the local environment in the protein. | 1. Monitor protein unfolding and aggregation using intrinsic and extrinsic fluorophores. 2. Evaluate the effect of tyrosine or tryptophan quenchers to evaluate solvent exposure |
| Steady-state anisotropy | Consists of the illumination of the sample with polarizers parallel to the Z axis. The fluorescence of the sample is collected firstly with the emission polarizer oriented (1) parallel to the excitation polarizer (I||) and (2) perpendicular to the excitation polarizer (I⊥). The following equation (5) describes how anisotropy is obtained: There is a direct relation with the correlation time where: is the anisotropy at time 0, is the lifetime and is the correlation time. | 1. Anisotropy measures the rotational displacement of a molecule during the lifetime of the excited state of the fluorophore. 2. It is sensitive to the size of the molecule and local mobility. In the first case, the value increases when increases the size of the particle |
| Time-resolved spectroscopy | The sample is excited with a light pulse of a specific wavelength, and the decay of the signal emission is detected over time. By this approach, the lifetime of the fluorophore (τ), is determined, which is the time that the emitted intensity decays to 1/e of the original value. The equation is: where is the intensity at time t, α is a normalization term, and τ is the lifetime. The same approach but using polarizers for the excitation and emission as it was described above could let study the dynamic of the fluorophore known as an anisotropy decay. | 1. It determines the origins of the changes detected by steady-state anisotropy. 2. It shows the presence of different species in the sample, for example, different oligomeric species that binds to a fluorophore. |
| Fluorescence correlation Spectroscopy | The sample is settled in a coverlid, and it is illuminated by the laser source of a confocal microscope. The molecule of interest must be in a low concentration to have a small number of particles. In this approach, the fluctuations of the fluorescence due to diffusion are recorded. The results obtained are reported as an autocorrelation function, described as below where is defined the intensity at time t, F(t), and the intensity at a later time and F(t + τ) is the Fluorescence at a time point t + τ, averaged over a large number of measurement. For protein oligomerization, there are two approaches photon counting histogram (PCH) and the fluorescence-intensity distribution analysis (FIDA). In both, the essential parameter is the inherent molecular brightness (B) of the fluorophore, which is the counts per second per molecule (CPSM). The idea is to measure the signal from a standard fluorophore, and then, with the same instrumentation settings, the sample is analyzed. The brightness parameter is proportional to the number of molecules of fluorophores under observation. | 1. It determines the diffusion coefficients and hence the size distribution of the aggregates. (the equivalent of dynamic light scattering) 2. In PCH and FIDA the oligomerization state of the sample is obtained |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignataro, M.F.; Herrera, M.G.; Dodero, V.I. Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods. Molecules 2020, 25, 4854. https://doi.org/10.3390/molecules25204854
Pignataro MF, Herrera MG, Dodero VI. Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods. Molecules. 2020; 25(20):4854. https://doi.org/10.3390/molecules25204854
Chicago/Turabian StylePignataro, María Florencia, María Georgina Herrera, and Verónica Isabel Dodero. 2020. "Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods" Molecules 25, no. 20: 4854. https://doi.org/10.3390/molecules25204854
APA StylePignataro, M. F., Herrera, M. G., & Dodero, V. I. (2020). Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods. Molecules, 25(20), 4854. https://doi.org/10.3390/molecules25204854