
Discovery of Potential Chemical Probe as Inhibitors of CXCL12 Using Ligand-Based Virtual Screening and Molecular Dynamic Simulation




Abstract
1. Introduction
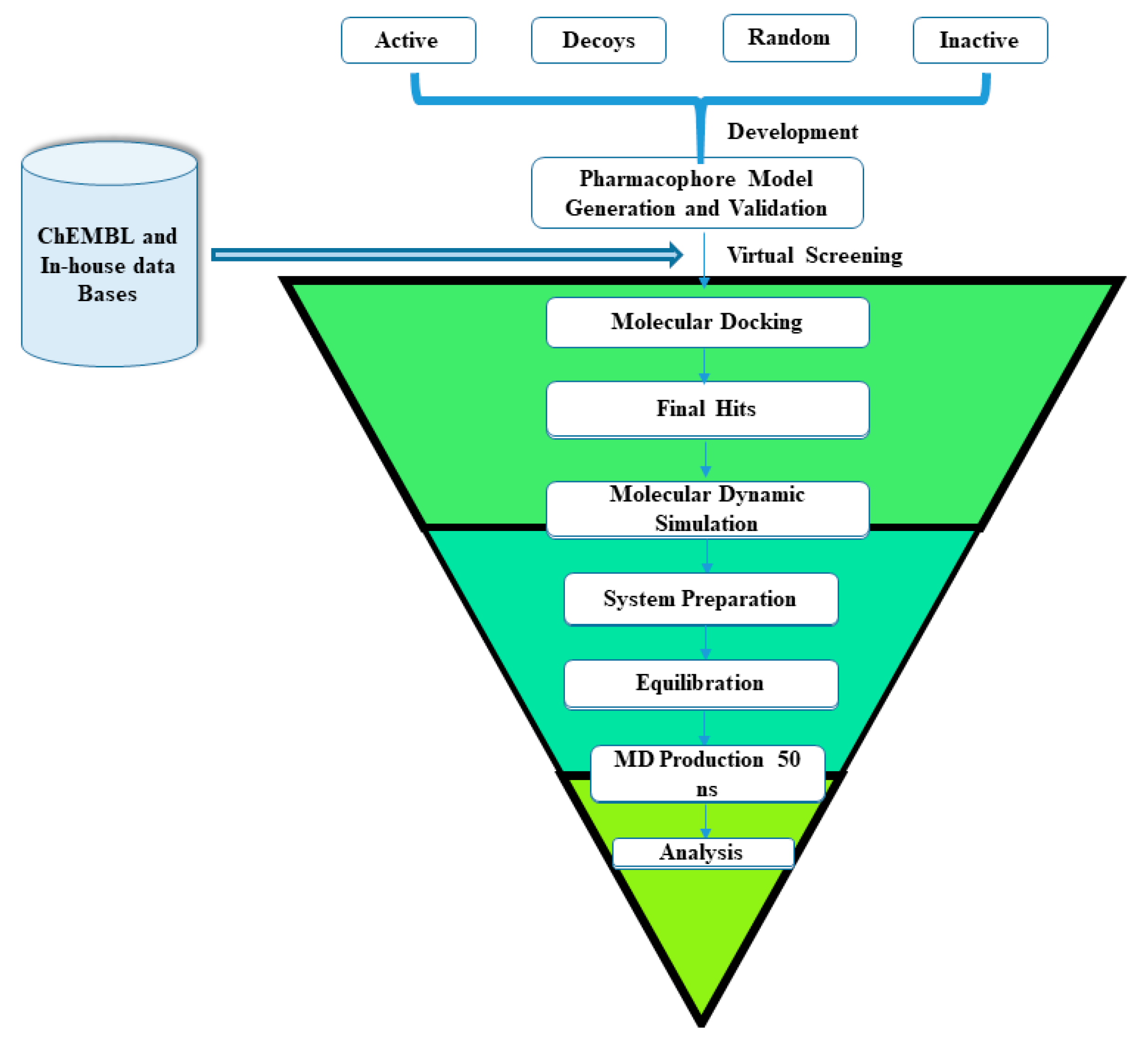
2. Results and Discussion
2.1. Validation of Docking Software via Re-Docking Run Approach
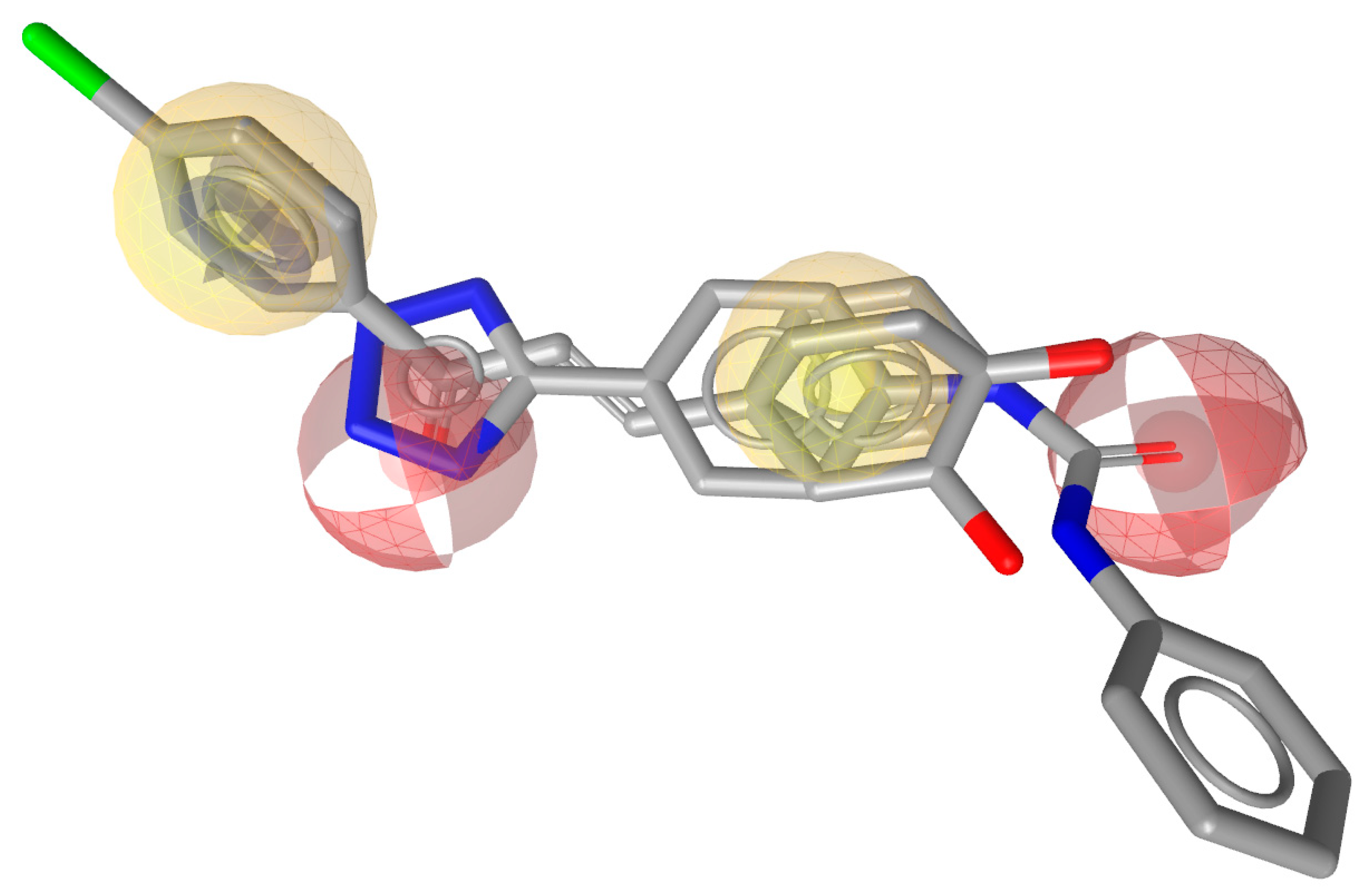
2.2. Pharmacophore Model Generation and Validation
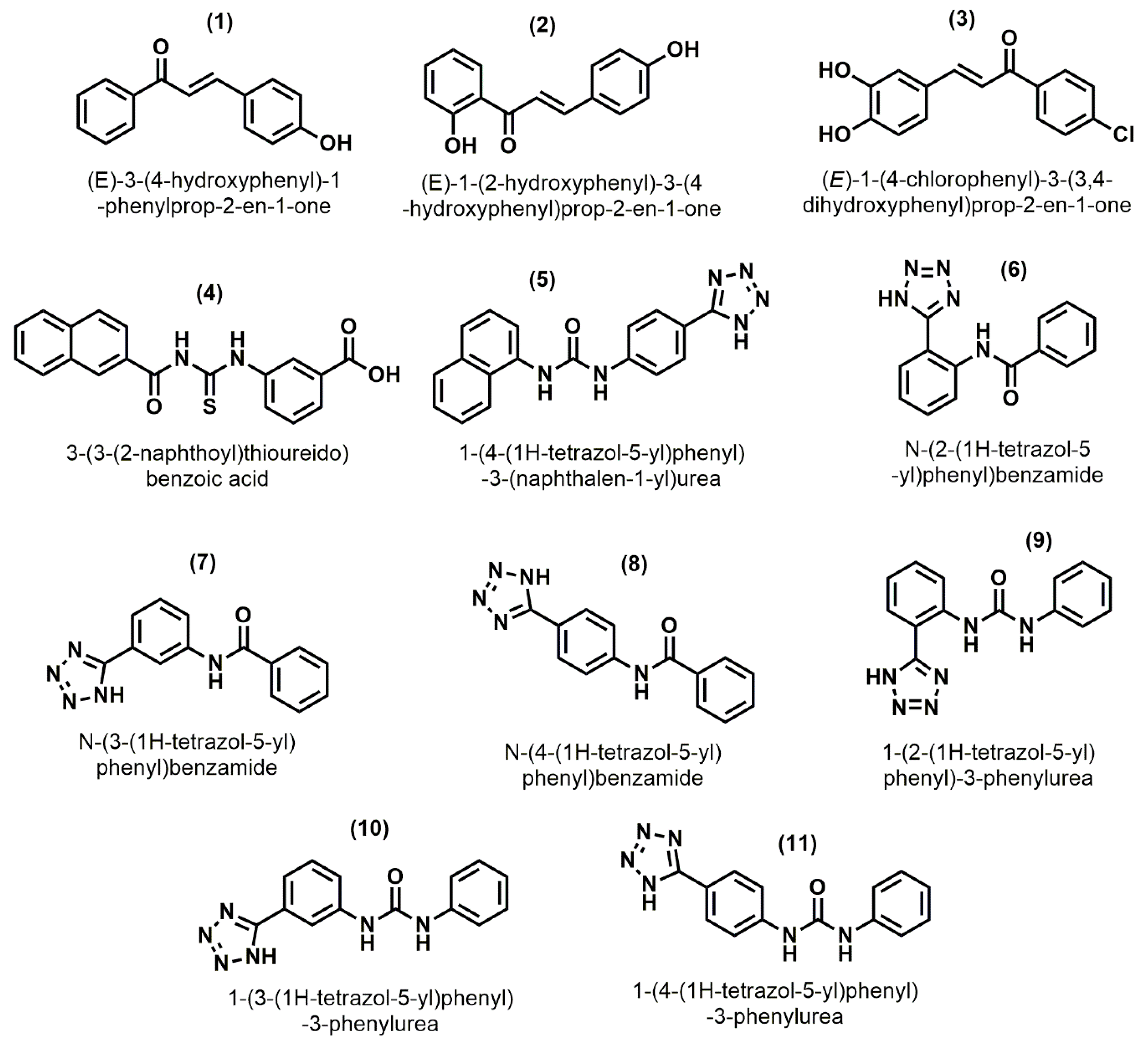
2.3. Pharmacophore Based Database Screening
2.4. Molecular Docking
2.5. Molecular Dynamic Simulation
2.6. Analysis and Visual Inspection of Molecular Dynamic Simulation
2.6.1. Root Mean Square Deviation (RMSD)
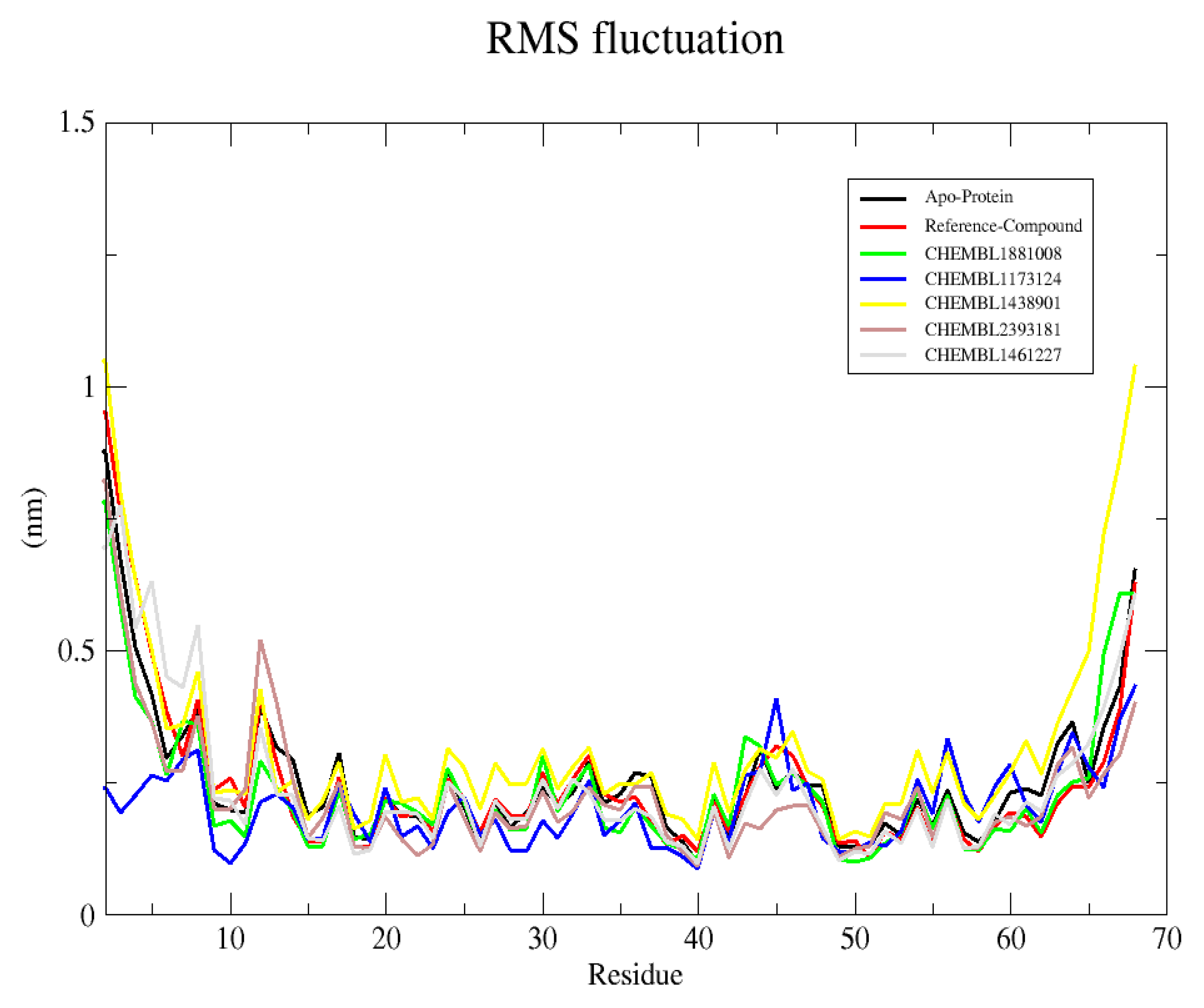
2.6.2. Root Mean Square Fluctuation (RMSF)
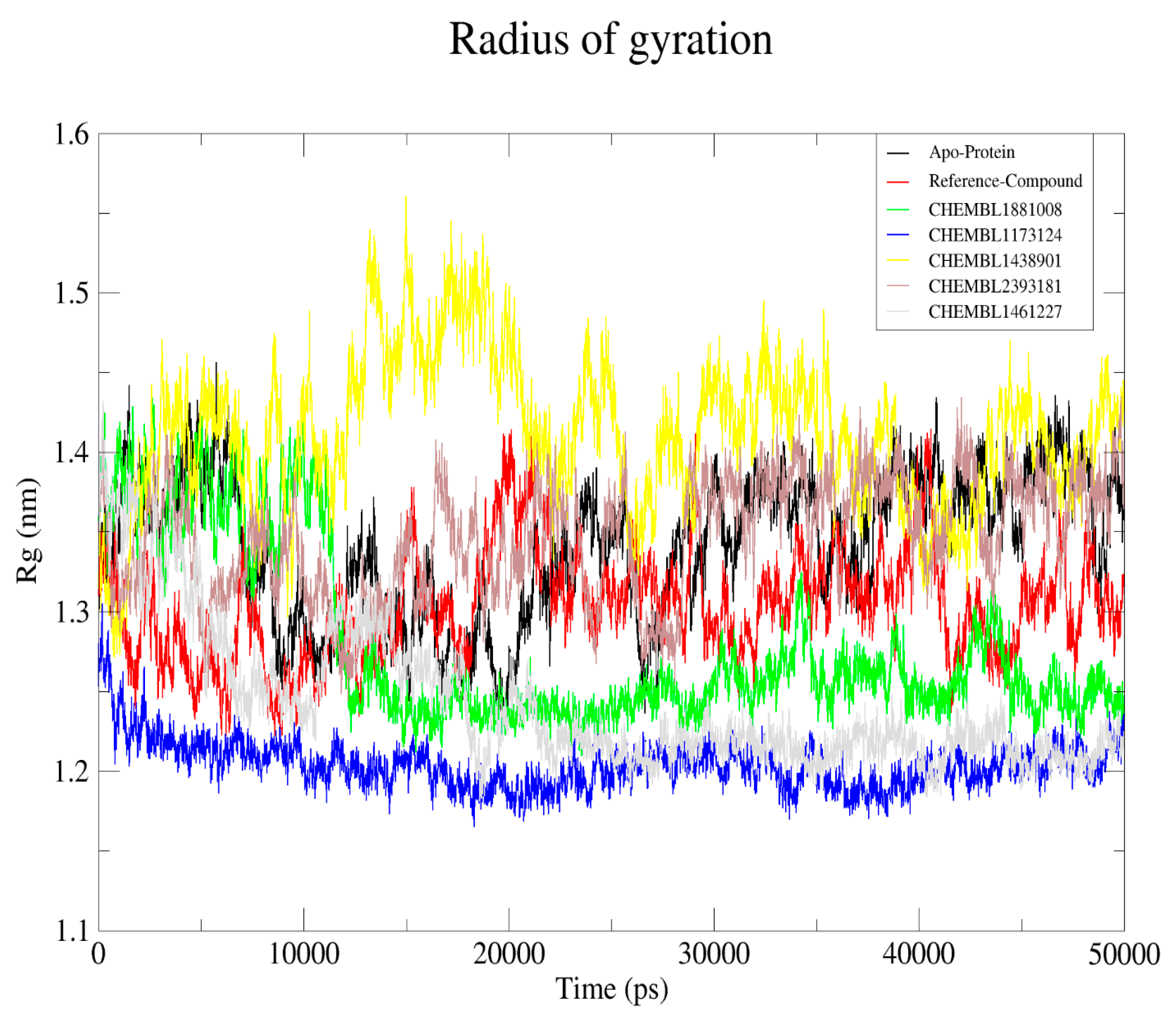
2.6.3. Radius of Gyration
3. Materials and Methods
3.1. Preparation of Dataset
3.2. Receptor Preparation
3.3. Re-Docking Experiment
3.4. Pharmacophore Model Generation
3.5. Pharmacophore Validation
3.6. Pharmacophore-Based Virtual Screening
3.7. Molecular Docking
3.8. Molecular Dynamic Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data
Abbreviations
| CXCL12 | C-X-C motif chemokine 12 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| SDF-1 | Stromal Derived Factor-1 |
| JNK | Janus kinase |
| MAPK | Mitogen Activated Protein Kinase |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| Rg | Radius of gyration |
| VS | Virtual Screening |
| ERK | Extracellular Signal-Regulated Kinase |
| AKT | Protein kinase B |
| MMFF94 | Merck Molecular Force Field 94 |
| PLIF | Protein Ligand Profiling Fingerprinting |
| NVT | Constant Number of particles, Volume and Temperature |
| NPT | Number of Particles, Pressure and Temperature |
| LINCS | Linear Constrain Solver |
| PME | Particle Mesh Ewald |
References
- Hachet-Haas, M.; Balabanian, K.; Rohmer, F.; Pons, F.; Franchet, C.; Lecat, S.; Chow, K.Y.; Dagher, R.; Gizzi, P.; Didier, B. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J. Biol. Chem. 2008, 283, 23189–23199. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J. Mol. Med. 2014, 92, 433–439. [Google Scholar] [CrossRef] [PubMed]
- García-Cuesta, E.M.; Santiago, C.A.; Vallejo, J.; Juarranz, Y.; Rodriguez Frade, J.M.; Mellado, M. The role of the CXCL12/CXCR4/ACKR3 axis in autoimmune diseases. Front. Endocrinol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Cecil, J.; Peng, S.-B.; Schrementi, J.; Kovacevic, S.; Paul, D.; Su, E.W.; Wang, J. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene 2006, 374, 174–179. [Google Scholar] [CrossRef]
- Farnsworth, R.H.; Karnezis, T.; Maciburko, S.J.; Mueller, S.N.; Stacker, S.A. The interplay between lymphatic vessels and chemokines. Front. Immunol. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Lee, R.L.; Westendorf, J.; Gold, M.R. Differential role of reactive oxygen species in the activation of mitogen-activated protein kinases and Akt by key receptors on B-lymphocytes: CD40, the B cell antigen receptor, and CXCR4. J. Cell Commun. Signal. 2007, 1, 33–43. [Google Scholar] [CrossRef]
- Roland, J.; Murphy, B.J.; Ahr, B.; Robert-Hebmann, V.R.; Delauzun, V.; Nye, K.E.; Devaux, C.; Biard-Piechaczyk, M. Role of the intracellular domains of CXCR4 in SDF-1–mediated signaling. Blood J. Am. Soc. Hematol. 2003, 101, 399–406. [Google Scholar] [CrossRef]
- Phillips, A.J.; Taleski, D.; Koplinski, C.A.; Getschman, A.E.; Moussouras, N.A.; Richard, A.M.; Peterson, F.C.; Dwinell, M.B.; Volkman, B.F.; Payne, R.J. CCR7 Sulfotyrosine enhances CCL21 binding. Int. J. Mol. Sci. 2017, 18, 1857. [Google Scholar] [CrossRef]
- Kufareva, I.; Gustavsson, M.; Zheng, Y.; Stephens, B.S.; Handel, T.M. What do structures tell us about chemokine receptor function and antagonism? Annu. Rev. Biophys. 2017, 46, 175–198. [Google Scholar] [CrossRef] [PubMed]
- Dillenburg-Pilla, P.; Patel, V.; Mikelis, C.M.; Zárate-Bladés, C.R.; Doçi, C.L.; Amornphimoltham, P.; Wang, Z.; Martin, D.; Leelahavanichkul, K.; Dorsam, R.T. SDF-1/CXCL12 induces directional cell migration and spontaneous metastasis via a CXCR4/G α i/mTORC1 axis. FASEB J. 2015, 29, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.S.; Keating, S.M.; Abdel-Mohsen, M.; Gibb, S.L.; Heitman, J.W.; Inglis, H.C.; Martin, J.N.; Zhang, J.; Kaidarova, Z.; Deng, X. Cytokines elevated in HIV elite controllers reduce HIV replication in vitro and modulate HIV restriction factor expression. J. Virol. 2017, 91, e02051-16. [Google Scholar] [CrossRef] [PubMed]
- Basini, G.; Ragionieri, L.; Bussolati, S.; Di Lecce, R.; Cacchioli, A.; Dettin, M.; Cantoni, A.; Grolli, S.; La Bella, O.; Zamuner, A. Expression and function of the stromal cell-derived factor-1 (SDF-1) and CXC chemokine receptor 4 (CXCR4) in the swine ovarian follicle. Domest. Anim. Endocrinol. 2020, 71, 106404. [Google Scholar] [CrossRef]
- Stone, M.; Hayward, J.; Huang, C.; Huma, Z.E.; Sanchez, J. Mechanisms of regulation of the chemokine-receptor network. Int. J. Mol. Sci. 2017, 18, E342. [Google Scholar] [CrossRef]
- Ziarek, J.J.; Liu, Y.; Smith, E.; Zhang, G.; Peterson, F.C.; Chen, J.; Yu, Y.; Chen, Y.; Volkman, B.F.; Li, R. Fragment-based optimization of small molecule CXCL12 inhibitors for antagonizing the CXCL12/CXCR4 interaction. Curr. Top. Med. Chem. 2012, 12, 2727–2740. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- El-Zahabi, H.S.; Khalifa, M.M.; Gado, Y.M.; Farrag, A.M.; Elaasser, M.M.; Safwat, N.A.; AbdelRaouf, R.R.; Arafa, R.K. New thiobarbituric acid scaffold-based small molecules: Synthesis, cytotoxicity, 2D-QSAR, pharmacophore modelling and in-silico ADME screening. Eur. J. Pharm. Sci. 2019, 130, 124–136. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Lorthioir, O.; Valenzuela, A.; Magerus, A.; Thelen, M.; Montes, M.; Virelizier, J.-L.; Delepierre, M.; Baleux, F.; Lortat-Jacob, H. Stromal cell-derived factor-1α associates with heparan sulfates through the first β-strand of the chemokine. J. Biol. Chem. 1999, 274, 23916–23925. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Alamri, M.A. Pharmacophore and docking-based sequential virtual screening for the identification of novel Sigma 1 receptor ligands. Bioinformation 2019, 15, 586. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Farooq, U.; Khan, S.; Sarwar, R.; Mabkhot, Y.N.; Saeed, M.; Alsayari, A.; Muhsinah, A.B.; Ul-Haq, Z. Pharmacophore Model Based Virtual Screening, Docking, Biological Evaluation and Molecular Dynamics Simulations for Inhibitors Discovery against α-Tryptophan synthase from Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.K.; Jain, C.K. Computational Drug Discovery Based on Natural Products against Acinetobacter Baumannii. J. Mater. Sci. Surf. Eng. 2019, 6, 895–898. [Google Scholar]
- Khan, H.; Zafar, M.; Patel, S.; Shah, S.M.; Bishayee, A. Pharmacophore studies of 1, 3, 4-oxadiazole nucleus: Lead compounds as α-glucosidase inhibitors. Food Chem. Toxicol. 2019, 130, 207–218. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Bernardi, A.; Faller, R.; Reith, D.; Kirschner, K.N. ACPYPE update for nonuniform 1–4 scale factors: Conversion of the GLYCAM06 force field from AMBER to GROMACS. SoftwareX 2019, 10, 100241. [Google Scholar] [CrossRef]
- Shukla, R.; Shukla, H.; Sonkar, A.; Pandey, T.; Tripathi, T. Structure-based screening and molecular dynamics simulations offer novel natural compounds as potential inhibitors of Mycobacterium tuberculosis isocitrate lyase. J. Biomol. Struct. Dyn. 2018, 36, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zheng, X.; Fang, D.; Zhou, S.; Wu, W.; Zheng, K. Identifying hQC inhibitors of Alzheimer’s disease by effective customized pharmacophore-based virtual screening, molecular dynamic simulation, and binding free energy analysis. Appl. Biochem. Biotechnol. 2019, 187, 1173–1192. [Google Scholar] [CrossRef]
- Kumar, K.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol. Biosyst. 2014, 10, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Rampogu, S.; Son, M.; Park, C.; Kim, H.-H.; Suh, J.-K.; Lee, K.W. Sulfonanilide derivatives in identifying novel aromatase inhibitors by applying docking, virtual screening, and MD simulations studies. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Libraries | Total No | Picked Out | Hit Rate% |
|---|---|---|---|---|
| 1 | Actives | 11 | 8 | 72.78 |
| 2 | Inactive | 05 | 0 | 0 |
| 3 | Decoys | 550 | 20 | 3.61 |
| 4 | Random | 115 | 18 | 15.65 |
| Compound ID | Docking Score | H-Bonds | Hydrophobic | Salt Bridges | Pi-Stacking |
|---|---|---|---|---|---|
| Reference compound | −5.970 | Glu15, Ala19, Asn22, Asn44, Arg47 | Val18, Leu42 | ---- | ---- |
| CHEMBL1881008 (A) | −6.1680 | Glu15, Ala19, Asn22, Asn44, Asn45, Arg47 | Val18, Leu42, Val49 | ---- | ---- |
| CHEMBL1173124 (B) | −6.1645 | Ala19, Asn22, Asn44, Arg47, Gln48, Cys50 | Phe13, His17 | ---- | ---- |
| CHEMBL1438901 (C) | −5.8470 | Glu15,Ala19, Asn22, Arg47 | Leu42 | ---- | ---- |
| CHEMBL2393181 (D) | −5.6232 | Glu15, Ala19, Asn22, Asn44, Arg47 | ---- | ---- | ---- |
| CHEMBL1461227 (E) | −5.2401 | Glu15,Ala19, Asn22, Asn44, Arg47 | Ala19 | ---- | ---- |
Sample Availability: Structure of the compounds and trajectories are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haider, S.; Barakat, A.; Ul-Haq, Z. Discovery of Potential Chemical Probe as Inhibitors of CXCL12 Using Ligand-Based Virtual Screening and Molecular Dynamic Simulation. Molecules 2020, 25, 4829. https://doi.org/10.3390/molecules25204829
Haider S, Barakat A, Ul-Haq Z. Discovery of Potential Chemical Probe as Inhibitors of CXCL12 Using Ligand-Based Virtual Screening and Molecular Dynamic Simulation. Molecules. 2020; 25(20):4829. https://doi.org/10.3390/molecules25204829
Chicago/Turabian StyleHaider, Sajjad, Assem Barakat, and Zaheer Ul-Haq. 2020. "Discovery of Potential Chemical Probe as Inhibitors of CXCL12 Using Ligand-Based Virtual Screening and Molecular Dynamic Simulation" Molecules 25, no. 20: 4829. https://doi.org/10.3390/molecules25204829
APA StyleHaider, S., Barakat, A., & Ul-Haq, Z. (2020). Discovery of Potential Chemical Probe as Inhibitors of CXCL12 Using Ligand-Based Virtual Screening and Molecular Dynamic Simulation. Molecules, 25(20), 4829. https://doi.org/10.3390/molecules25204829