
Docking and QSAR of Aminothioureas at the SARS-CoV-2 S-Protein–Human ACE2 Receptor Interface



Abstract
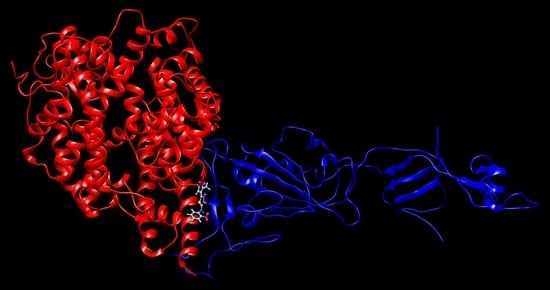
1. Introduction
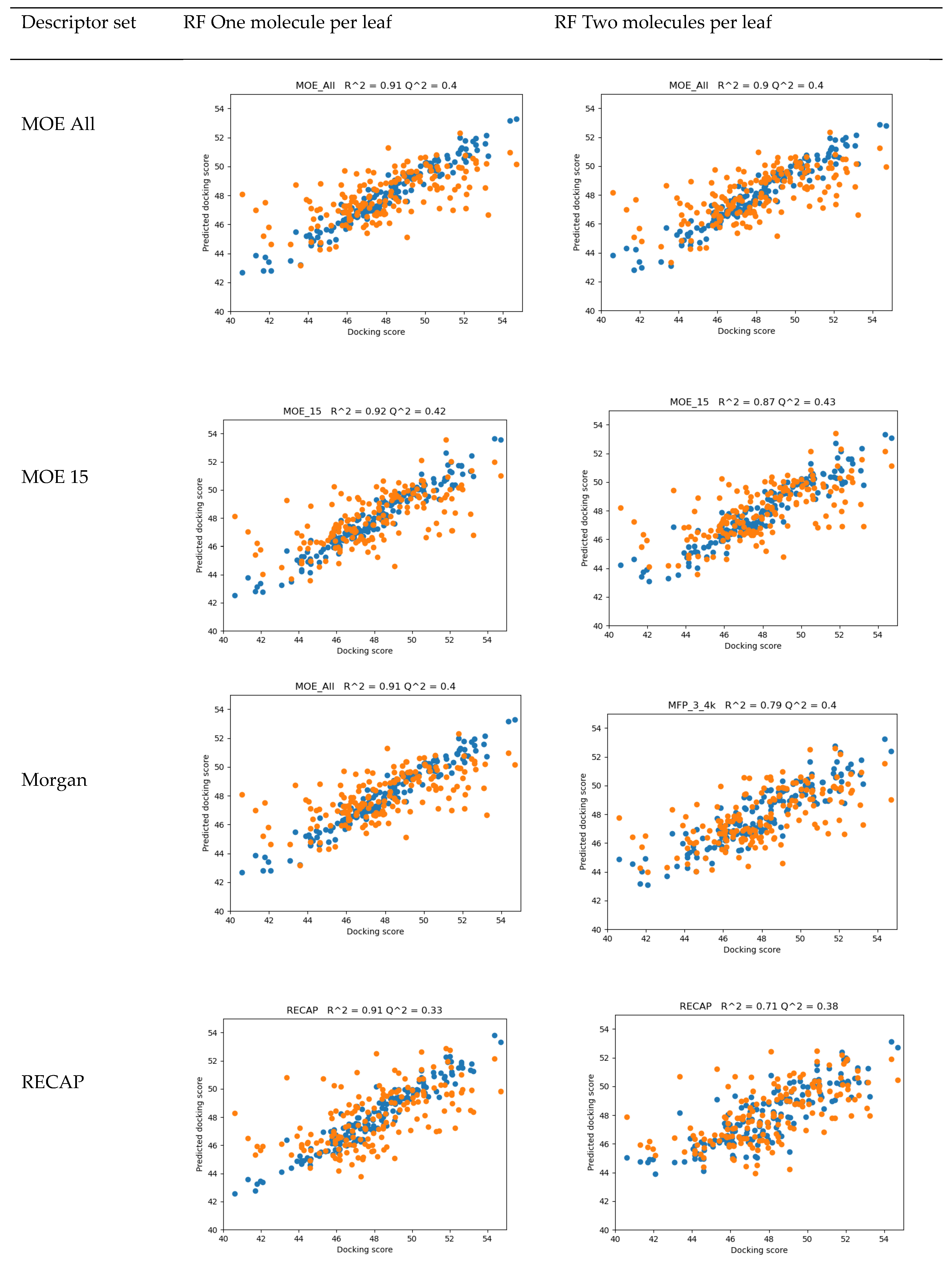
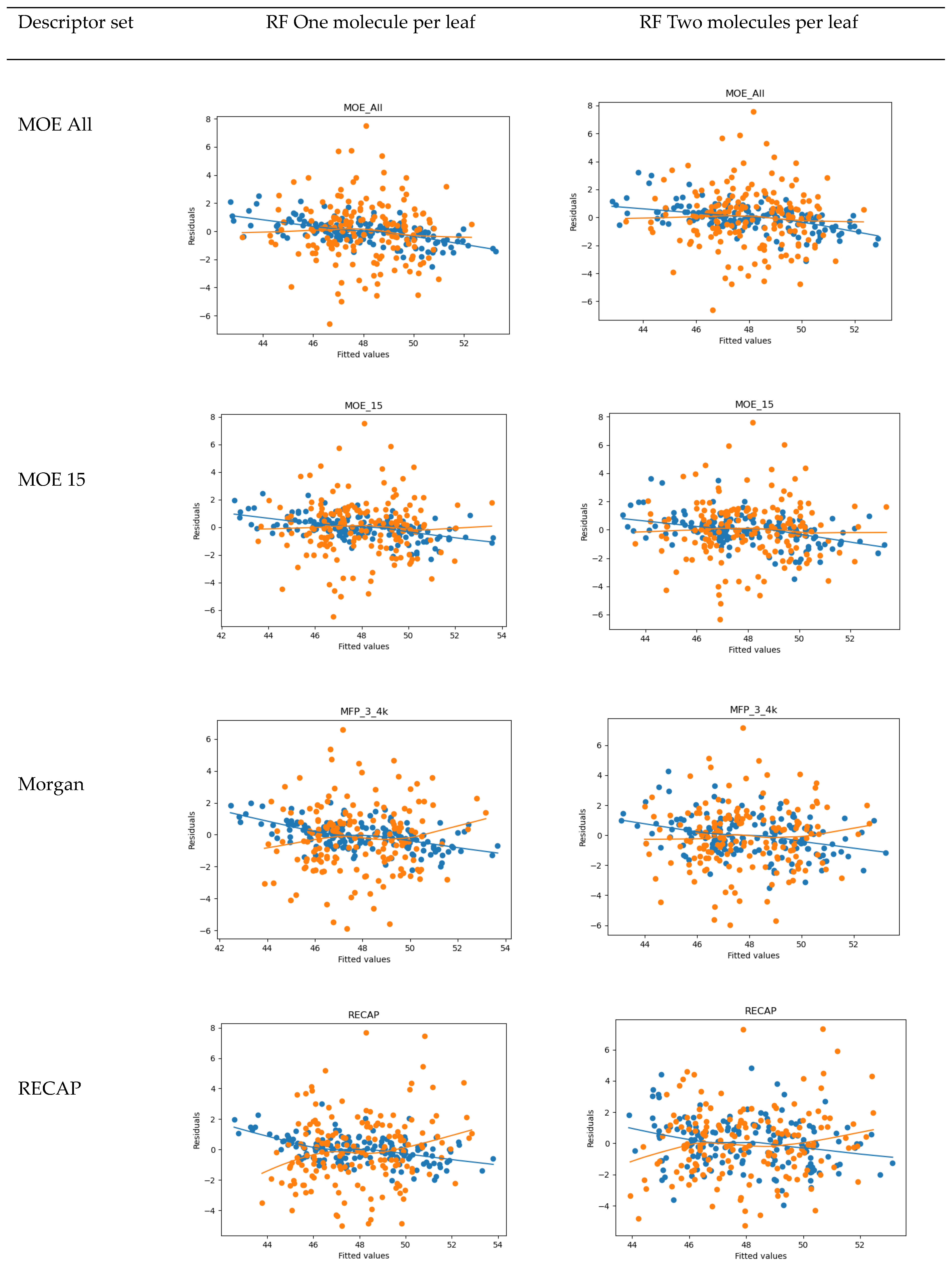
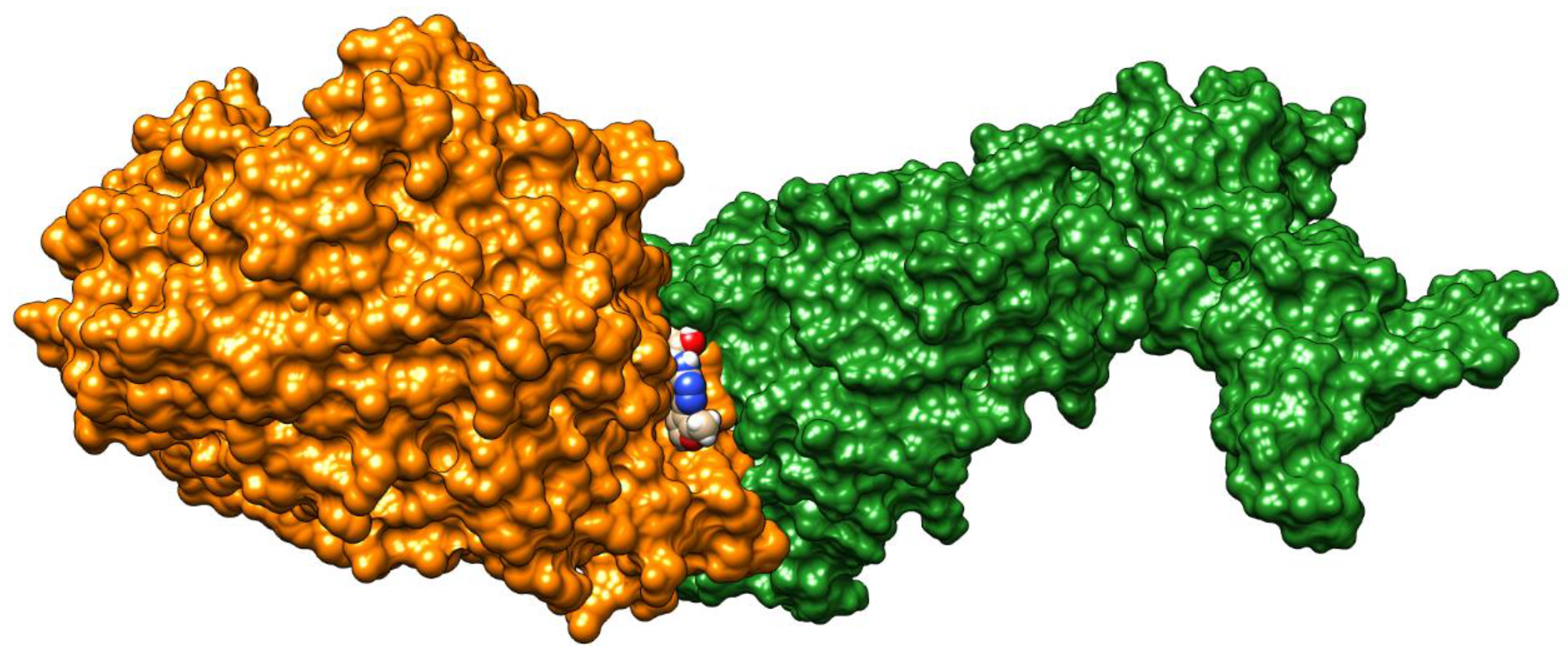
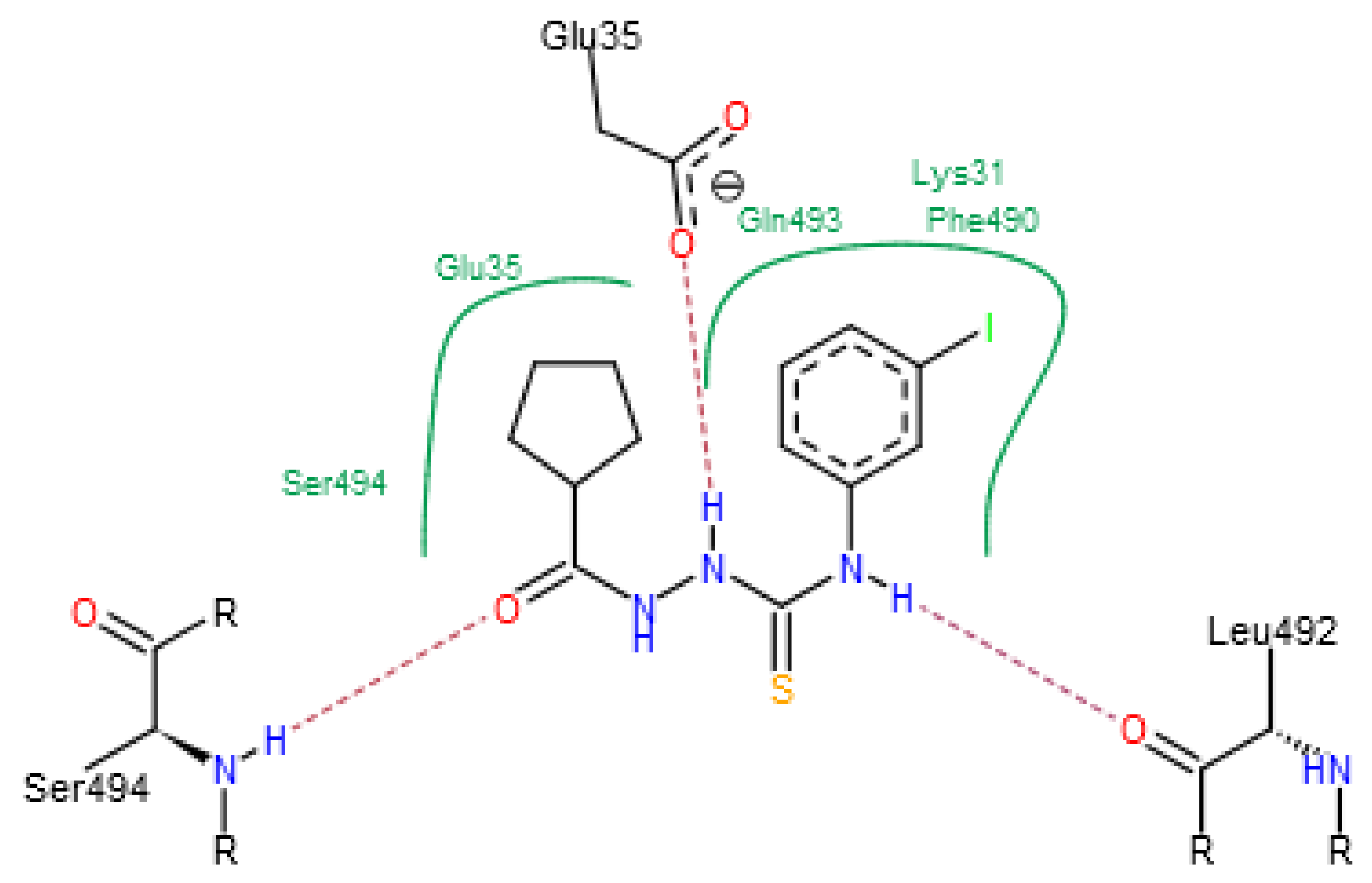
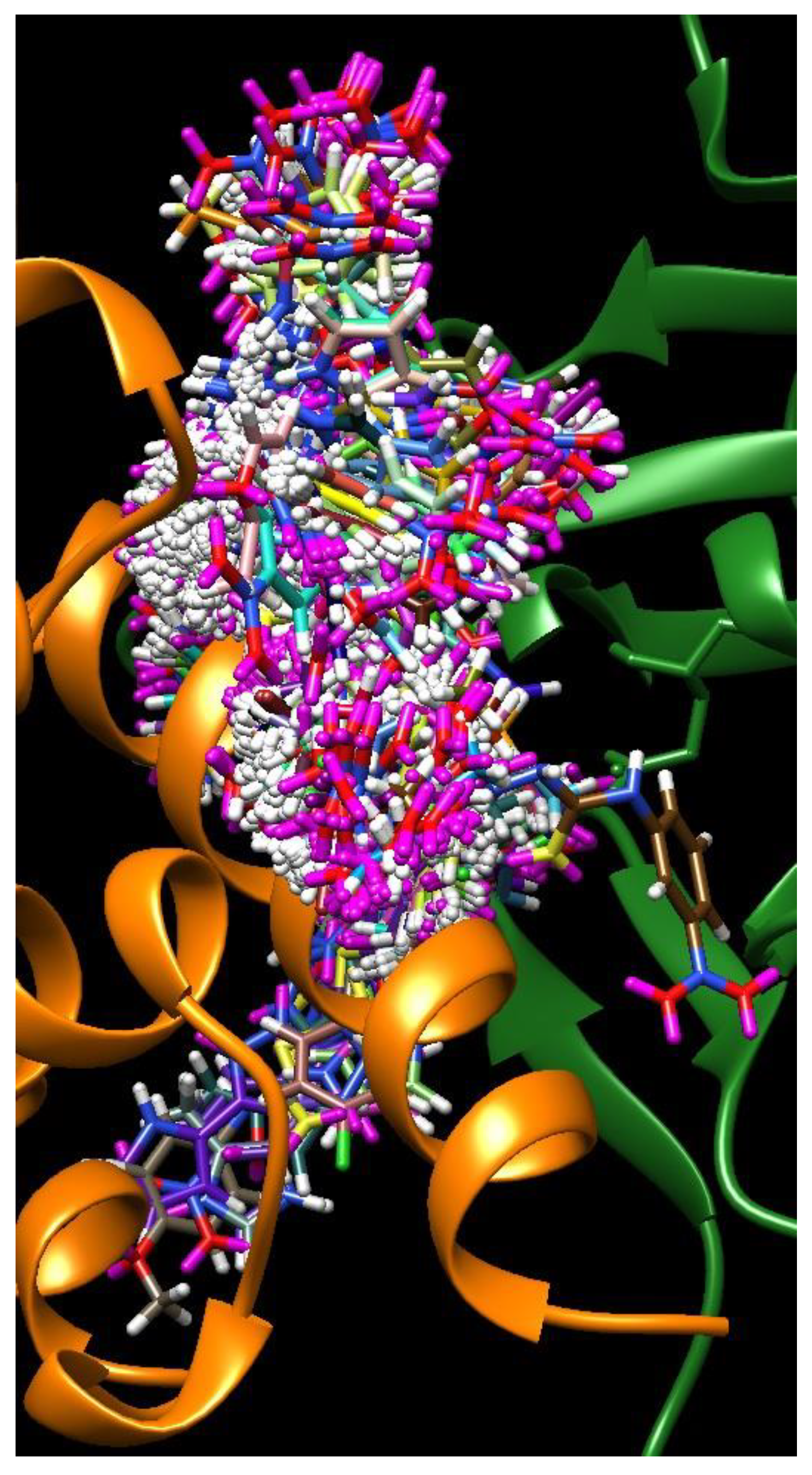
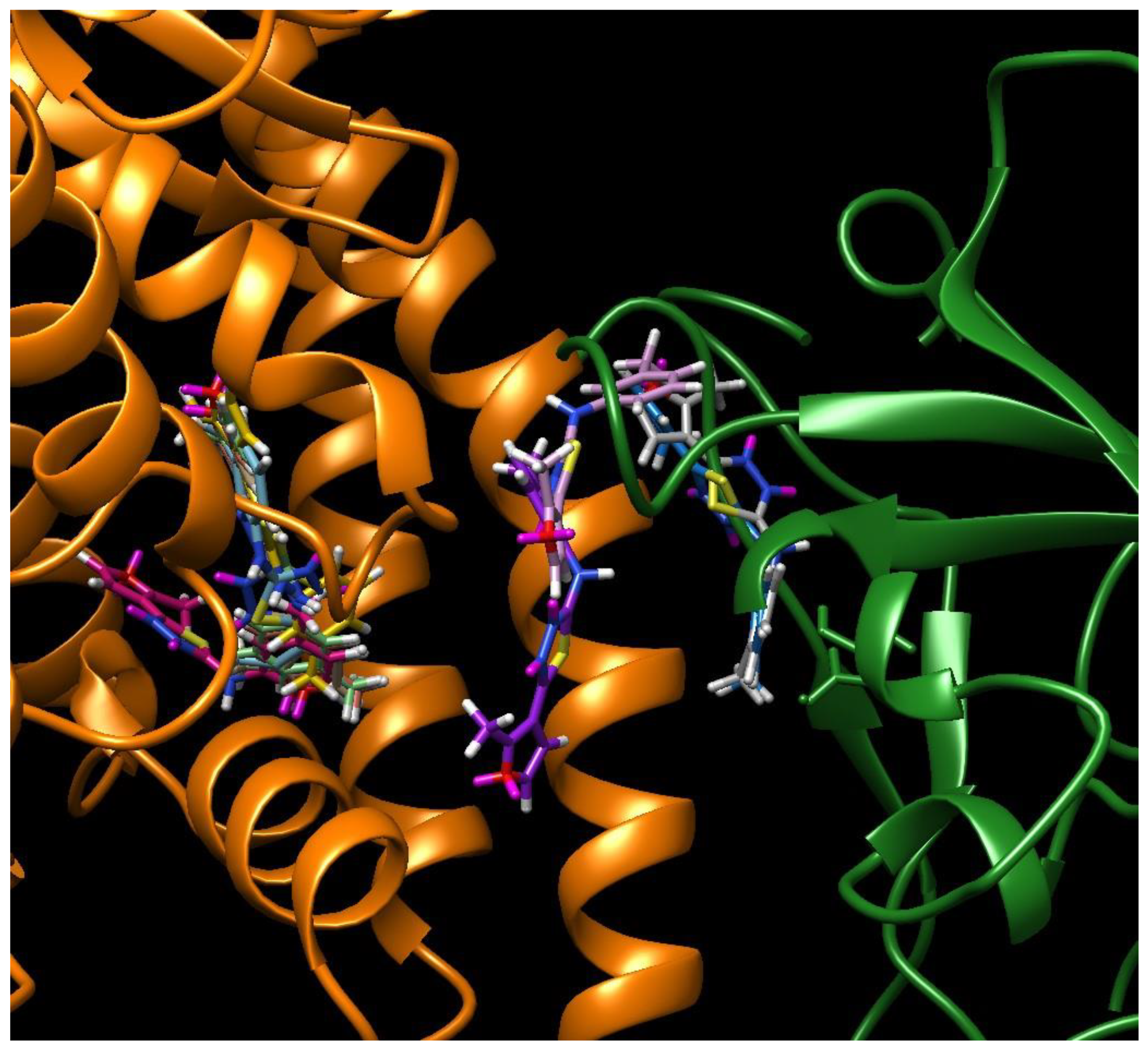
2. Results
3. Discussion
4. Materials and Methods
4.1. Docking
4.2. QSAR
- 15 MOE 2D descriptors selected by Particle Swarm Optimization using Fujitsu ADMEWORKS ModelBuilder software [57] (‘“ASA_H”, “a_acc”, “a_nH”, “a_nN”, “E_ele”, “E_oop”, “GCUT_PEOE_3”, “GCUT_SLOGP_1”, “opr_violation”, “PEOE_VSA_PPOS”, “rsynth”, “SlogP_VSA6”, “SMR_VSA1”, “vsurf_CW5”, “vsurf_W5”), which were found to correlate best with activity in a linear combination, as a low-count set of general properties
- Morgan Fingerprints [58] with Radius = 3 and bit length of 4096 as general structural features
- Counts of fragments frequently appearing in our compound set obtained by the RECAP algorithm using Fujitsu ADMEWORKS ModelBuilder software, as features most specific to our set, see Table S4
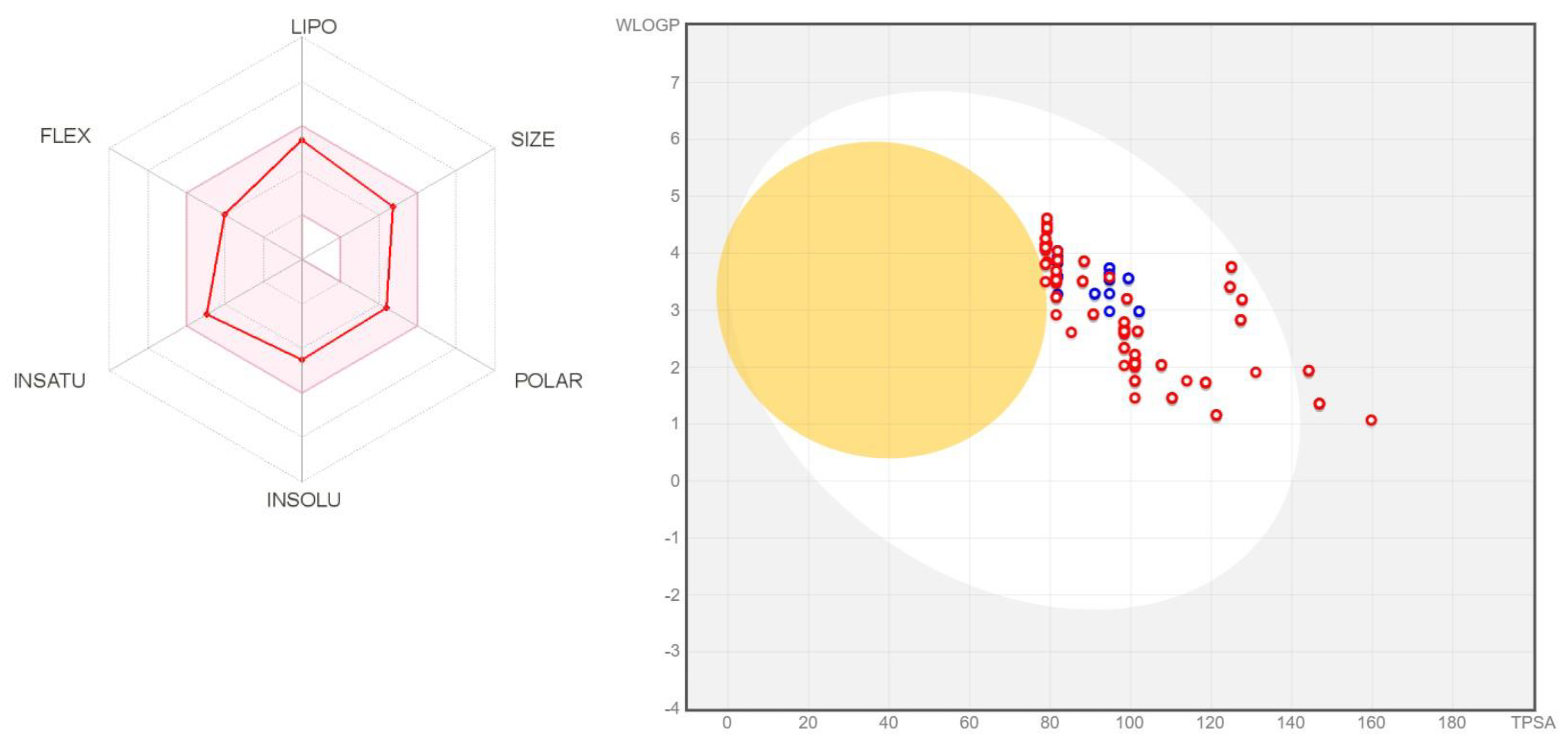
4.3. ADMET
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guarner, J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus COVID-19 Global Cases by the Center for Systems Science and Engineering at Johns Hopkins University. Available online: https://www.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6 (accessed on 1 October 2020).
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Mercurio, I.; Tragni, V.; Busto, F.; De Grassi, A.; Pierri, C.L. Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: From conformational changes to novel neutralizing antibodies. Cell. Mol. Life Sci. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Lu, Y.; Jin, X.; Zhang, L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020, 526, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 2020, 10, 7448–7464. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Lim, P.L.; Lee, T.H.; Rowe, E.K. Middle East Respiratory Syndrome coronavirus (MERS CoV): Update 2013. Curr. Infect. Dis. Rep. 2013, 15, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Forrest, J.C.; Zhang, X. A screen of the NIH Clinical Collection small molecule library identifies potential anti-coronavirus drugs. Antivir. Res. 2015, 114, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Paneth, A.; Paneth, P. Binding repurposed drugs and aminothioureas derivatives to SARS-CoV-2 enzymes—A docking perspective. Sci. Rep. 2020. unpublihed work. [Google Scholar] [CrossRef]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Armstrong, J.; Davenport, A.P.; Davies, J.; Faccenda, E.; Harding, S.D.; Levi-Schaffer, F.; Maguire, J.J.; Pawson, A.J.; Southan, C.; et al. A rational roadmap for SARS-CoV-2/COVID-19 pharmacotherapeutic research and development. Brit. J. Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.-T.; Wong, A.Y.-L.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.-H.; Huang, X.; et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Tung, Y.; Lee, K.; Chen, T.; Hsiao, Y.; Chang, H.; Hsieh, T.; Su, C.; Wang, S.; Yu, J.; et al. Potential Therapeutic Agents for COVID-19 Based on the Analysis of Protease and RNA Polymerase Docking. Preprints 2020, unpublished work. [Google Scholar] [CrossRef]
- Smith, M.; Smith, J.C. Repurposing therapeutics for COVID-19: Supercomputer-based docking to the SARS CoV-2 viral spike protein and viral spike protein-human ACE2 interface. ChemRxiv 2020, unpublished work. [Google Scholar] [CrossRef]
- Novick, P.A.; Ortiz, O.F.; Poelman, J.; Abdulhay, A.Y.; Pande, V.S. SWEETLEAD: An In Silico Database of Approved Drugs, Regulated Chemicals, and Herbal Isolates for Computer-Aided Drug Discovery. PloS ONE 2013, 8, e79568. [Google Scholar] [CrossRef]
- Bai, C.; Warshel, A. Critical Differences between the Binding Features of the Spike Proteins of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. B 2020, 124, 5907–5912. [Google Scholar] [CrossRef]
- Paneth, A.; Węglińska, L.; Bekier, A.; Stefaniszyn, E.; Wujec, M.; Trotsko, N.; Hawrył, A.; Hawrył, M.; Dzitko, K. Discovery of Potent and Selective Halogen-Substituted Imidazole-Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro via Structure-Based Design. Molecules 2019, 24, 1618. [Google Scholar] [CrossRef] [PubMed]
- Paneth, A.; Węglińska, L.; Bekier, A.; Stefaniszyn, E.; Wujec, M.; Trotsko, N.; Dzitko, K. Systematic Identification of Thiosemicarbazides for Inhibition of Toxoplasma gondii Growth In Vitro. Molecules 2019, 24, 614. [Google Scholar] [CrossRef] [PubMed]
- Dzitko, K.; Paneth, A.; Plech, T.; Pawełczyk, J.; Węglińska, L.; Paneth, P. Triazole-Based Compound as a Candidate To Develop Novel Medicines To Treat Toxoplasmosis. Antimicrob. Agents Chemother. 2014, 58, 7583–7585. [Google Scholar] [CrossRef]
- Dzitko, K.; Paneth, A.; Plech, T.; Pawełczyk, J.; Stączek, P.; Stefańska, J.; Paneth, P. 1,4-Disubstituted Thiosemicarbazide Derivatives are Potent Inhibitors of Toxoplasma gondii Proliferation. Molecules 2014, 19, 9926–9943. [Google Scholar] [CrossRef] [PubMed]
- Frączek, T.; Paneth, A.; Kamiński, R.; Krakowiak, A.; Paneth, P. Searching for novel scaffold of triazole non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Enzym. Inhib. Med. Chem. 2015, 31, 1–9. [Google Scholar] [CrossRef]
- Kaproń, B.; Czarnomysy, R.; Paneth, A.; Wujec, M.; Bielawski, K.; Bielawska, A.; Swiatek, L.; Rajtar, B.; Polz-Dacewicz, M.; Plech, T. Dual antibacterial and anticancer activity of 4-benzoyl-1-dichlorobenzoylthiosemicarbazide derivatives. Anti-Cancer Agents Med. Chem. 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Paneth, A.; Stączek, P.; Plech, T.; Strzelczyk, A.; Janowska, D.; Stefańska, J.; Dzitko, K.; Wujec, M.; Kosiek, S.; Paneth, P. Synthesis and antibacterial activity of 1,4-dibenzoylthiosemicarbazide derivatives. Biomed. Pharm. 2017, 88, 1235–1242. [Google Scholar] [CrossRef]
- Paneth, A.; Stączek, P.; Plech, T.; Strzelczyk, A.; Dzitko, K.; Wujec, M.; Kusmierz, E.; Kosikowska, U.; Grzegorczyk, A.; Paneth, P. Biological evaluation and molecular modelling study of thiosemicarbazide derivatives as bacterial type IIA topoisomerases inhibitors. J. Enzym. Inhib. Med. Chem. 2015, 31, 1–9. [Google Scholar] [CrossRef]
- Paneth, A.; Plech, T.; Kaproń, B.; Hagel, D.; Kosikowska, U.; Kusmierz, E.; Dzitko, K.; Paneth, P. Design, synthesis and biological evaluation of 4-benzoyl-1-dichlorobenzoylthiosemicarbazides as potent Gram-positive antibacterial agents. J. Enzym. Inhib. Med. Chem. 2015, 31, 1–7. [Google Scholar] [CrossRef]
- Plech, T.; Kaproń, B.; Paneth, A.; Kosikowska, U.; Malm, A.; Strzelczyk, A.; Stączek, P.; Świątek, Ł.; Rajtar, B.; Polz-Dacewicz, M. Search for factors affecting antibacterial activity and toxicity of 1,2,4-triazole-ciprofloxacin hybrids. Eur. J. Med. Chem. 2015, 97, 94–103. [Google Scholar] [CrossRef]
- Plech, T.; Paneth, A.; Kaproń, B.; Kosikowska, U.; Malm, A.; Strzelczyk, A.; Stączek, P. Structure-activity Relationship Studies of Microbiologically Active Thiosemicarbazides Derived from Hydroxybenzoic Acid Hydrazides. Chem. Biol. Drug Des. 2014, 85, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Plech, T.; Kaproń, B.; Paneth, A.; Wujec, M.; Czarnomysy, R.; Bielawska, A.; Bielawski, K.; Trotsko, N.; Kusmierz, E.; Paneth, P. Search for human DNA topoisomerase II poisons in the group of 2,5-disubstituted-1,3,4-thiadiazoles. J. Enzym. Inhib. Med. Chem. 2015, 30, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Siwek, A.; Bielawska, A.; Maciorkowska, E.; Lepiarczyk, M.; Bielawski, K.; Trotsko, N.; Wujec, M. Cytotoxicity and topoisomerase I/II inhibition activity of novel 4-aryl/alkyl-1-(piperidin-4-yl)-carbonylthiosemicarbazides and 4-benzoylthiosemicarbazides. J. Enzym. Inhib. Med. Chem. 2013, 29, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Siwek, A.; Stączek, P.; Wujec, M.; Bielawski, K.; Bielawska, A.; Paneth, P. Cytotoxic effect and molecular docking of 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide—A novel topoisomerase II inhibitor. J. Mol. Model. 2012, 19, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Kaproń, B.; Luszczki, J.J.; Paneth, A.; Wujec, M.; Siwek, A.; Karcz, T.; Mordyl, B.; Głuch-Lutwin, M.; Gryboś, A.; Nowak, G.; et al. Molecular mechanism of action and safety of 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione—A novel anticonvulsant drug candidate. Int. J. Med. Sci. 2017, 14, 741–749. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Plech, T.; Kaproń, B.; Luszczki, J.J.; Paneth, A.; Siwek, A.; Kołaczkowski, M.; Żołnierek, M.; Nowak, G. Studies on the anticonvulsant activity of 4-alkyl-1,2,4-triazole-3-thiones and their effect on GABAergic system. Eur. J. Med. Chem. 2014, 86, 690–699. [Google Scholar] [CrossRef]
- Plech, T.; Kaproń, B.; Luszczki, J.J.; Wujec, M.; Paneth, A.; Siwek, A.; Kołaczkowski, M.; Żołnierek, M.; Nowak, G. Studies on the Anticonvulsant Activity and Influence on GABA-ergic Neurotransmission of 1,2,4-Triazole-3-thione- Based Compounds. Molecules 2014, 19, 11279–11299. [Google Scholar] [CrossRef]
- Paneth, A.; Wujec, M.; Plech, T.; Ku͇mierz, E.; Hagel, D.; Kosiek, S.; Jagiello-Wójtowicz, E.; Piątkowska-Chmiel, I.; Herbet, A.S.M. Preliminary Pharmacological Screening of Some Thiosemicarbazide, s-triazole, and Thiadiazole Derivatives. Cns Neurol. Disord.—Drug Targets 2016, 15, 730–739. [Google Scholar] [CrossRef]
- Wujec, M.; Kedzierska, E.; Kusmierz, E.; Plech, T.; Wróbel, A.; Paneth, A.; Orzelska-Górka, J.; Fidecka, S.; Paneth, P. Pharmacological and Structure-Activity Relationship Evaluation of 4-aryl-1-Diphenylacetyl(thio)semicarbazides. Molecules 2014, 19, 4745–4759. [Google Scholar] [CrossRef]
- Paneth, A.; Płonka, W.; Paneth, P. What do docking and QSAR tell us about the design of HIV-1 reverse transcriptase nonnucleoside inhibitors? J. Mol. Model. 2017, 23, 317. [Google Scholar] [CrossRef]
- Paneth, A.; Plonka, W.; Paneth, P. Assessment of Nonnucleoside Inhibitors Binding to HIV-1 Reverse Transcriptase Using HYDE Scoring. Pharmaceuticals 2019, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lan, J.; Ge, J.; Yu, J.; Shan, S. Crystal structure of SARS-CoV-2 spike receptor-binding domain bound with ACE2. Protein Data Bank 2020. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins Struct. Funct. Bioinform. 1999, 37, 228–241. [Google Scholar] [CrossRef]
- LeadIT 2.3.2 Program; BioSolveIT GmbH: Augustin, Germany, 2017.
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible docking using tabu search and an empirical estimate of binding affinity. Proteins 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Hyperchem 8.0.8 Program; HyperCube, Inc.: Florida, FL, USA, 2010.
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- ADMEWORKS/ModelBuilder 7.9.1 Enterprise Edition, Fujitdu Kyushu Systems Limited, Japan, 2019. Available online: https://www.fujitsu.com/jp/group/kyushu/en/solutions/industry/lifescience/admeworks/modelbuilder/ (accessed on 20 May 2020).
- RDKit, Q2. Available online: https://www.rdkit.org (accessed on 28 September 2020).
- Anaconda Software Distribution. Computer Software. Version 2020.02. Available online: https://anaconda.com (accessed on 11 April 2020).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- SwissADME, SIB, Switzerland, 2019. Available online: http://swissadme.ch/index.php (accessed on 26 September 2020).
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- PreADMET, BMDRC, Korea, 2017. Available online: https://preadmet.bmdrc.kr/toxicity/ (accessed on 1 October 2020).
- Lee, S.K.; Lee, I.H.; Kim, H.J.; Chang, G.S.; Chung, J.E.; No, K.T. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. In EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions; Blackwell Publishing: Massachusetts, MA, USA, 2003; pp. 418–420. [Google Scholar]
- Jain, A.K.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A.K. Models and Methods for in Vitro Toxicology. In Vitro Toxicology; Dhavan, A., Kwon, S., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 45–65. [Google Scholar]
- Keramagi, A.R.; Skariyachan, S. Prediction of binding potential of natural leads against the prioritized drug targets of chikungunya and dengue viruses by computational screening. Biotech. 2018, 8, 274. [Google Scholar] [CrossRef]
- Teixidó, E.; Leuthold, D.; Crozé, N.; Léonard, M.; Scholz, S. Comparative Assessment of the Sensitivity of Fish Early-Life Stage, Daphnia, and Algae Tests to the Chronic Ecotoxicity of Xenobiotics: Perspectives for Alternatives to Animal Testing. Env. Toxicol. Chem. 2019, 39, 30–41. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C-substituents R1 | Core | N-substitutents R2 |
|---|---|---|
 2-methylfuran-3-yl (F)  4-methyl-imidazol-5-yl (I)  pyrrol-2-yl (P)  cyclopentyl (C) |  carbonylthiosemicarbazide (C)  1,3,4-thiadiazole (S)  1,2,4-triazole-3-thione (T) |  R3 = H, ortho, meta, or para: F, Cl, Br, I, OH, OMe, Me, NO2 |
| Compound | Interface | Virus S-Protein | Human ACE2 Receptor | TA100/TA1535 Ames Tests | Carcino (Mouse) Test | Daphnia Test (vs. Chloroquine) |
|---|---|---|---|---|---|---|
| FSoOH | 54.71 | 39.89 | 53.16 | −+++ | negative | 3.0 |
| PSoF | 53.96 | 49.01 | 47.68 | all positive | positive | 1.8 |
| FCmOH | 53.67 | 46.07 | 51.68 | −+++ | negative | 3.3 |
| PSoMe | 52.09 | 49.05 | 47.45 | +−−+ | negative | 1.3 |
| CCmI | 53.25 | 41.24 | 52.6 | +−++ | negative | 0.9 |
| FCpF | 46.48 | 49.01 | 45.56 | all positive | positive | 2.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Płonka, W.; Paneth, A.; Paneth, P. Docking and QSAR of Aminothioureas at the SARS-CoV-2 S-Protein–Human ACE2 Receptor Interface. Molecules 2020, 25, 4645. https://doi.org/10.3390/molecules25204645
Płonka W, Paneth A, Paneth P. Docking and QSAR of Aminothioureas at the SARS-CoV-2 S-Protein–Human ACE2 Receptor Interface. Molecules. 2020; 25(20):4645. https://doi.org/10.3390/molecules25204645
Chicago/Turabian StylePłonka, Wojciech, Agata Paneth, and Piotr Paneth. 2020. "Docking and QSAR of Aminothioureas at the SARS-CoV-2 S-Protein–Human ACE2 Receptor Interface" Molecules 25, no. 20: 4645. https://doi.org/10.3390/molecules25204645
APA StylePłonka, W., Paneth, A., & Paneth, P. (2020). Docking and QSAR of Aminothioureas at the SARS-CoV-2 S-Protein–Human ACE2 Receptor Interface. Molecules, 25(20), 4645. https://doi.org/10.3390/molecules25204645