
Mitotic Poisons in Research and Medicine





Abstract
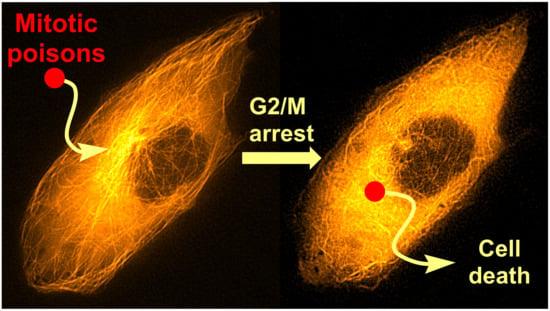
1. Introduction
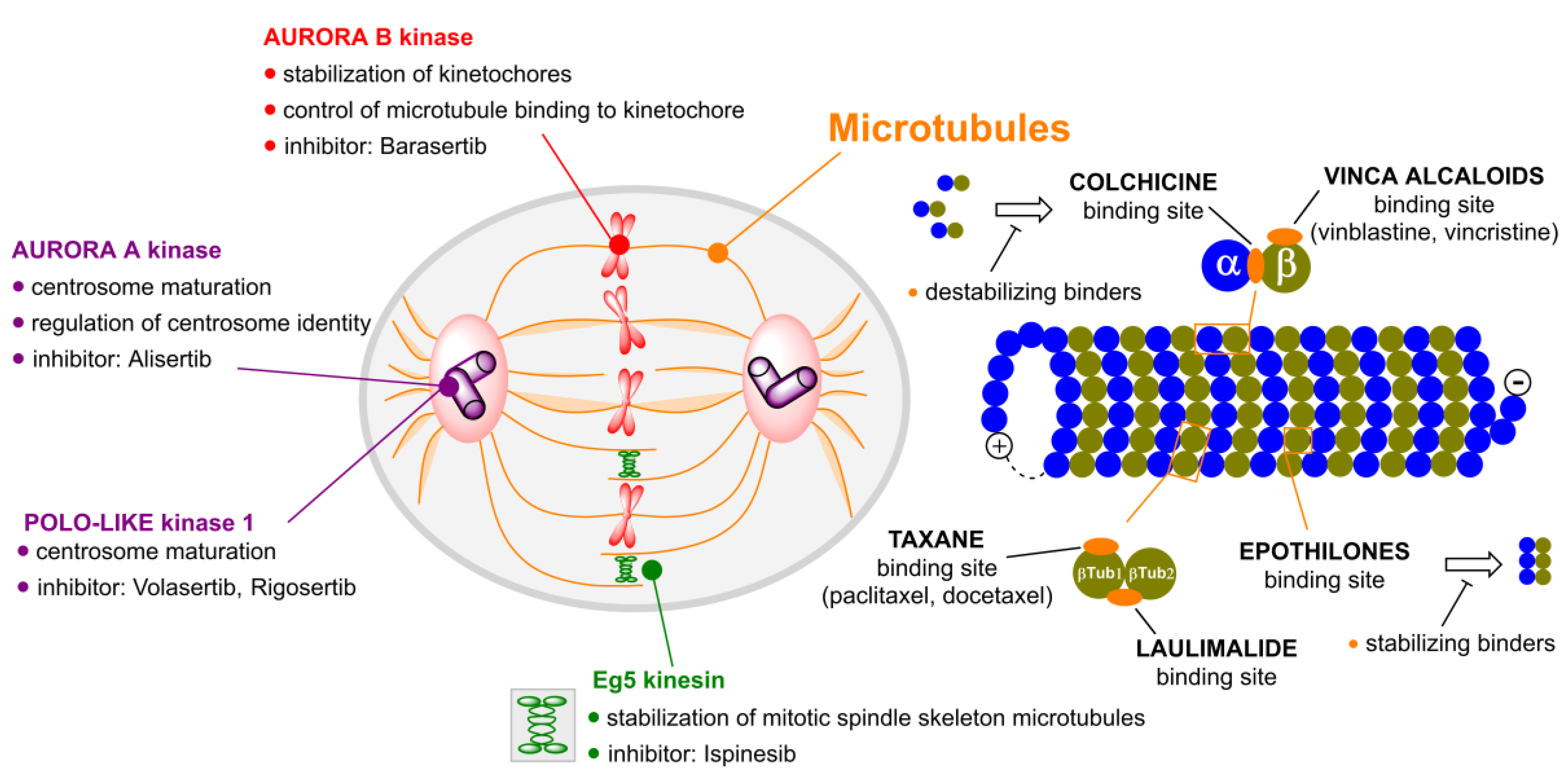
2. Microtubule Inhibitors
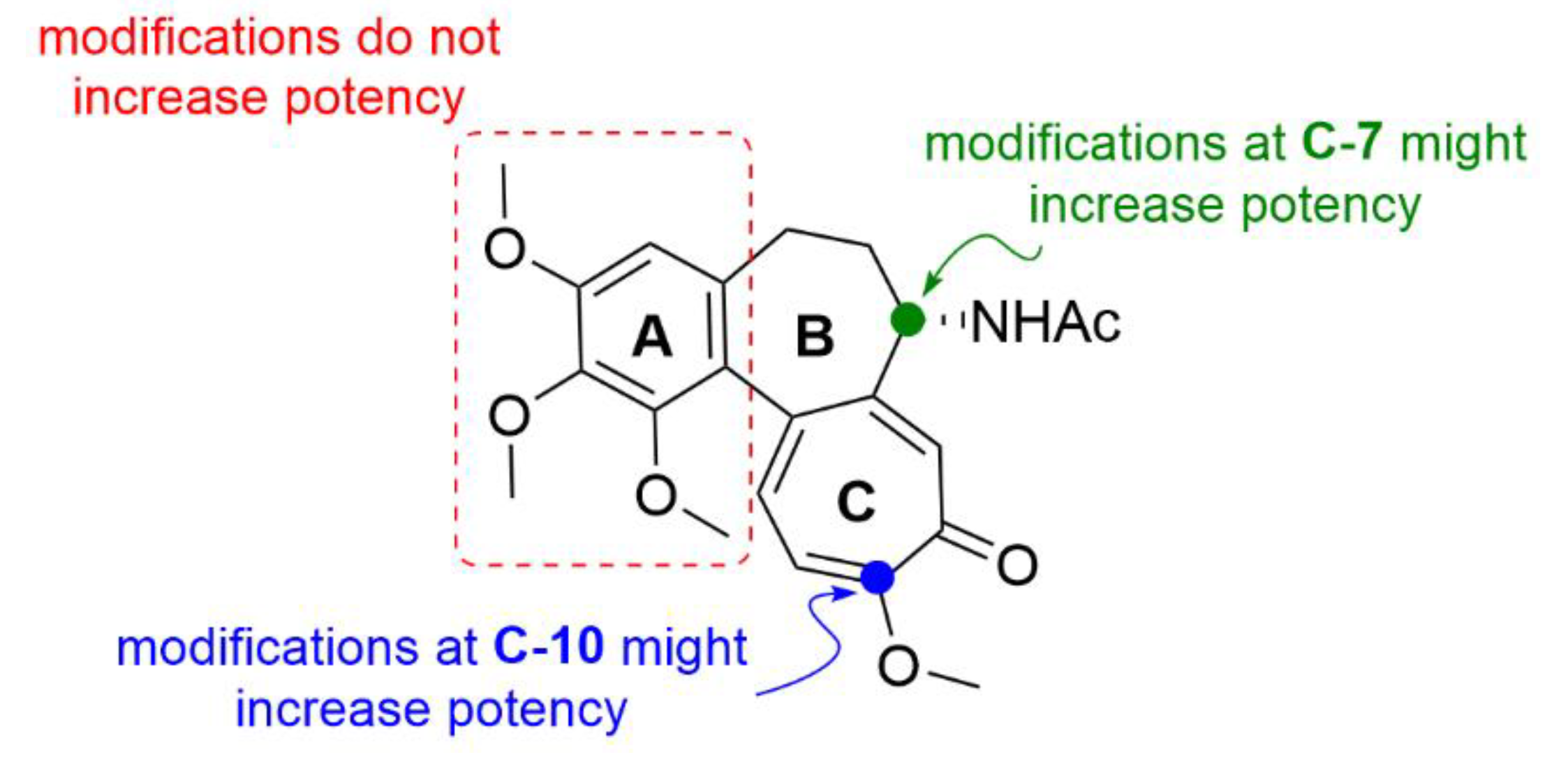
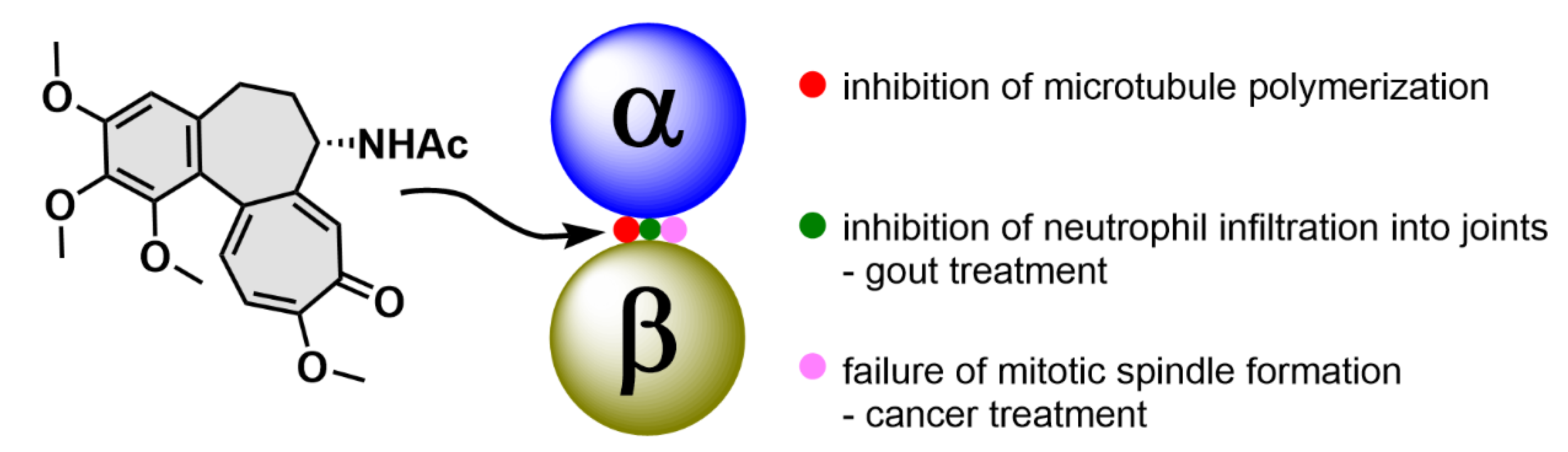
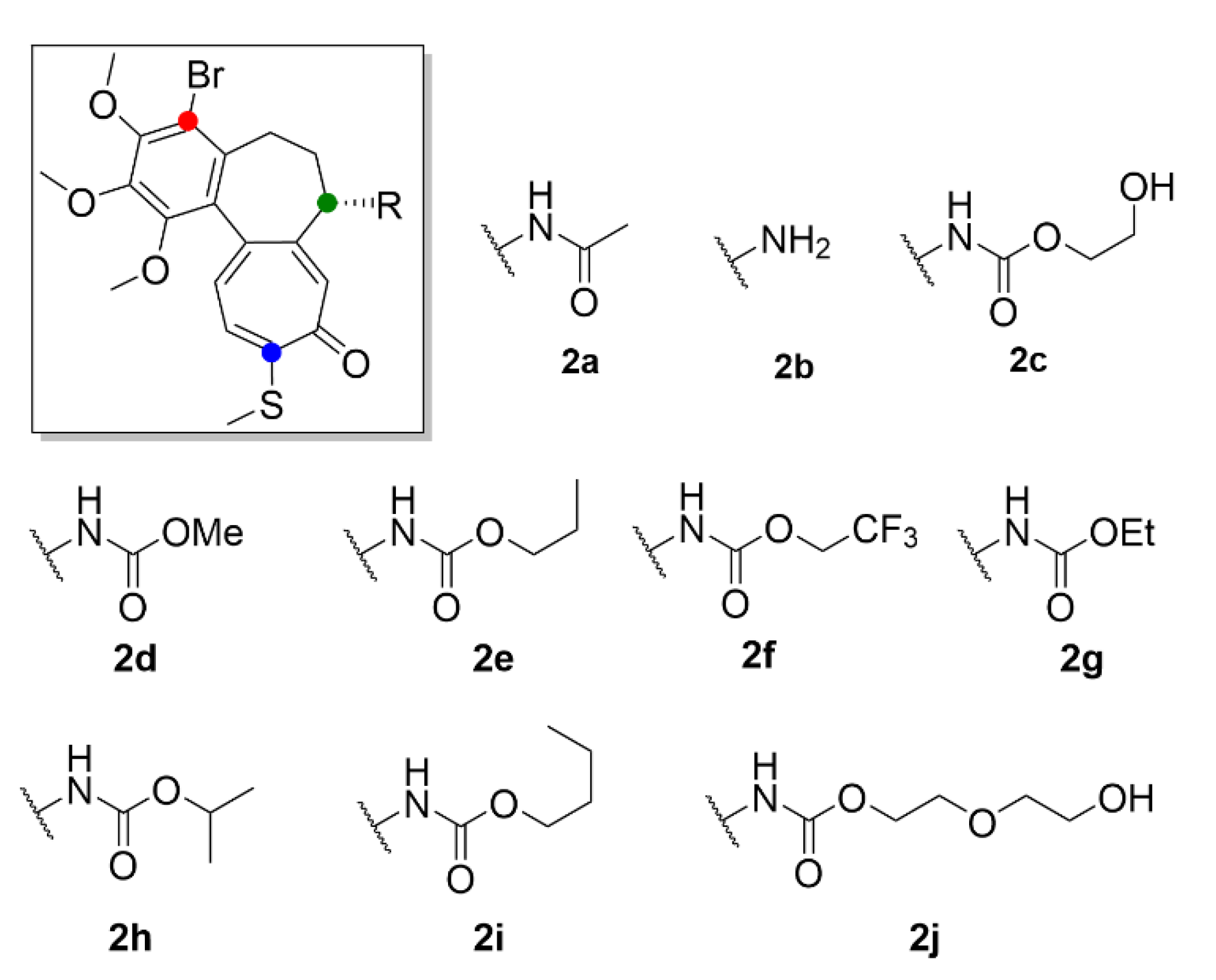
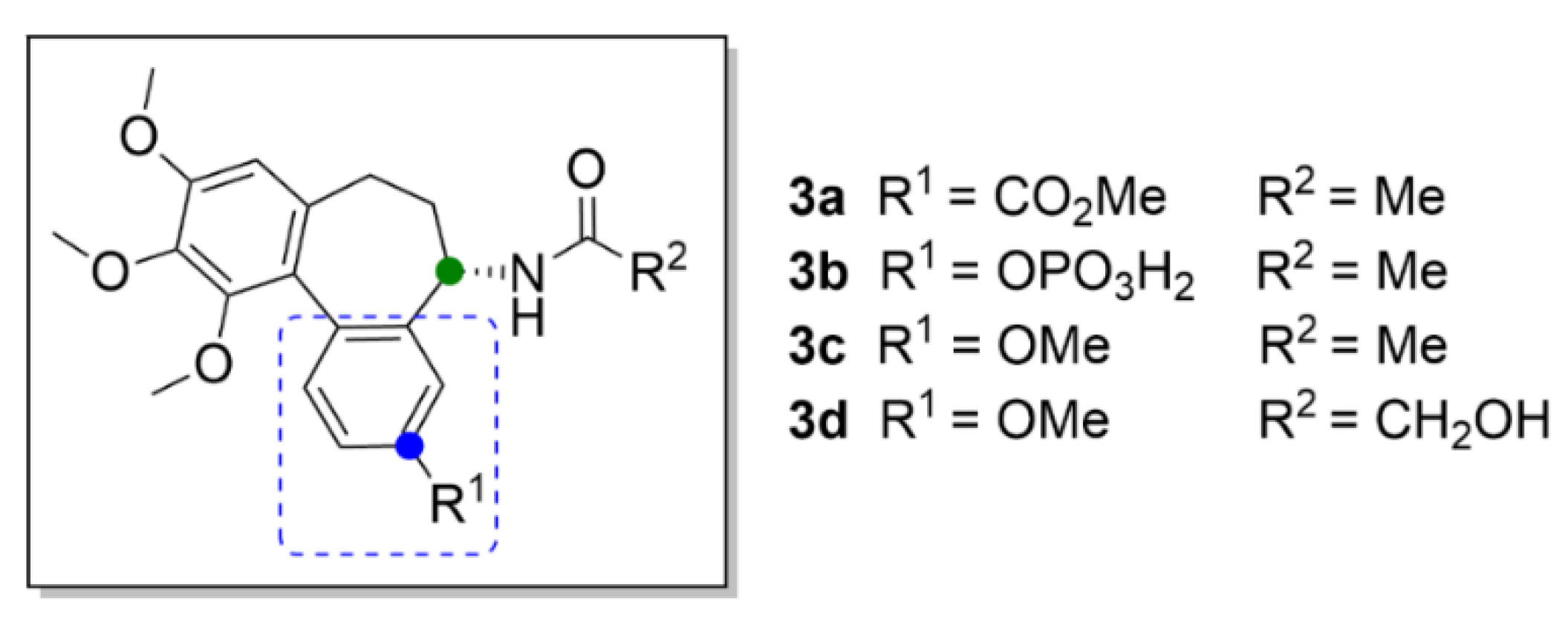
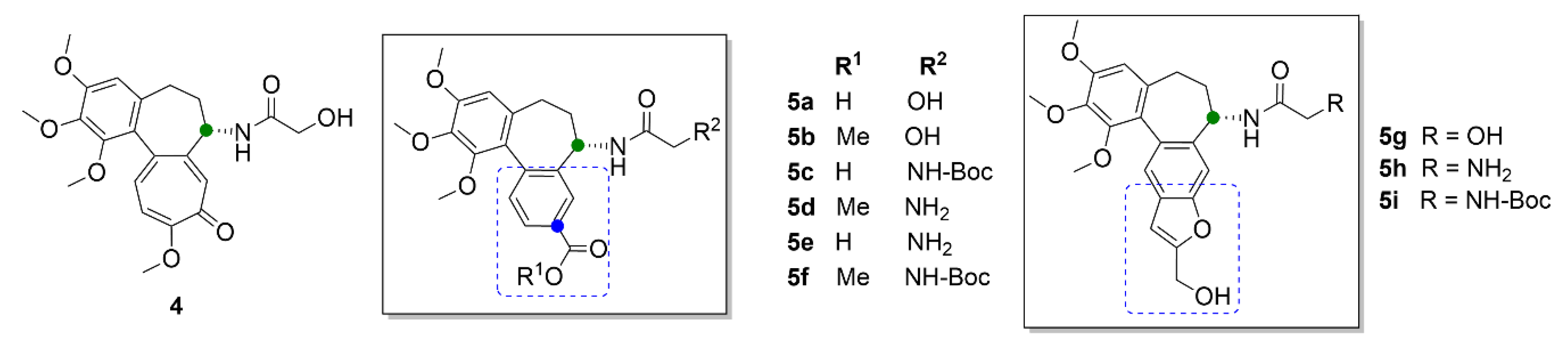
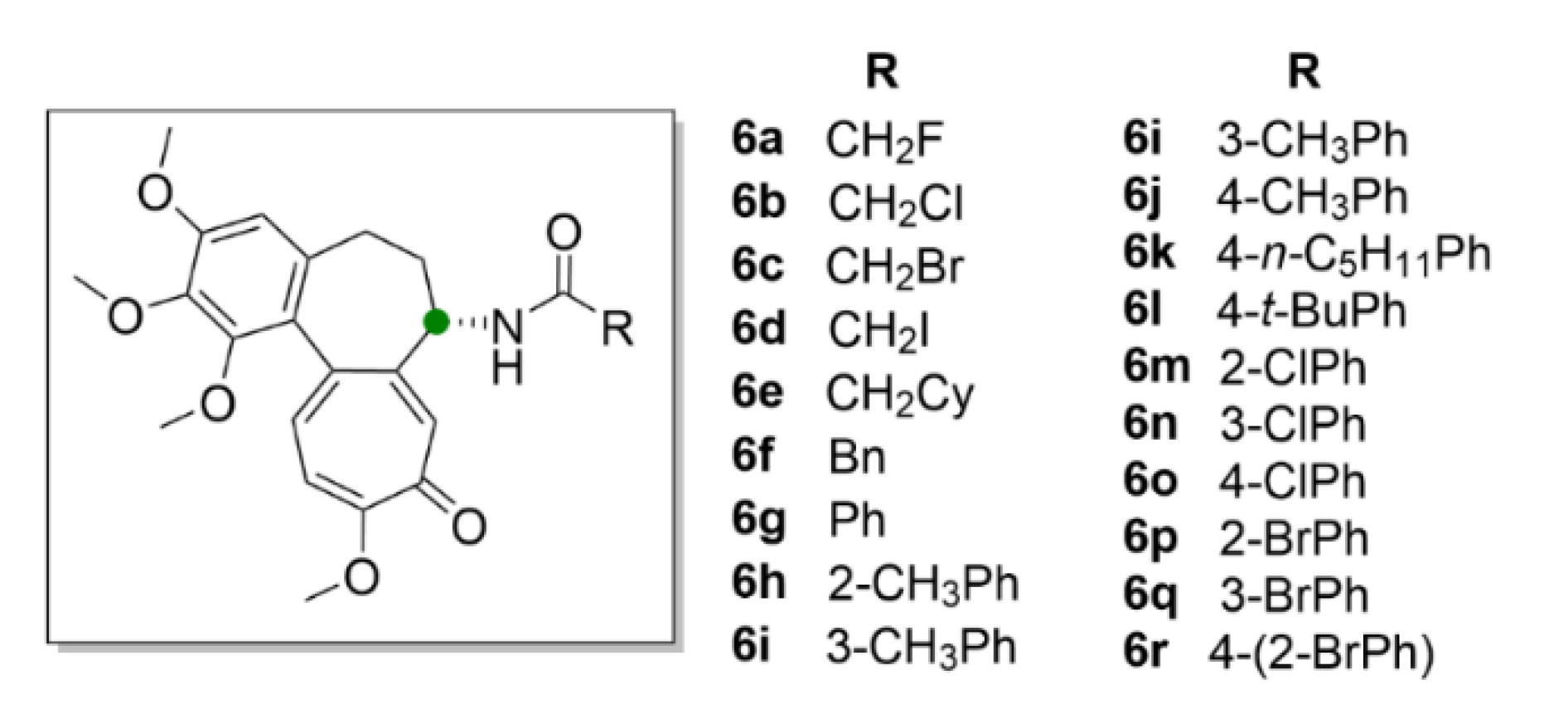
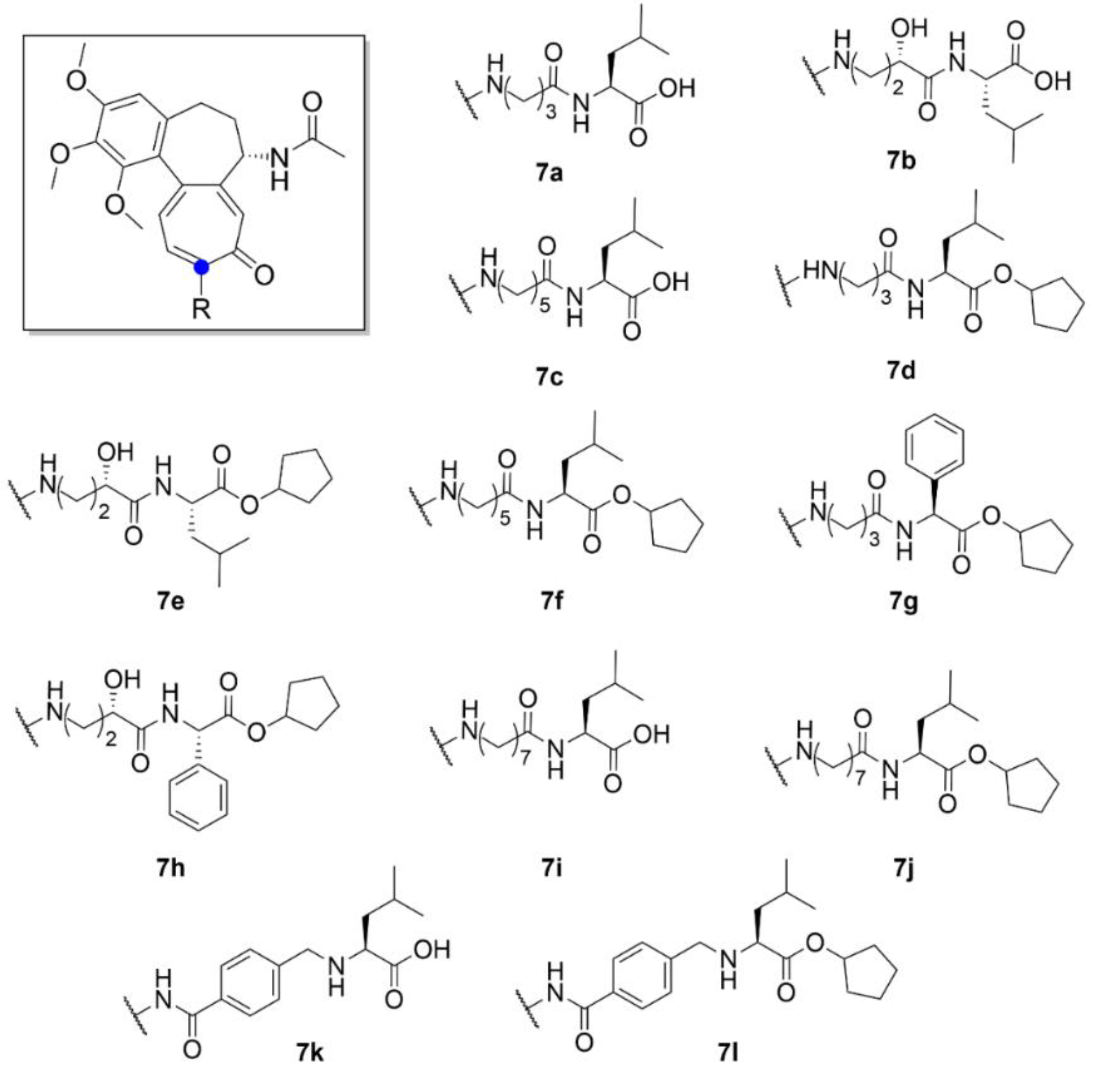
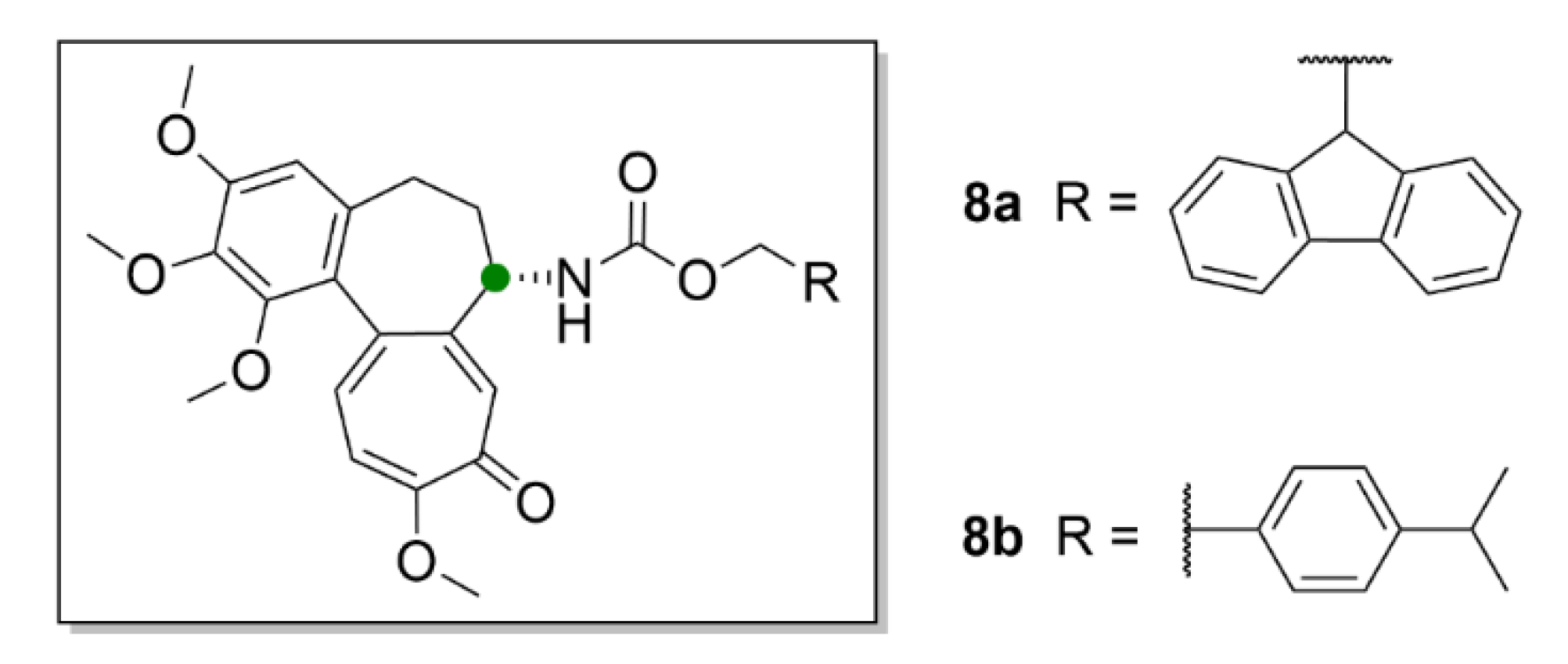
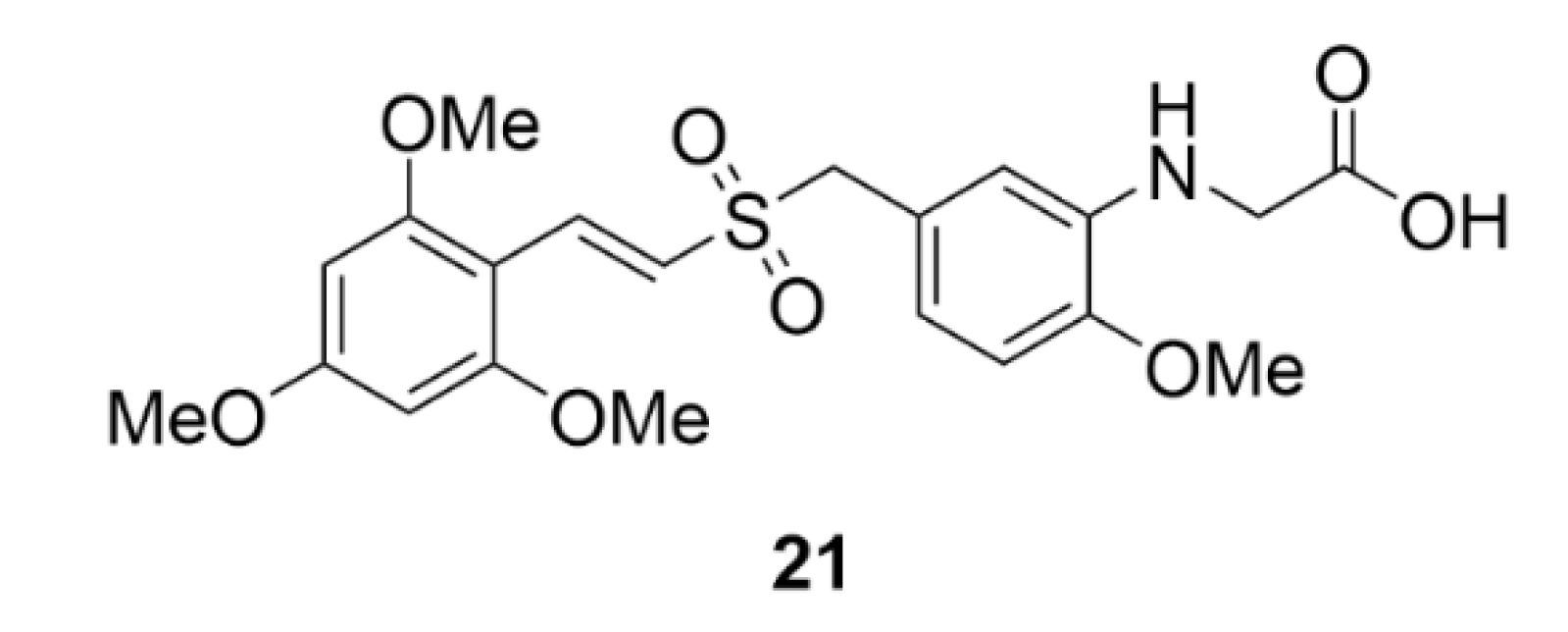
2.1. Colchicine
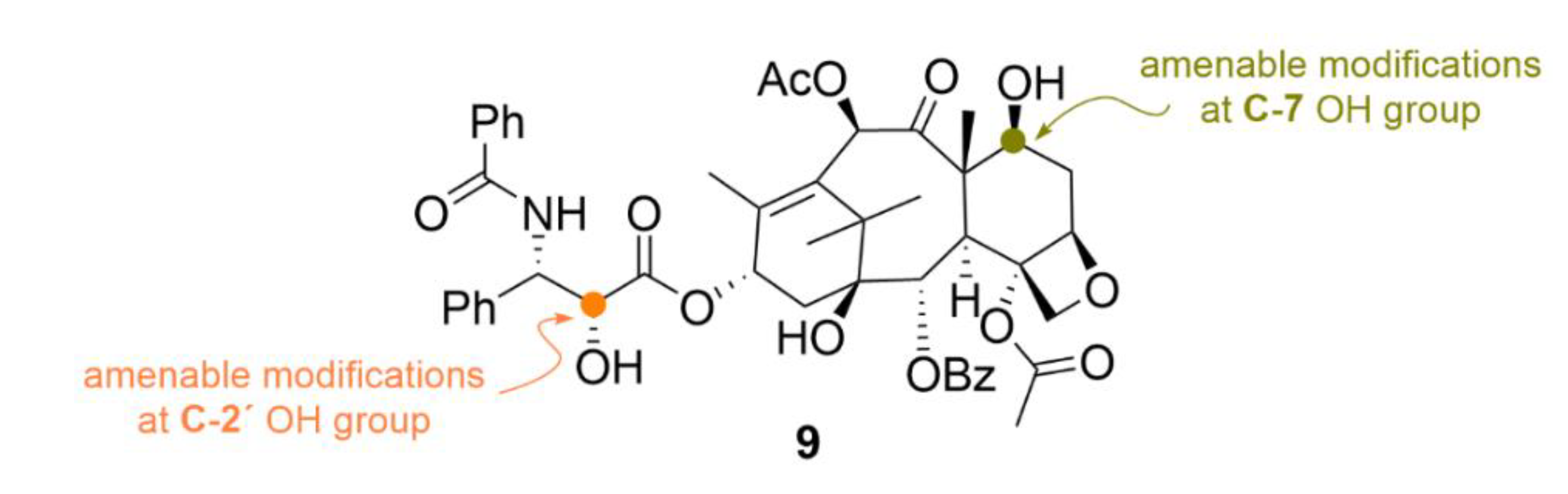
2.2. Taxanes
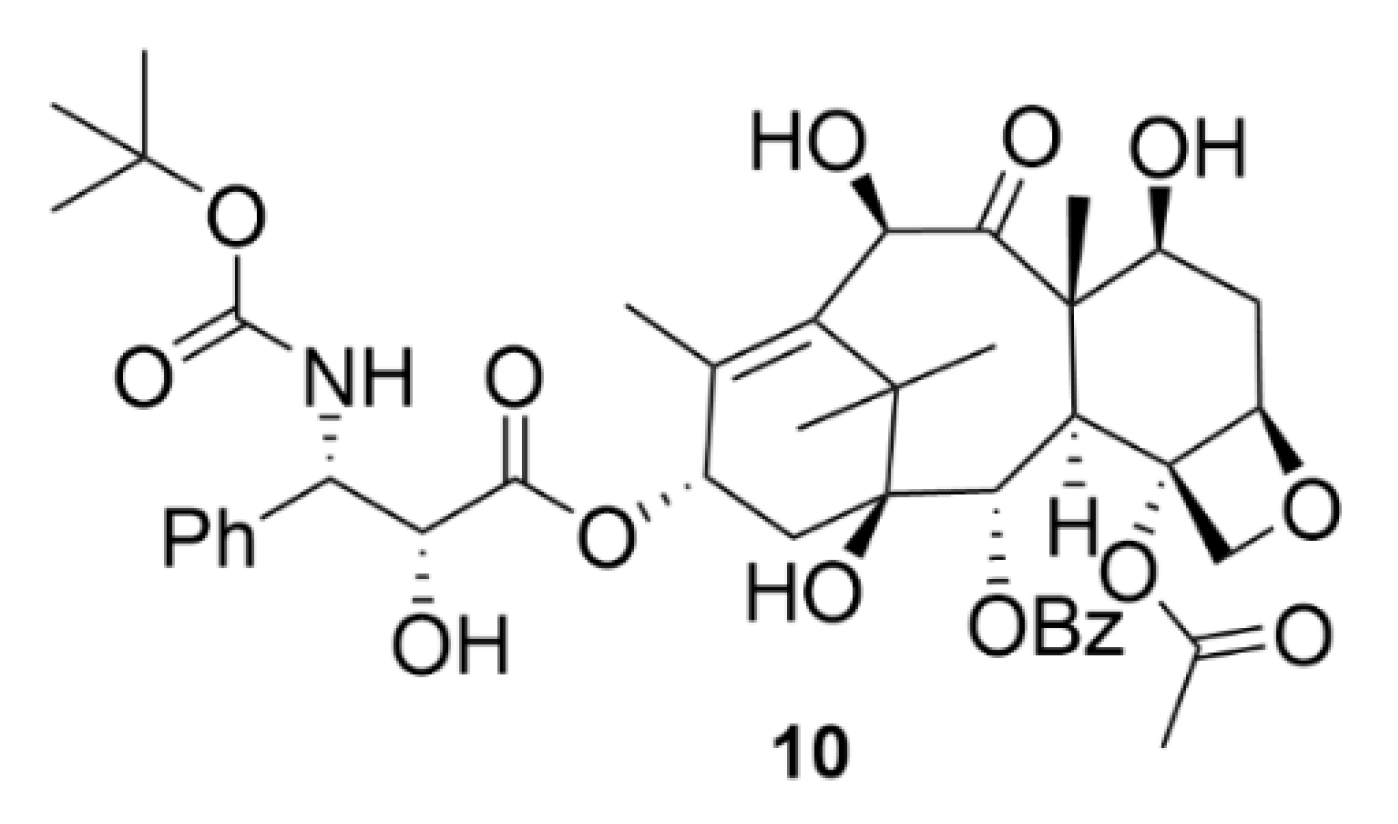
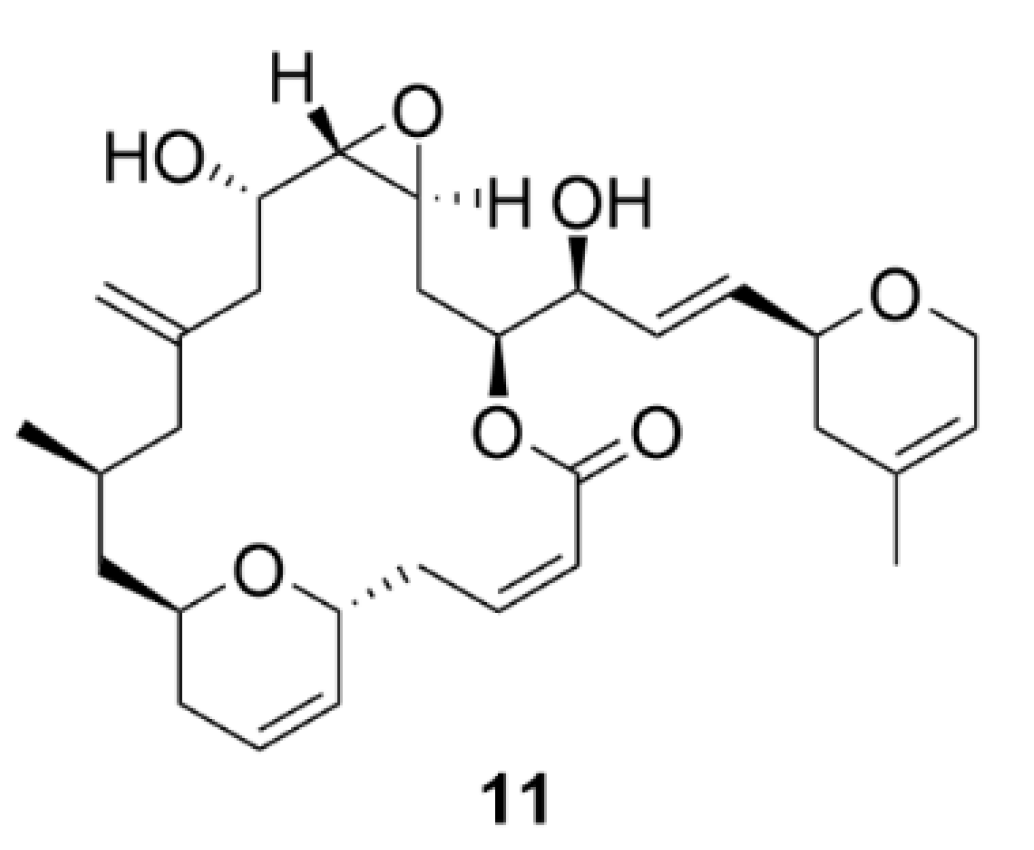
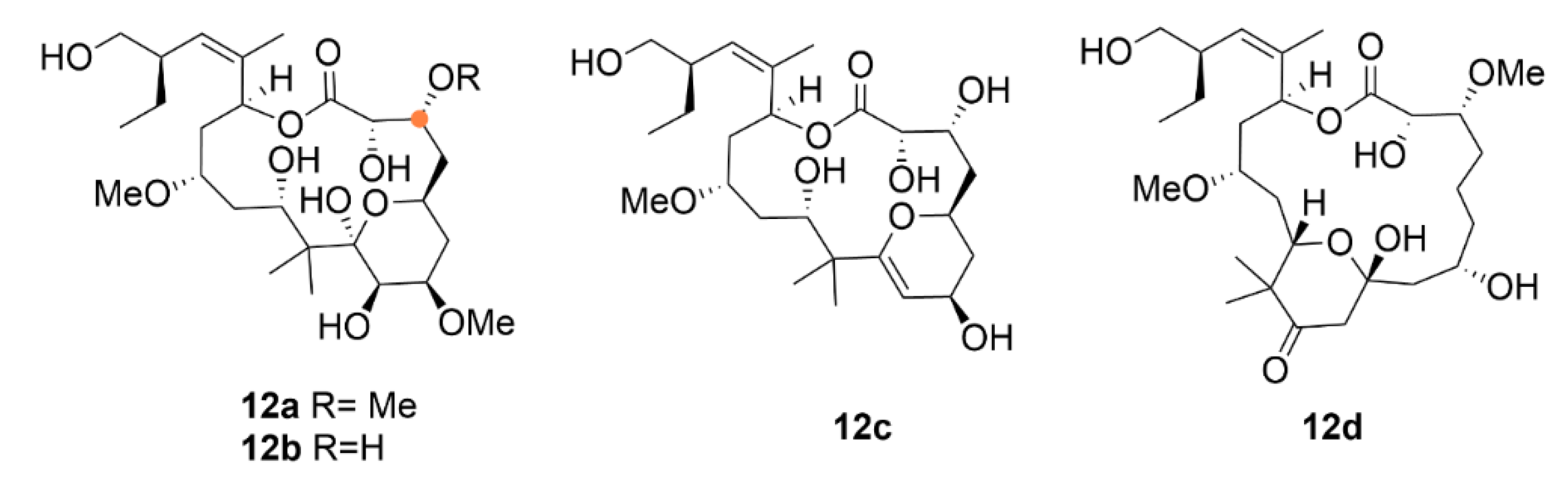
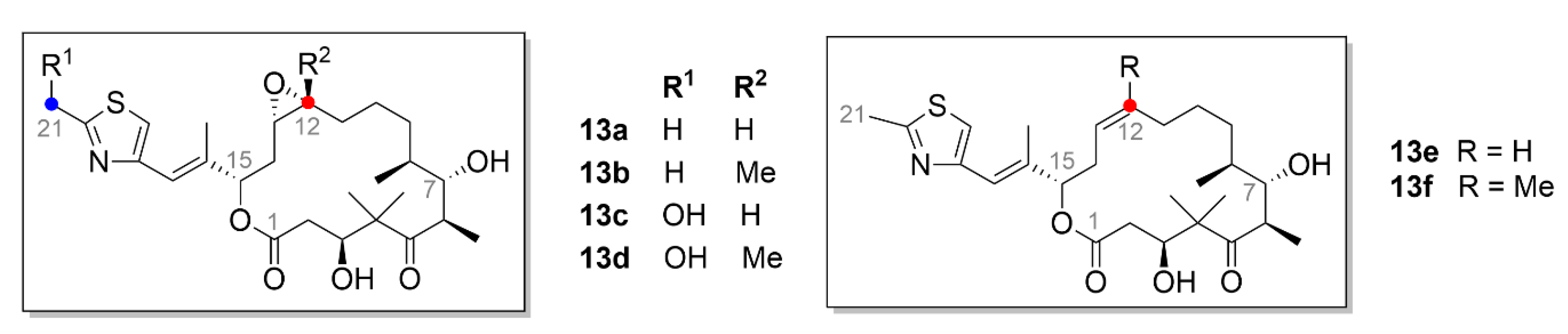
2.3. Laulimalide
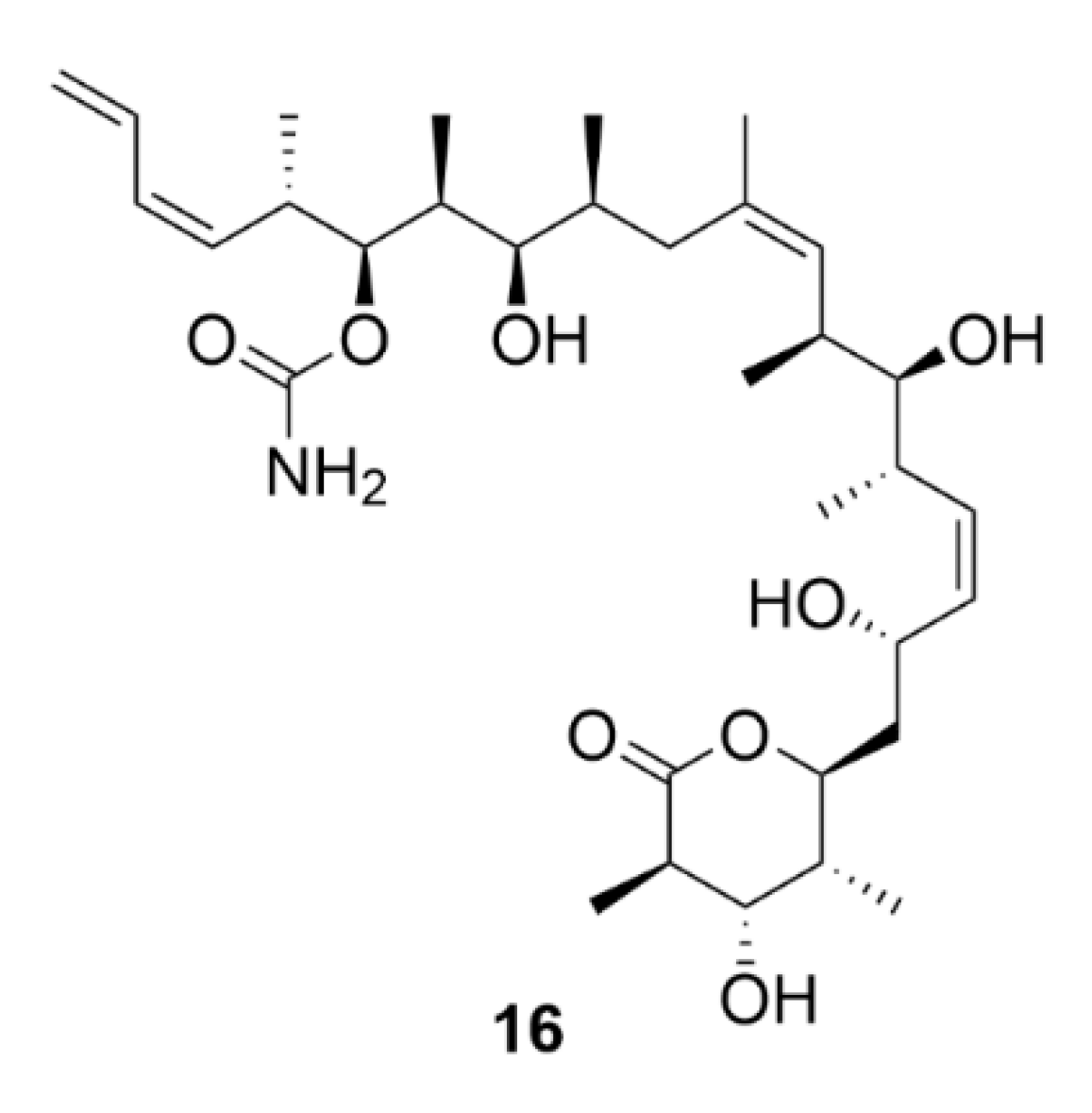
2.4. Peloruside A
2.5. Epothilones
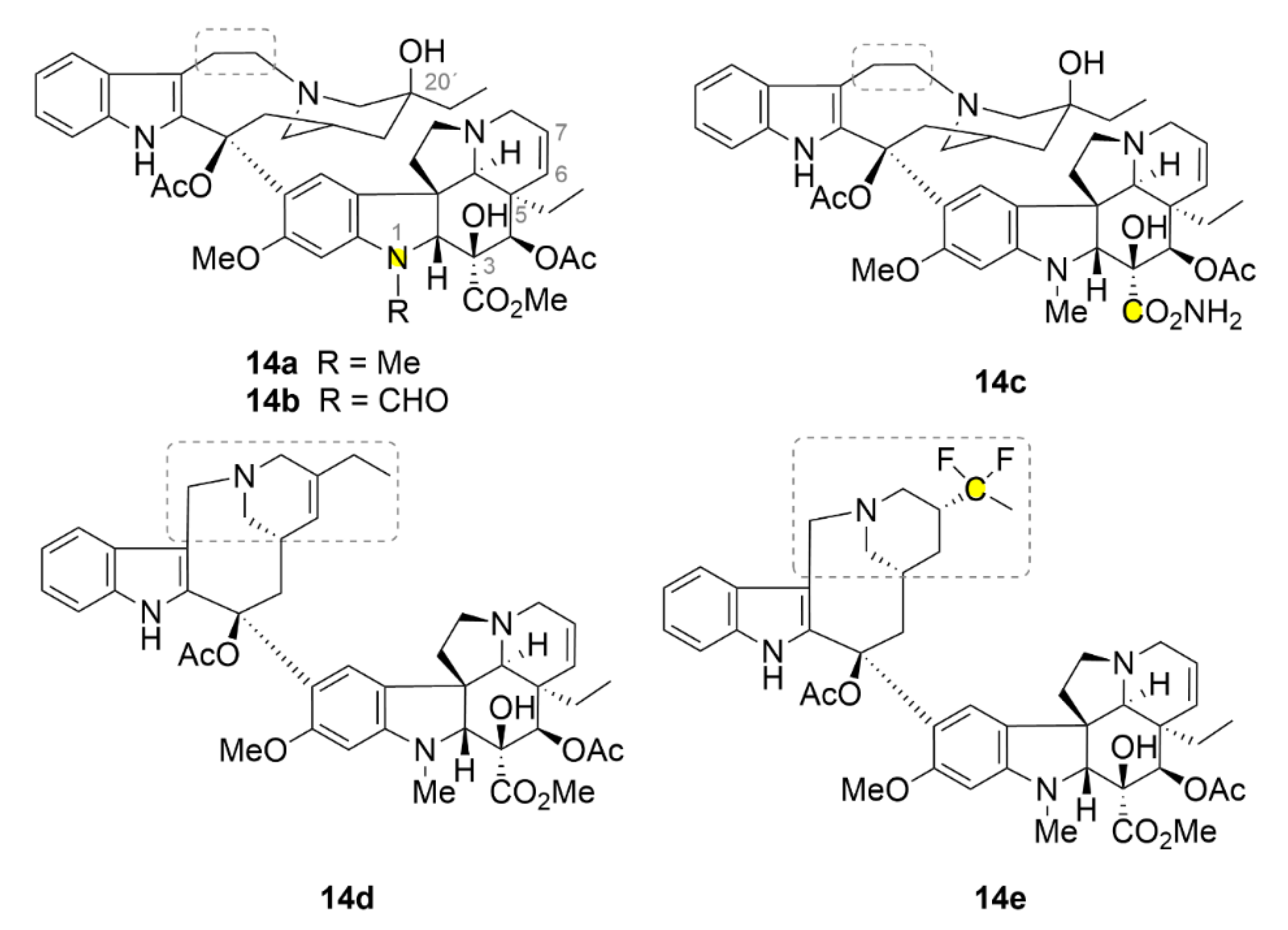
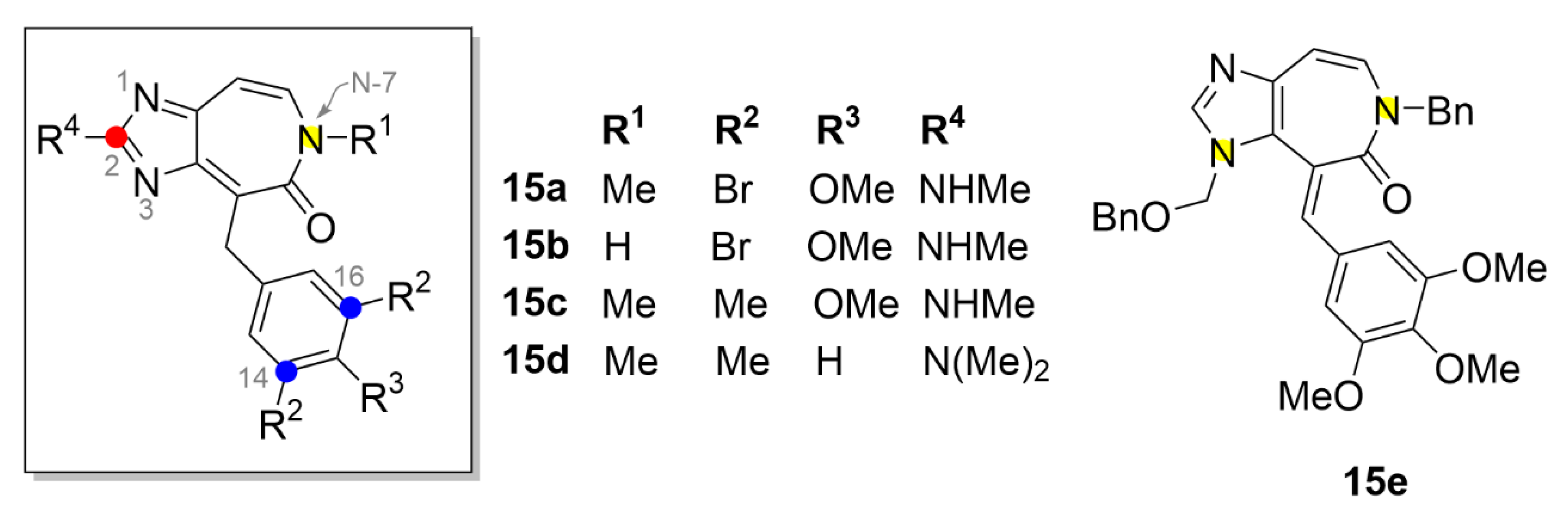
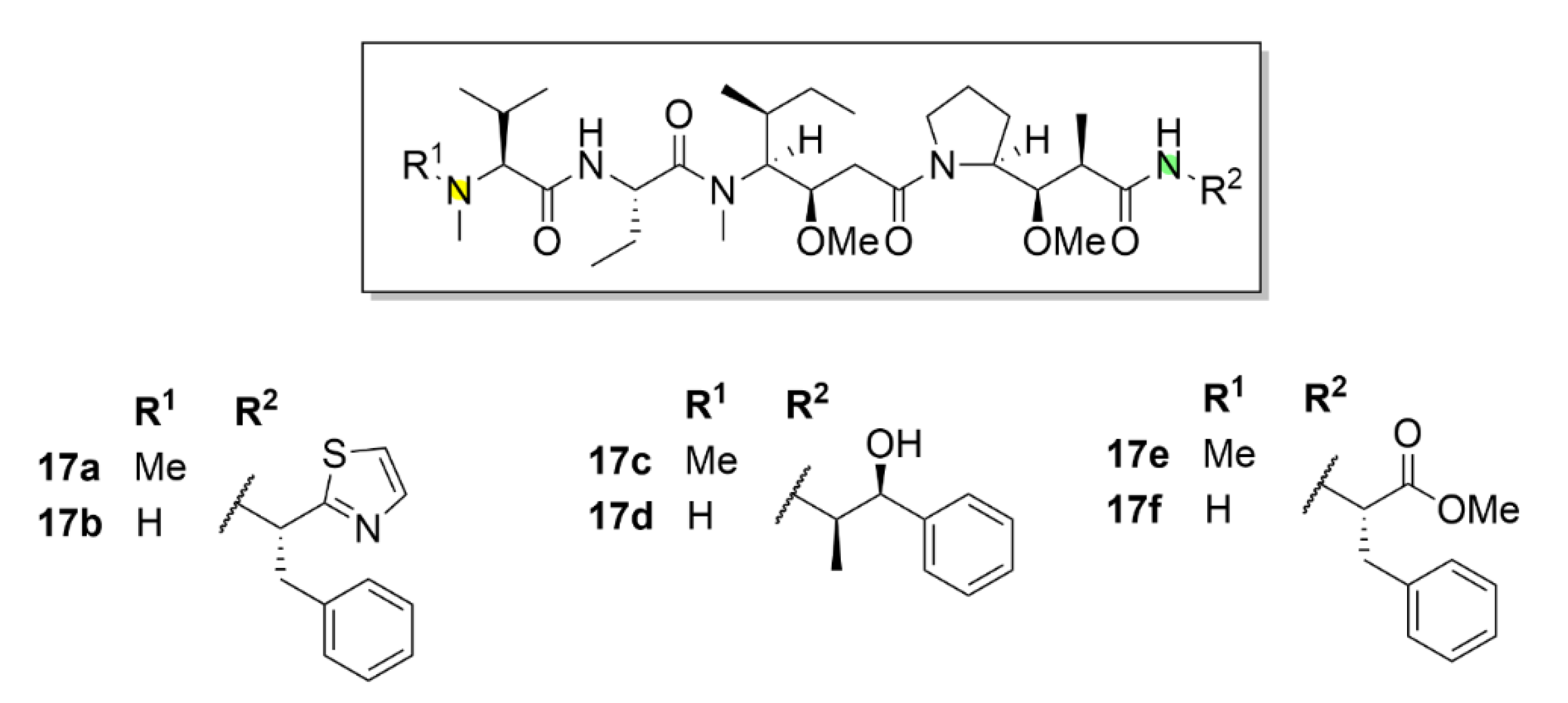
2.6. Vinca Alkaloids
2.7. Other Selected Tubulin-Binding Mitotic Poisons
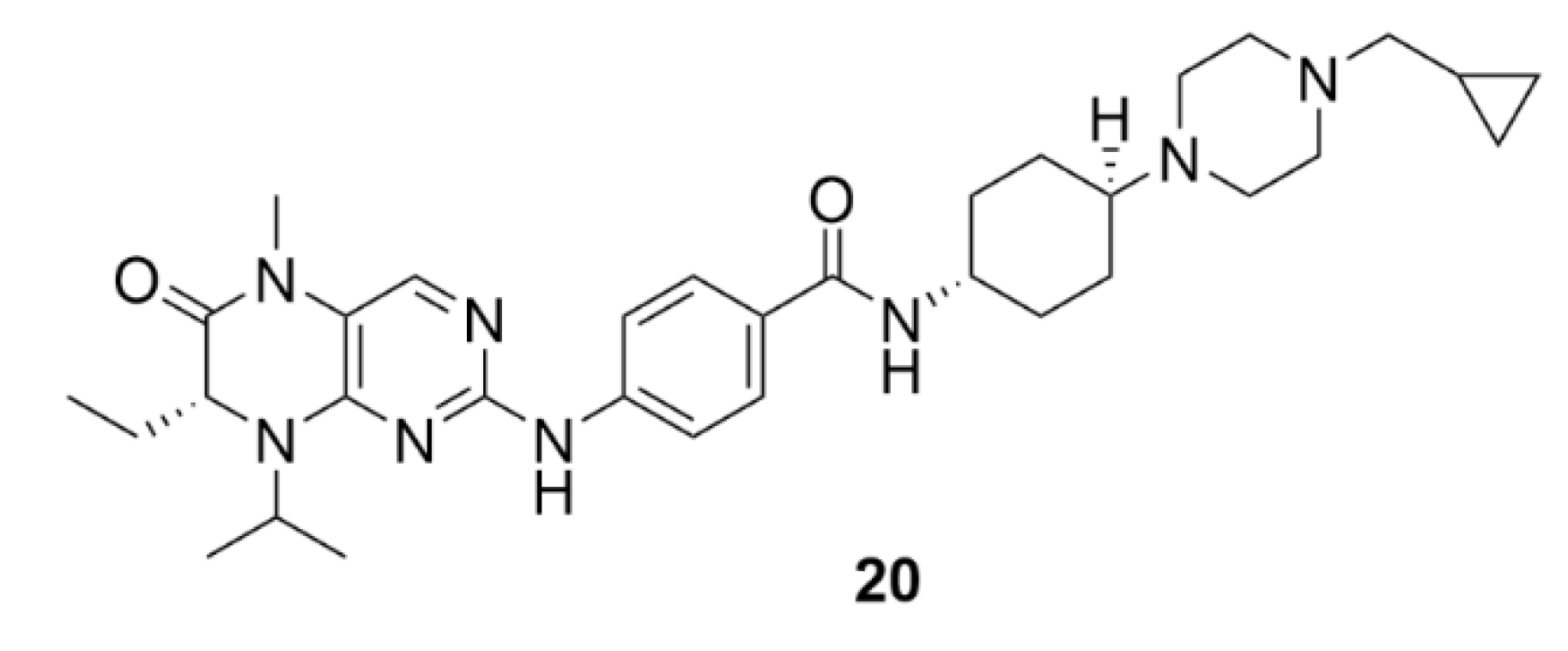
3. Aurora Kinases Inhibitors
3.1. Alisertib
3.2. Barasertib
4. Polo-like Kinases Inhibitors
4.1. Volasertib
4.2. Rigosertib
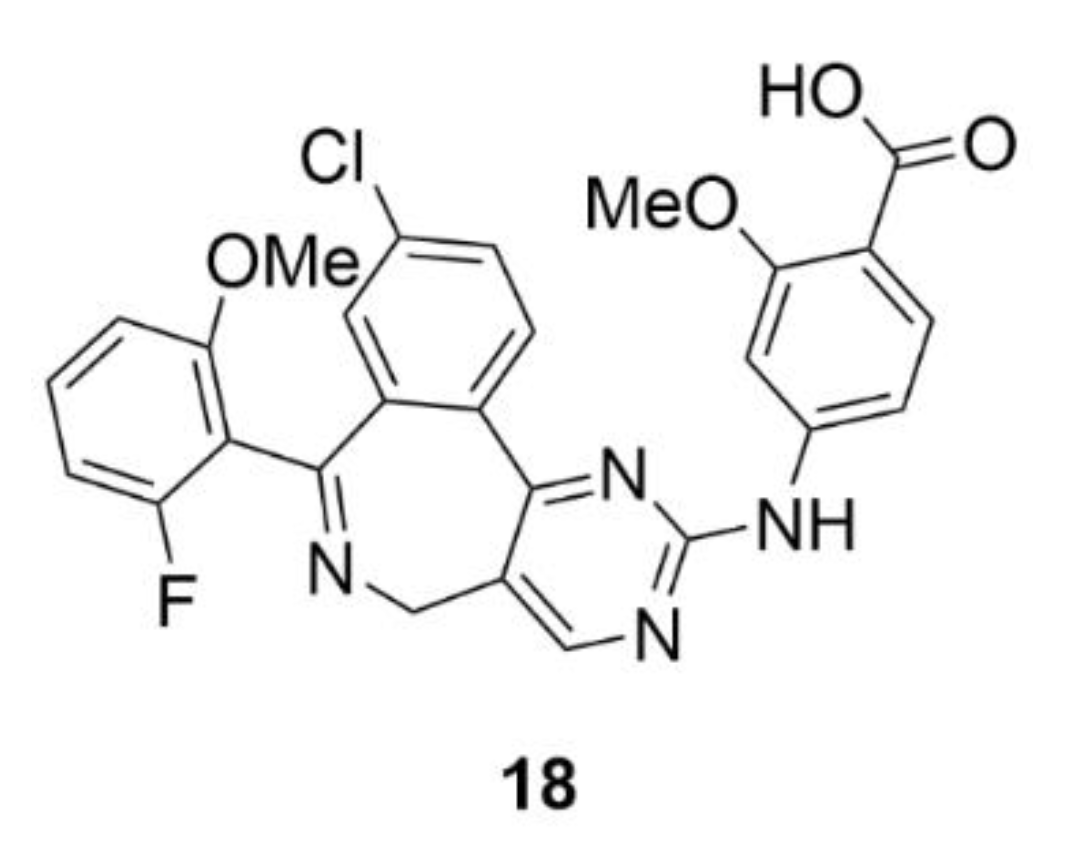
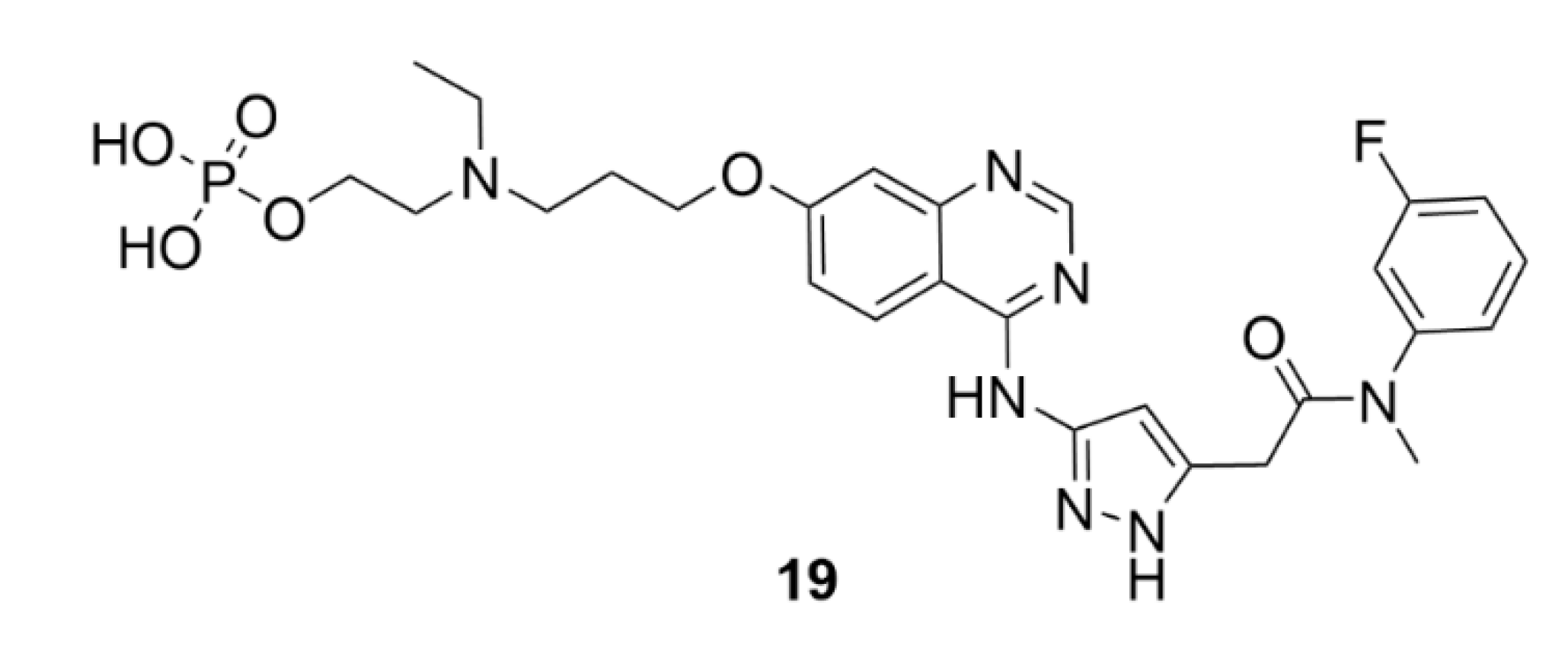
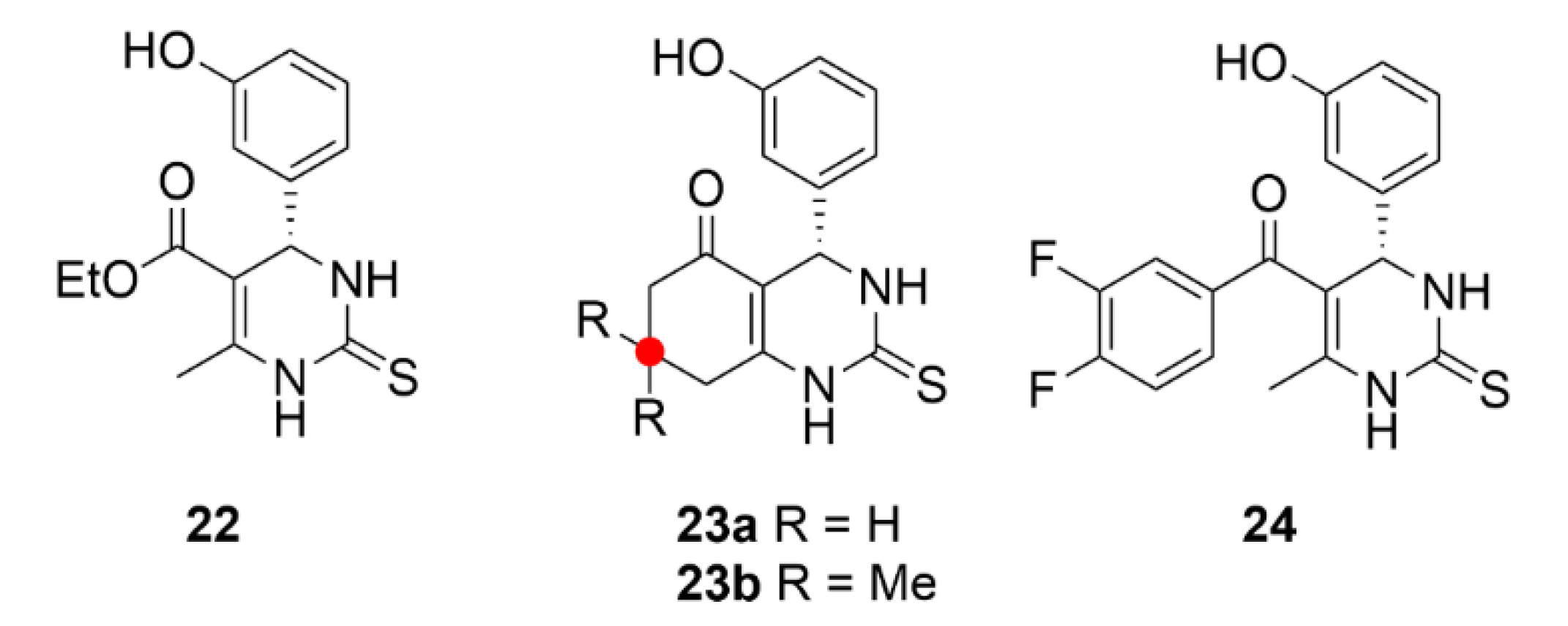
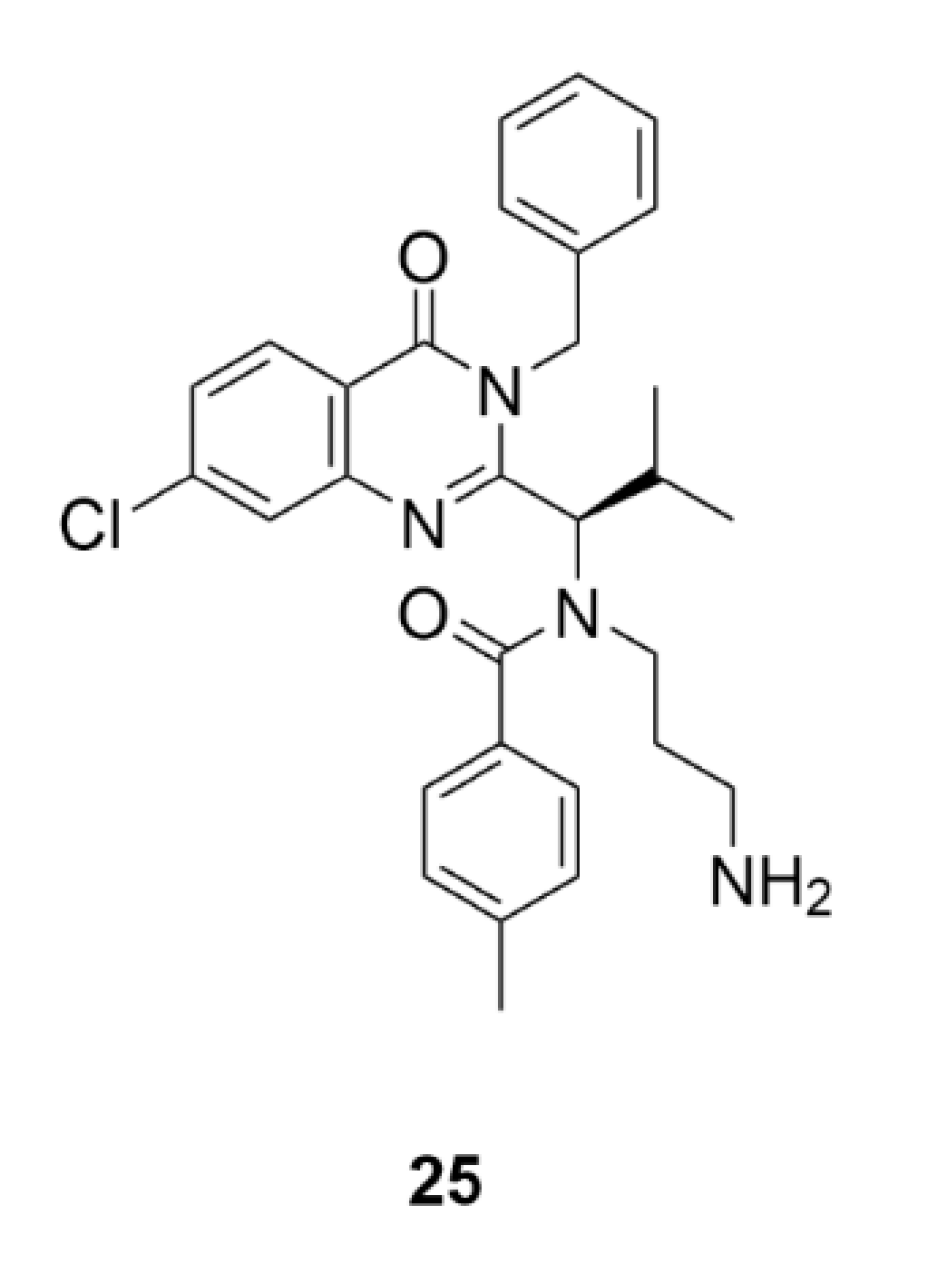
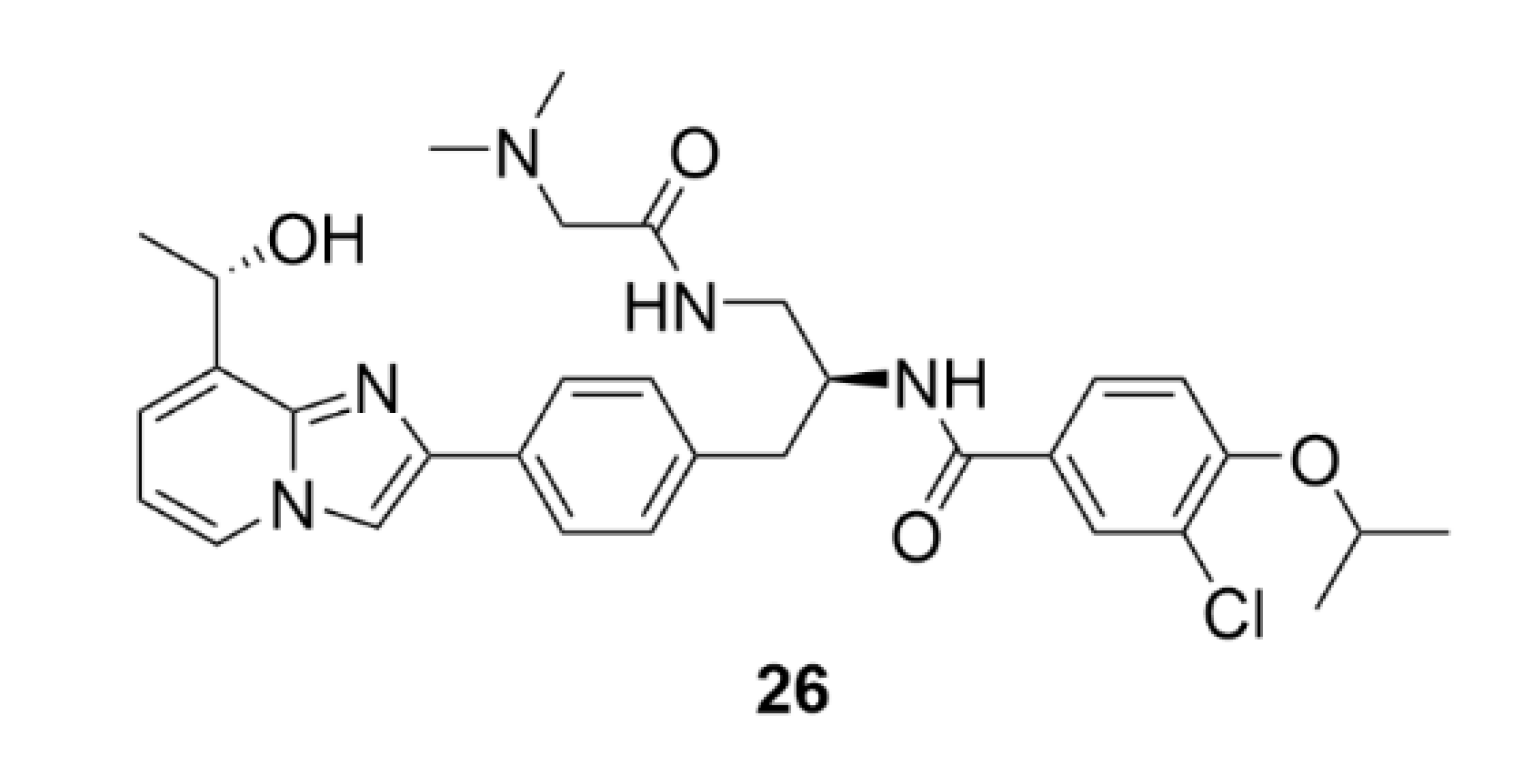
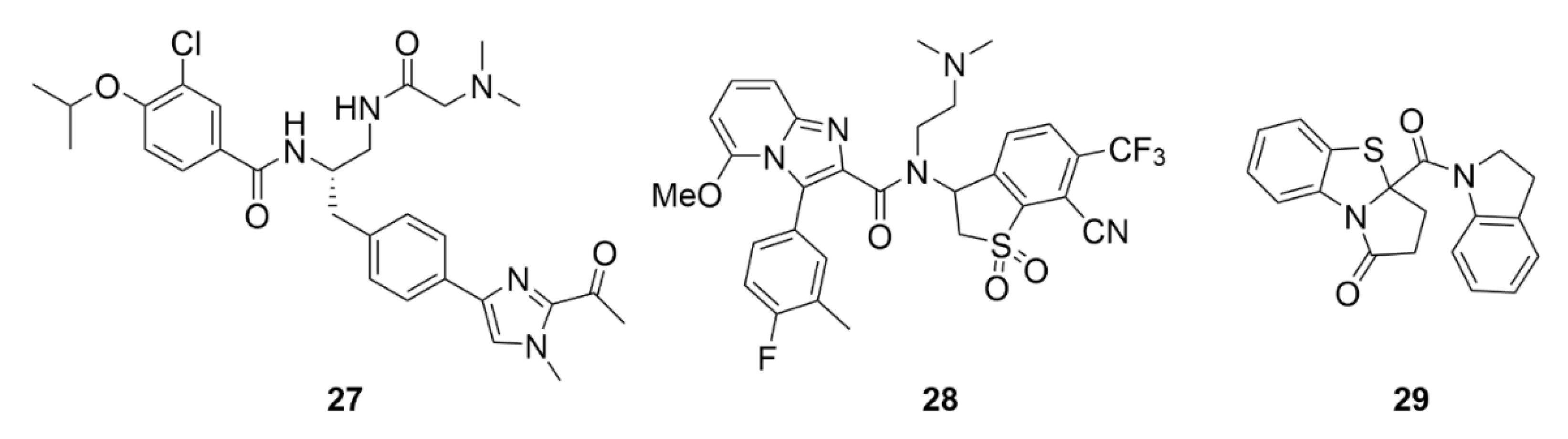
5. Kinesins Inhibitors
5.1. Kinesin-5 Inhibitors
5.2. CENP-E Inhibitors
6. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 7 October 2020).
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy-leveraging a knowledge base for clinical decision-making. Cold Spring Harb. Mol. Case Stud. 2018, 4, a001578. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/medicines/publications/essentialmedicines/en/ (accessed on 7 October 2020).
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Widmer, L.A.; Stangier, M.M.; Steinmetz, M.O.; Stelling, J.; Barral, Y. Remote control of microtubule plus-end dynamics and function from the minus-end. eLife 2019, 8, e48627. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; Goodson, H.V.; Alber, M.S. Microtubule dynamic instability: The role of cracks between protofilaments. Soft Matter. 2014, 10, 2069–2080. [Google Scholar] [CrossRef]
- Strothman, C.; Farmer, V.; Arpağ, G.; Rodgers, N.; Podolski, M.; Norris, S.; Ohi, R.; Zanic, M. Microtubule minus-end stability is dictated by the tubulin off-rate. J. Cell Biol. 2019, 218, 2841–2853. [Google Scholar] [CrossRef]
- Ferro, L.S.; Can, S.; Turner, M.A.; ElShenawy, M.M.; Yildiz, A. Kinesin and dynein use distinct mechanisms to bypass obstacles. eLife 2019, 8, e48629. [Google Scholar] [CrossRef]
- Stearns, T.; Evans, L.; Kirschner, M. γ-Tubulin is a highly conserved component of the centrosome. Cell 1991, 65, 825–836. [Google Scholar] [CrossRef]
- Findeisen, P.; Mühlhausen, S.; Dempewolf, S.; Hertzog, J.; Zietlow, A.; Carlomagno, T.; Kollmar, M. Six subgroups and extensive recent duplications characterize the evolution of the eukaryotic tubulin protein family. Genome Biol. Evol. 2014, 6, 2274–2288. [Google Scholar] [CrossRef]
- Ross, I.; Clarissa, C.; Giddings, T.H., Jr.; Winey, M. ε-tubulin is essential in Tetrahymena thermophila for the assembly and stability of basal bodies. J. Cell Sci. 2013, 126, 3441–3451. [Google Scholar] [CrossRef]
- Garreau de Loubresse, N.; Ruiz, F.; Beisson, J.; Klotz, C. Role of delta-tubulin and the C-tubule in assembly of Paramecium basal bodies. BMC Cell Biol. 2001, 2, 4. [Google Scholar] [CrossRef]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials. Available online: http://clinicaltrials.gov (accessed on 13 July 2020).
- Hartung, E.F. History of the use of colchicum and related medicaments in gout; with suggestions for further research. Ann. Rheum. Dis. 1954, 13, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Graham, W.; Roberts, J.B. Intravenous colchicine in the management of gouty arthritis. Ann. Rheum. Dis. 1953, 12, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Eigsti, O.J.; Dustin, P., Jr.; Gay-winn, N. On the discovery of the action of colchicine on mitosis in 1889. Science 1949, 110, 692. [Google Scholar] [CrossRef] [PubMed]
- Spasevska, I.; Ayoub, A.T.; Winter, P.; Preto, J.; Wong, G.K.-S.; Dumontet, C.; Tuszynski, J.A. Modeling the Colchicum autumnale tubulin and a comparison of its interaction with colchicine to human tubulin. Int. J. Mol. Sci. 2017, 18, 1676. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008, 28, 155–183. [Google Scholar] [CrossRef]
- Ateş, F.B.; Özmen, N.; Sezginer, E.K.; Kurt, E.E. Effects of colchicine on cell cycle arrest and MMP-2 mRNA expression in MCF 7 breast adenocarcinoma cells. Turk. Bull. Hyg. Exp. Biol. 2018, 75, 239–244. [Google Scholar] [CrossRef]
- McCarty, D.J. Urate crystals, inflammation, and colchicine. Arthritis Rheum. 2008, 58, S20–S24. [Google Scholar] [CrossRef]
- Paschke, S.; Weidner, A.F.; Paust, T.; Marti, O.; Beil, M.; Ben-Chetrit, E. Technical advance: Inhibition of neutrophil chemotaxis by colchicine is modulated through viscoelastic properties of subcellular compartments. J. Leukoc. Biol. 2013, 94, 1091–1096. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Wolff, S.M.; Goldfinger, S.E.; Dale, D.C.; Alling, D.W. Colchicine therapy for familial mediterranean fever. A double-blind trial. N. Engl. J. Med. 1974, 291, 934–937. [Google Scholar] [CrossRef]
- Filkenstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine poisoning: The dark side of an ancient drug. Clin. Toxicol. 2010, 48, 407–414. [Google Scholar]
- Bhattacharya, S.; Das, A.; Datta, S.; Ganguli, A.; Chakrabarti, G. Colchicine induces autophagy and senescence in lung cancer cells at clinically admissible concentration: Potential use of colchicine in combination with autophagy inhibitor in cancer therapy. Tumor Biol. 2016, 37, 10653–10664. [Google Scholar] [CrossRef] [PubMed]
- Majcher, U.; Klejborowska, G.; Kaik, M.; Maj, E.; Wietrzyk, J.; Moshari, M.; Preto, J.; Tuszynski, J.A.; Huczyński, A. Synthesis and biological evaluation of novel triple-modified colchicine derivatives as potent tubulin-targeting anticancer agents. Cells 2018, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.J.; Getahun, Z.; Muzaffar, A.; Brossi, A.; Hamel, E. N-acetylcolchinol O-methyl ether and thiocolchicine, potent analogs of colchicine modified in the C ring. Evaluation of the mechanistic basis for their enhanced biological properties. J. Biol. Chem. 1990, 265, 10255–10259. [Google Scholar] [PubMed]
- Yasobu, N.; Kitajima, M.; Kogure, N.; Shishido, Y.; Matsuzaki, T.; Nagaoka, M.; Takayama, H. Design, synthesis, and antitumor activity of 4-halocolchicines and their pro-drugs activated by cathepsin B. ACS Med. Chem. Lett. 2011, 2, 348–352. [Google Scholar] [CrossRef]
- Larocque, K.; Ovadje, P.; Djurdjevic, S.; Mehdi, M.; Green, J.; Pandey, S. Novel analogue of colchicine induces selective pro-death autophagy and necrosis in human cancer cells. PLoS ONE 2014, 9, e87064. [Google Scholar] [CrossRef]
- Davis, P.D.; Dougherty, G.J.; Blakey, D.C.; Galbraith, S.M.; Tozer, G.M.; Holder, A.L.; Naylor, M.A.; Nolan, J.; Stratford, M.R.L.; Chaplin, D.J.; et al. ZD6126, A novel vascular-targeting agent that causes selective destruction of tumor vasculature. Cancer Res. 2002, 62, 7247–7253. [Google Scholar]
- Gracheva, I.A.; Svirshchevskaya, E.V.; Faerman, V.I.; Beletskaya, I.P.; Fedorov, A.Y. Synthesis and antiproliferative properties of bifunctional allocolchicine derivatives. Synthesis 2018, 50, 2753–2760. [Google Scholar]
- Lippert, J.W., 3rd. Vascular disrupting agents. Bioorg. Med. Chem. 2007, 15, 605–615. [Google Scholar] [CrossRef]
- Marzo-Mas, A.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Effects on tubulin polymerization and down-regulation of c-Myc, hTERT and VEGF genes by colchicine haloacetyl and haloaroyl derivatives. Eur. J. Med. Chem. 2018, 150, 591–600. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural characterization of zika virus replication factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Boldescu, V.; Graf, D.; Streicher, F.; Dimoglo, A.; Bartenschlager, R.; Klein, C.D. Synthesis, biological evaluation, and molecular docking of combretastatin and colchicine derivatives and their hCE1-activated prodrugs as antiviral agents. ChemMedChem 2019, 14, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Wee, Y.M.; Kim, Y.H.; Shin, S.; Yoo, S.E.; Han, D.J. Novel colchicine derivatives enhance graft survival after transplantation via suppression of T-cell differentiation and activity. J. Cell Biochem. 2019, 120, 12436–12449. [Google Scholar] [CrossRef]
- Van Tamelen, E.E.; Spencer, T.A., Jr.; Allen, D.S., Jr.; Orvis, R.L. The total synthesis of colchicine. J. Am. Chem. Soc. 1959, 81, 6341–6342. [Google Scholar] [CrossRef]
- Schreiber, J.; Leimgruber, W.; Pesaro, M.; Schudel, P.; Eschenmoser, A. Synthese des colchicins. Angew. Chem. 1959, 71, 637–640. [Google Scholar] [CrossRef]
- Chen, B.; Liu, X.; Hu, Y.J.; Zhang, D.M.; Deng, L.; Lu, J.; Min, L.; Ye, W.C.; Li, C.C. Enantioselective total synthesis of (−)-colchicine, (+)-demecolcinone and metacolchicine: Determination of the absolute configurations of the latter two alkaloids. Chem. Sci. 2017, 8, 4961–4966. [Google Scholar] [CrossRef]
- Pandey, D.K.; Banik, R.M. Optimization of extraction conditions for colchicine from Gloriosa superba tubers using response surface methodology. J. Agric. Technol. 2012, 8, 1301–1315. [Google Scholar]
- Kefi, S. A novel approach for production of colchicine as a plant secondary metabolite by in vitro plant cell and tissue cultures. J. Agric. Sci. Technol. A 2018, 8, 121–128. [Google Scholar]
- Sivakumar, G.; Alba, K.; Phillips, G.C. Biorhizome: A biosynthetic platform for colchicine biomanufacturing. Front. Plant Sci. 2017, 8, 1137. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Parness, J.; Horwitz, S.B. Taxol binds to polymerized tubulin in vitro. J. Cell Biol. 1981, 91, 479–487. [Google Scholar] [CrossRef]
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell-proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B.; Donehower, R.C.; Wiernik, P.H.; Ohnuma, T.; Gralla, R.J.; Trump, D.L.; Baker, J.R., Jr.; Van Echo, D.A.; Von Hoff, D.D.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. Int. J. Nanomedicine. 2009, 4, 99–105. [Google Scholar] [PubMed]
- Du, X.; Yin, S.; Xu, L.; Ma, J.; Yu, H.; Wang, G.; Li, J. Polylysine and cysteine functionalized chitosan nanoparticle as an efficient platform for oral delivery of paclitaxel. Carbohydr. Polym. 2020, 229, 115484. [Google Scholar] [CrossRef]
- Zhao, M.; Li, H.; Fan, L.; Ma, Y.; Gong, H.; Lai, W.; Fang, Q.; Hu, Z. Quantitative proteomic analysis to the first commercialized liposomal paclitaxel nano-platform Lipusu revealed the molecular mechanism of the enhanced anti-tumor effect. Artif. Cells Nanomed. Biotechnol. 2018, 46, S147–S155. [Google Scholar] [CrossRef]
- Ranade, A.A.; Joshi, D.A.; Phadke, G.K.; Patil, P.P.; Kasbekar, R.B.; Apte, T.G.; Dasare, R.R.; Mengde, S.D.; Parikh, P.M.; Bhattacharyya, G.S.; et al. Clinical and economic implications of the use of nanoparticle paclitaxel (Nanoxel) in India. Ann. Oncol. 2013, 24, v6–v12. [Google Scholar] [CrossRef]
- Galletti, E.; Magnani, M.; Renzulli, M.L.; Botta, M. Paclitaxel and docetaxel resistance: Molecular mechanisms and development of new generation taxanes. ChemMedChem 2007, 2, 920–942. [Google Scholar] [CrossRef]
- Clarke, S.J.; Rivory, L.P. Clinical pharmacokinetics of docetaxel. Clin. Pharmacokinet. 1999, 36, 99–114. [Google Scholar] [CrossRef]
- Valero, V.; Jones, S.E.; Von Hoff, D.D.; Booser, D.J.; Mennel, R.G.; Ravdin, P.M.; Holmes, F.A.; Rahman, Z.; Schottstaedt, M.W.; Erban, J.K.; et al. A phase II study of docetaxel in patients with paclitaxel-resistant metastatic breast cancer. J. Clin. Oncol. 1998, 16, 3362–3368. [Google Scholar] [CrossRef]
- Leonelli, F.; La Bella, A.; Migneco, L.M.; Bettolo, R.M. Design, synthesis and applications of hyaluronic acid-paclitaxel bioconjugates. Molecules 2008, 13, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Safavy, A.; Raisch, K.P.; Khazaeli, M.B.; Buchsbaum, D.J.; Bonner, J.A. Paclitaxel derivatives for targeted therapy of cancer: Toward the development of smart taxanes. J. Med. Chem. 1999, 42, 4919–4924. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Nakajima, N.; Matsumura, K.; Hyon, S.H. Water-soluble taxol conjugates with dextran and targets tumor cells by folic acid immobilization. Anticancer Res. 2010, 30, 903–909. [Google Scholar]
- Darmostuk, M.; Rimpelova, S.; Gbelcova, H.; Ruml, T. Current approaches in SELEX: An update to aptamer selection technology. Biotechnol. Adv. 2015, 33, 1141–1161. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Liu, J.; Liang, C.; Wang, M.; Wang, L.; Li, D.; Yao, H.; Zhang, Q.; Wen, J.; et al. A water-soluble nucleolin aptamer-paclitaxel conjugate for tumor-specific targeting in ovarian cancer. Nat. Commun. 2017, 8, 1390. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, S.; Zhi, D.; Zhao, Y.; Cui, S.; Cui, J. Co-delivery of paclitaxel and survivin siRNA with cationic liposome for lung cancer therapy. Colloid Surf. A-Physicochem. Eng. Asp. 2020, 585, 124054. [Google Scholar] [CrossRef]
- Lee, J.; Chae, S.W.; Ma, L.J.; Lim, S.Y.; Alnajjar, S.; Choo, H.Y.P.; Lee, H.J.; Rhie, S.J. Pharmacokinetic alteration of paclitaxel by ferulic acid derivative. Pharmaceutics 2019, 11, 593. [Google Scholar] [CrossRef]
- Kubo, T.; Nogami, N.; Bessho, A.; Morita, A.; Ikeo, S.; Yokoyama, T.; Ishihara, M.; Honda, T.; Fujimoto, N.; Murakami, S.; et al. Phase II trial of carboplatin, nab-paclitaxel and bevacizumab for advanced non-squamous non-small cell lung cancer (CARNAVAL study; TORG1424/OLCSG1402). Ann. Oncol. 2019, 30, ix172–ix173. [Google Scholar] [CrossRef]
- Redondo, A.; Colombo, N.; Dreosti, L.M.; McCormack, M.; Nogeira Rodrigues, A.; Scambia, G.; Roszak, A.; Donica, M.; Ulker, B.; González Martín, A. Primary results from CECILIA, a global single-arm phase II study evaluating bevacizumab (BEV), carboplatin (C) and paclitaxel (P) for advanced cervical cancer (aCC). Ann. Oncol. 2019, 30, v403–v434. [Google Scholar] [CrossRef]
- González Martín, A.; Oza, A.M.; Embleton, A.C.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Bertrand, M.A.; Beale, P.; Cervantes, A.; et al. ICON7 investigators. Exploratory outcome analyses according to stage and/or residual disease in the ICON7 trial of carboplatin and paclitaxel with or without bevacizumab for newly diagnosed ovarian cancer. Gynecol. Oncol. 2019, 152, 53–60. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, E.R.; Ahmed, A.S.; Hassan, I.A.; Ismaiel, A.A.; El-Din, A.A.K. Strain improvement and immobilization technique for enhanced production of the anticancer drug paclitaxel by Aspergillus fumigatus and Alternaria tenuissima. Appl. Microbiol. Biotechnol. 2019, 103, 8923–8935. [Google Scholar] [CrossRef] [PubMed]
- Holton, R.A.; Somoza, C.; Kim, H.B.; Liang, F.; Biediger, R.J.; Boatman, D.; Shindo, M.; Smith, C.C.; Kim, S.; Nadizadeh, H.; et al. First total synthesis of taxol. 1. Functionalization of the B ring. J. Am. Chem. Soc. 1994, 116, 1597–1598. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Yang, Z.; Liu, J.J.; Ueno, H.; Nantermet, P.G.; Guy, R.K.; Claiborne, C.F.; Renaud, J.; Couladouros, E.A.; Paulvannan, K.; et al. Total synthesis of taxol. Nature 1994, 367, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Danishefsky, S.J.; Masters, J.J.; Young, W.B.; Link, J.T.; Snyder, L.B.; Magee, T.V.; Jung, D.K.; Isaacs, R.C.A.; Bornmann, W.G.; Alaimo, C.A.; et al. Total synthesis of baccatin III and taxol. J. Am. Chem. Soc. 1996, 118, 2843–2859. [Google Scholar] [CrossRef]
- Wender, P.A.; Badham, N.F.; Conway, S.P.; Floreancig, P.E.; Glass, T.E.; Gränicher, C.; Houze, J.B.; Jänichen, J.; Lee, D.; Marquess, D.G.; et al. The pinene path to taxanes. 5. Stereocontrolled synthesis of a versatile taxane precursor. J. Am. Chem. Soc. 1997, 119, 2755–2756. [Google Scholar] [CrossRef]
- Morihira, K.; Hara, R.; Kawahara, S.; Nishimori, T.; Nakamura, N.; Kusama, H.; Kuwajima, I. Enantioselective total synthesis of taxol. J. Am. Chem. Soc. 1998, 120, 12980–12981. [Google Scholar] [CrossRef]
- Isamu, S.; Hayato, I.; Hiroki, S.; Masatoshi, H.; Yu-ichirou, T.; Teruaki, M. A new method for the synthesis of baccatin III. Chem. Lett. 1998, 27, 1–2. [Google Scholar]
- Doi, T.; Fuse, S.; Miyamoto, S.; Nakai, K.; Sasuga, D.; Takahashi, T. A formal total synthesis of taxol aided by an automated synthesizer. Chem. Asian J. 2006, 1, 370–383. [Google Scholar] [CrossRef]
- Fukaya, K.; Kodama, K.; Tanaka, Y.; Yamazaki, H.; Sugai, T.; Yamaguchi, Y.; Watanabe, A.; Oishi, T.; Sato, T.; Chida, N. Synthesis of paclitaxel. 2. Construction of the ABCD ring and formal synthesis. Org. Lett. 2015, 17, 2574–2577. [Google Scholar] [CrossRef]
- Hirai, S.; Utsugi, M.; Iwamoto, M.; Nakada, M. Formal total synthesis of (−)-taxol through Pd-catalyzed eight-membered carbocyclic ring formation. Chem. Eur. J. 2015, 21, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.N. Tour de paclitaxel: Biocatalysis for semisynthesis. Annu. Rev. Microbiol. 1998, 52, 361–395. [Google Scholar] [CrossRef] [PubMed]
- Ganem, B.; Franke, R.R. Paclitaxel from primary taxanes: A perspective on creative invention in organozirconium chemistry. J. Org. Chem. 2007, 72, 3981–3987. [Google Scholar] [CrossRef]
- Kumar, P.; Singh, B.; Thakur, V.; Thakur, A.; Thakur, N.; Pandey, D.; Chand, D. Hyper-production of taxol from Aspergillus fumigatus, an endophytic fungus isolated from Taxus sp. of the Northern Himalayan region. Biotechnol. Rep. 2019, 24, e00395. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.S.M.; Raizada, M.N. Darkness: A crucial factor in fungal taxol production. Front. Microbiol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar] [PubMed]
- Churchill, C.D.M.; Klobukowski, M.; Tuszynski, J.A. The unique binding mode of laulimalide to two tubulin protofilaments. Chem. Biol. Drug Des. 2015, 86, 190–199. [Google Scholar] [CrossRef]
- Castro-Alvarez, A.; Pineda, O.; Vilarrasa, J. Further insight into the interactions of the cytotoxic macrolides laulimalide and peloruside A with their common binding site. ACS Omega 2018, 3, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Pryor, D.E.; O’Brate, A.; Bilcer, G.; Díaz, J.F.; Wang, Y.; Wang, Y.; Kabaki, M.; Jung, M.K.; Andreu, J.M.; Ghosh, A.K.; et al. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 2002, 41, 9109–9115. [Google Scholar] [CrossRef]
- Clark, E.A.; Hills, P.M.; Davidson, B.S.; Wender, P.A.; Mooberry, S.L. Laulimalide and synthetic laulimalide analogues are synergistic with paclitaxel and 2-methoxyestradiol. Mol. Pharm. 2006, 3, 457–467. [Google Scholar] [CrossRef]
- Liu, J.; Towle, M.J.; Cheng, H.; Saxton, P.; Reardon, C.; Wu, J.; Murphy, E.A.; Kuznetsov, G.; Johannes, C.W.; Tremblay, M.R.; et al. In vitro and in vivo anticancer activities of synthetic (−)-laulimalide, a marine natural product microtubule stabilizing agent. Anticancer Res. 2007, 27, 1509–1518. [Google Scholar] [PubMed]
- Paterson, I.; Menche, D.; Håkansson, A.E.; Longstaff, A.; Wong, D.; Barasoain, I.; Buey, R.M.; Díaz, J.F. Design, synthesis and biological evaluation of novel, simplified analogues of laulimalide: Modification of the side chain. Bioorg. Med. Chem. Lett. 2005, 15, 2243–2247. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Bergmann, H.; Menche, D.; Berkessel, A. Synthesis of novel 11-desmethyl analogues of laulimalide by Nozaki-Kishi coupling. Org. Lett. 2004, 6, 1293–1295. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L.; Randall-Hlubek, D.A.; Leal, R.M.; Hegde, S.G.; Hubbard, R.D.; Zhang, L.; Wender, P.A. Microtubule-stabilizing agents based on designed laulimalide analogues. Proc. Natl. Acad. Sci. USA 2004, 101, 8803–8808. [Google Scholar] [CrossRef]
- Wender, P.A.; Hegde, S.G.; Hubbard, R.D.; Zhang, L.; Mooberry, S.L. Synthesis and biological evaluation of (−)-laulimalide analogues. Org. Lett. 2003, 5, 3507–3509. [Google Scholar] [CrossRef]
- Gallagher, B.M., Jr.; Fang, F.G.; Johannes, C.W.; Pesant, M.; Tremblay, M.R.; Zhao, H.; Akasaka, K.; Li, X.; Liu, J.; Littlefield, B.A. Synthesis and biological evaluation of (−)-laulimalide analogues. Bioorg. Med. Chem. Lett. 2004, 14, 575–579. [Google Scholar] [CrossRef]
- Ahmed, A.; Hoegenauer, E.K.; Enev, V.S.; Hanbauer, M.; Kaehlig, H.; Öhler, E.; Mulzer, J. Total synthesis of the microtubule stabilizing antitumor agent laulimalide and some nonnatural analogues: The power of sharpless’ asymmetric epoxidation. J. Org. Chem. 2003, 68, 3026–3042. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Stanton, M.G.; Allwein, S.P. Asymmetric total synthesis of (−)-laulimalide: Exploiting the asymmetric glycolate alkylation reaction. J. Am. Chem. Soc. 2002, 124, 5958–5959. [Google Scholar] [CrossRef]
- Enev, V.S.; Kaehlig, H.; Mulzer, J. Macrocyclization via allyl transfer: Total synthesis of laulimalide. J. Am. Chem. Soc. 2001, 123, 10764–10765. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Wang, Y. Total synthesis of (−)-laulimalide. J. Am. Chem. Soc. 2000, 122, 11027–11028. [Google Scholar] [CrossRef]
- Mulzer, J.; Hanbauer, M. Total synthesis of the antitumor agent (−)-laulimalide. Tetrahedron Lett. 2002, 43, 3381–3383. [Google Scholar] [CrossRef]
- Nelson, S.G.; Cheung, W.S.; Kassick, A.J.; Hilfiker, M.A. A de novo enantioselective total synthesis of (−)-laulimalide. J. Am. Chem. Soc. 2002, 124, 13654–13655. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; De Savi, C.; Tudge, M. Total synthesis of the microtubule-stabilizing agent (−)-laulimalide. Org. Lett. 2001, 3, 3149–3152. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Seganish, W.M.; Chung, C.K.; Amans, D. Total synthesis of laulimalide: Synthesis of the Northern and Southern fragments. Chem. Eur. J. 2012, 18, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Uenishi, J.; Ohmi, M. Total synthesis of (−)-laulimalide: Pd-catalyzed stereospecific ring construction of the substituted 3,6-dihydro [2H] pyran units. Angew. Chem.-Int. Ed. 2005, 44, 2756–2760. [Google Scholar] [CrossRef]
- Wender, P.A.; Hegde, S.G.; Hubbard, R.D.; Zhang, L. Total synthesis of (−)-laulimalide. J. Am. Chem. Soc. 2002, 124, 4956–4957. [Google Scholar] [CrossRef]
- Williams, D.R.; Mi, L.; Mullins, R.J.; Stites, R.E. Synthesis of (−)-laulimalide: An agent for microtubule stabilization. Tetrahedron Lett. 2002, 43, 4841–4844. [Google Scholar] [CrossRef]
- Bennett, M.J.; Chan, G.K.; Rattner, J.B.; Schriemer, D.C. Low-dose laulimalide represents a novel molecular probe for investigating microtubule organization. Cell Cycle 2012, 11, 3045–3054. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Fernando Díaz, J.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem.-Int. Ed. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- West, L.M.; Northcote, P.T.; Battershill, C.N. Peloruside A: A potent cytotoxic macrolide isolated from the New Zealand marine sponge Mycale sp. J. Org. Chem. 2000, 65, 445–449. [Google Scholar] [CrossRef]
- Hood, K.A.; West, L.M.; Rouwé, B.; Northcote, P.T.; Berridge, M.V.; Wakefield, J.; Miller, J.H. Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res. 2002, 62, 3356–3360. [Google Scholar] [PubMed]
- Meyer, C.J.; Krauth, M.; Wick, M.J.; Shay, J.W.; Gellert, G.; De Brabander, J.K.; Northcote, P.T.; Miller, J.H. Peloruside A inhibits growth of human lung and breast tumor xenografts in an athymic nu/nu mouse model. Mol. Cancer Ther. 2015, 14, 1816–1823. [Google Scholar] [CrossRef]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Gilfillan, C.; Templeton, N.; Paterson, I.; Northcote, P.T.; Miller, J.H. Induction of accelerated senescence by the microtubule-stabilizing agent peloruside A. Investig. New Drugs 2017, 35, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Singh, A.J.; Northcote, P.T.; Miller, J.H. Inhibition of human vascular endothelial cell migration and capillary-like tube formation by the microtubule-stabilizing agent peloruside A. Investig. New Drugs 2015, 33, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Das, V.; Miller, J.H. Microtubule stabilization by peloruside A and paclitaxel rescues degenerating neurons from okadaic acid-induced tau phosphorylation. Eur. J. Neurosci. 2012, 35, 1705–1717. [Google Scholar] [CrossRef]
- O’Sullivan, D.; Miller, J.H.; Northcote, P.T.; La Flamme, A.C. Microtubule-stabilizing agents delay the onset of EAE through inhibition of migration. Immunol. Cell Biol. 2013, 91, 583–592. [Google Scholar] [CrossRef]
- Crume, K.P.; O’Sullivan, D.; Miller, J.H.; Northcote, P.T.; La Flamme, A.C. Delaying the onset of experimental autoimmune encephalomyelitis with the microtubule-stabilizing compounds, paclitaxel and Peloruside A. J. Leukoc. Biol. 2009, 86, 949–958. [Google Scholar] [CrossRef]
- Singh, A.J.; Razzak, M.; Teesdale-Spittle, P.; Gaitanos, T.N.; Wilmes, A.; Paterson, I.; Goodman, J.M.; Miller, J.H.; Northcote, P.T. Structure-activity studies of the pelorusides: New congeners and semi-synthetic analogues. Org. Biomol. Chem. 2011, 9, 4456–4466. [Google Scholar] [CrossRef]
- Chany, A.C.; Legros, F.; Haroun, H.; Kundu, U.K.; Biletskyi, B.; Torlak, S.; Mathé-Allainmat, M.; Lebreton, J.; Macé, A.; Carboni, B.; et al. Function-oriented synthesis toward peloruside A analogues. Org. Lett. 2019, 21, 2988–2992. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Xu, X.; Kim, J.H.; Xu, C.X. Enantioselective total synthesis of peloruside A: A potent microtubule stabilizer. Org. Lett. 2008, 10, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Jeon, J.; Kopel, L.C.; Ryba, T.D.; Tennakoon, M.A.; Wang, Y. Total synthesis of peloruside A through kinetic lactonization and relay ring-closing metathesis cyclization reactions. Angew. Chem. Int. Ed. 2010, 49, 6151–6155. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Taylor, R.E. Total synthesis of (+)-peloruside A. Org. Lett. 2005, 7, 1303–1305. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Wu, Y.; De Brabander, J.K. Total synthesis and absolute configuration of the novel microtubule-stabilizing agent peloruside A. Angew. Chem. Int. Ed. 2003, 42, 1648–1652. [Google Scholar] [CrossRef]
- McGowan, M.A.; Stevenson, C.P.; Schiffler, M.A.; Jacobsen, E.N. An enantioselective total synthesis of (+)-peloruside A. Angew. Chem. Int. Ed. 2010, 49, 6147–6150. [Google Scholar] [CrossRef]
- Evans, D.A.; Welch, D.S.; Speed, A.W.H.; Moniz, G.A.; Reichelt, A.; Ho, S. An aldol-based synthesis of (+)-peloruside A, a potent microtubule stabilizing agent. J. Am. Chem. Soc. 2009, 131, 3840–3841. [Google Scholar] [CrossRef]
- Brackovic, A.; Harvey, J.E. Synthetic, semisynthetic and natural analogues of peloruside A. Chem. Commun. 2015, 51, 4750–4765. [Google Scholar] [CrossRef]
- Ranade, A.R.; Higgins, L.A.; Markowski, T.W.; Glaser, N.; Kashin, D.; Bai, R.; Hong, K.H.; Hamel, E.; Höfle, G.; Georg, G.I. Characterizing the epothilone binding site on β-tubulin by photoaffinity labeling: Identification of β-tubulin peptides TARGSQQY and TSRGSQQY as targets of an epothilone photoprobe for polymerized tubulin. J. Med. Chem. 2016, 59, 3499–3514. [Google Scholar] [CrossRef]
- Hardt, I.H.; Steinmetz, H.; Gerth, K.; Sasse, F.; Reichenbach, H.; Höfle, G. New natural epothilones from Sorangium cellulosum, strains So ce90/B2 and So ce90/D13, Isolation, structure elucidation, and SAR studies. J. Nat. Prod. 2001, 64, 847–856. [Google Scholar] [CrossRef]
- Gerth, K.; Steinmetz, H.; Höfle, G.; Reichenbach, H. Studies on the biosynthesis of epothilones: The PKS and epothilone C/D monooxygenase. J. Antibiot. (Tokyo) 2001, 54, 144–148. [Google Scholar] [CrossRef][Green Version]
- Höfle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H. Epothilone A and B—novel 16-membered macrolides with cytotoxic activity: Isolation, crystal structure, and conformation in solution. Angew. Chem. Int. Ed. 1996, 35, 1567–1569. [Google Scholar] [CrossRef]
- Kowalski, R.J.; Giannakakou, P.; Hamel, E. Activities of the microtubule-stabilizing agents epothilones A and B with purified tubulin and in cells resistant to paclitaxel (Taxol(R)). J. Biol. Chem. 1997, 272, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, A.; Marczak, A. Therapeutic potential of patupilone in epithelial ovarian cancer and future directions. Life Sci. 2018, 205, 38–44. [Google Scholar] [CrossRef]
- Shen, S.; Kepp, O.; Martins, I.; Vitale, I.; Souquère, S.; Castedo, M.; Pierron, G.; Kroemer, G. Defective autophagy associated with LC3 puncta in epothilone-resistant cancer cells. Cell Cycle 2010, 9, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, A.; Gajek, A.; Marczak, A. Suppression of autophagy enhances preferential toxicity of epothilone A and epothilone B in ovarian cancer cells. Phytomedicine 2019, 61, 152847. [Google Scholar] [CrossRef]
- Luu, T.; Kim, K.P.; Blanchard, S.; Anyang, B.; Hurria, A.; Yang, L.; Beumer, J.H.; Somlo, G.; Yen, Y. Phase IB trial of ixabepilone and vorinostat in metastatic breast cancer. Breast Cancer Res. Treat. 2018, 167, 469–478. [Google Scholar] [CrossRef]
- Rugo, H.S.; Roche, H.; Thomas, E.; Chung, H.C.; Lerzo, G.L.; Vasyutin, I.; Patel, A.; Vahdat, L. Efficacy and safety of ixabepilone and capecitabine in patients with advanced triple-negative breast cancer: A pooled analysis from two large phase III, randomized clinical trials. Clin. Breast Cancer 2018, 18, 489–497. [Google Scholar] [CrossRef]
- Osborne, C.; Challagalla, J.D.; Eisenbeis, C.F.; Holmes, F.A.; Neubauer, M.A.; Koutrelakos, N.W.; Taboada, C.A.; Vukelja, S.J.; Wilks, S.T.; Allison, M.A.; et al. Ixabepilone and carboplatin for hormone receptor positive/HER2-neu negative and triple negative metastatic breast cancer. Clin. Breast Cancer 2018, 18, E89–E95. [Google Scholar] [CrossRef]
- Peethambaram, P.P.; Hartmann, L.C.; Jonker, D.J.; de Jonge, M.; Plummer, E.R.; Martin, L.; Konner, J.; Marshall, J.; Goss, G.D.; Teslenko, V.; et al. A phase I pharmacokinetic and safety analysis of epothilone folate (BMS-753493), a folate receptor targeted chemotherapeutic agent in humans with advanced solid tumors. Investig. New Drugs 2015, 33, 321–331. [Google Scholar] [CrossRef]
- Gaugaz, F.Z.; Chicca, A.; Redondo-Horcajo, M.; Barasoain, I.; Fernando Díaz, J.; Altmann, K.H. Synthesis, microtubule-binding affinity, and antiproliferative activity of new epothilone analogs and of an EGFR-targeted epothilone-peptide conjugate. Int. J. Mol. Sci. 2019, 20, 1113. [Google Scholar] [CrossRef]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.M.L.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Ruschel, J.; Hellal, F.; Flynn, K.C.; Dupraz, S.; Elliott, D.A.; Tedeschi, A.; Bates, M.; Sliwinski, C.; Brook, G.; Dobrindt, K.; et al. Axonal regeneration. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science 2015, 348, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Liu, T.; Zhu, M.; Zhang, W.; Huang, Z.; Li, S.; Li, H.; Kong, Y.; Chen, Y. An easy and efficient strategy for the enhancement of epothilone production mediated by TALE-TF and CRISPR/dcas9 systems in Sorangium cellulosum. Front. Bioeng. Biotechnol. 2019, 7, 334. [Google Scholar] [CrossRef] [PubMed]
- Julien, B.; Shah, S. Heterologous expression of epothilone biosynthetic genes in Myxococcus xanthus. Antimicrob. Agents Chemother. 2002, 46, 2772–2778. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Shah, S.; Chung, L.; Carney, J.; Katz, L.; Khosla, C.; Julien, B. Cloning and heterologous expression of the epothilone gene cluster. Science 2000, 287, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Mutka, S.C.; Carney, J.R.; Liu, Y.; Kennedy, J. Heterologous production of epothilone C and D in Escherichia coli. Biochemistry 2006, 45, 1321–1330. [Google Scholar] [CrossRef]
- Wenzel, S.C.; Müller, R. Recent developments towards the heterologous expression of complex bacterial natural product biosynthetic pathways. Curr. Opin. Biotechnol. 2005, 16, 594–606. [Google Scholar] [CrossRef]
- Lau, J.; Frykman, S.; Regentin, R.; Ou, S.; Tsuruta, H.; Licari, P. Optimizing the heterologous production of epothilone D in Myxococcus xanthus. Biotechnol. Bioeng. 2002, 78, 280–288. [Google Scholar] [CrossRef]
- Ye, W.; Zhang, W.; Chen, Y.; Li, H.; Li, S.; Pan, Q.; Tan, G.; Liu, T. A new approach for improving epothilone B yield in Sorangium cellulosum by the introduction of VGB epoF genes. J. Ind. Microbiol. Biotechnol. 2016, 43, 641–650. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Himes, R.H. Interactions of the catharanthus (Vinca) alkaloids with tubulin and microtubules. Pharmacol. Ther. 1991, 51, 257–267. [Google Scholar] [CrossRef]
- Lu, K.; Yap, H.Y.; Loo, T.L. Clinical pharmacokinetics of vinblastine by continuous intravenous infusion. Cancer Res. 1983, 43, 1405–1408. [Google Scholar] [PubMed]
- Deyell, R.J.; Wu, B.; Rassekh, S.R.; Tu, D.; Samson, Y.; Fleming, A.; Bouffet, E.; Sun, X.; Powers, J.; Seymour, L.; et al. Phase I study of vinblastine and temsirolimus in pediatric patients with recurrent or refractory solid tumors: Canadian cancer trials group study IND.218. Pediatr. Blood Cancer 2019, 66, e27540. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Brière, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Poeschel, V.; Held, G.; Ziepert, M.; Witzens-Harig, M.; Holte, H.; Thurner, L.; Borchmann, P.; Viardot, A.; Soekler, M.; Keller, U.; et al. Four versus six cycles of CHOP chemotherapy in combination with six applications of rituximab in patients with aggressive B-cell lymphoma with favourable prognosis (FLYER): A randomised, phase 3, non-inferiority trial. Lancet 2020, 394, 2271–2281. [Google Scholar] [CrossRef]
- Meyer, R.M.; Gospodarowicz, M.K.; Connors, J.M.; Pearcey, R.G.; Wells, W.A.; Winter, J.N.; Horning, S.J.; Dar, A.R.; Shustik, C.; Stewart, D.A.; et al. ABVD Alone versus Radiation-Based Therapy in Limited-Stage Hodgkin’s Lymphoma. N. Engl. J. Med. 2012, 366, 399–408. [Google Scholar] [CrossRef]
- Waters, E.; Dingle, B.; Rodrigues, G.; Vincent, M.; Ash, R.; Dar, R.; Inculet, R.; Kocha, W.; Malthaner, R.; Sanatani, M.; et al. Analysis of a novel protocol of combined induction chemotherapy and concurrent chemoradiation in unresected non-small-cell lung cancer: A ten-year experience with vinblastine, cisplatin, and radiation therapy. Clin. Lung Cancer 2010, 11, 243–250. [Google Scholar] [CrossRef]
- Zhigaltsev, I.V.; Maurer, N.; Akhong, Q.F.; Leone, R.; Leng, E.; Wang, J.; Semple, S.C.; Cullis, P.R. Liposome-encapsulated vincristine, vinblastine and vinorelbine: A comparative study of drug loading and retention. J. Control. Release 2005, 104, 103–111. [Google Scholar] [CrossRef]
- Ling, G.; Zhang, P.; Zhang, W.; Sun, J.; Meng, X.; Qin, Y.; Deng, Y.; He, Z. Development of novel self-assembled DS-PLGA hybrid nanoparticles for improving oral bioavailability of vincristine sulfate by P-gp inhibition. J. Control. Release 2010, 148, 241–248. [Google Scholar] [CrossRef]
- Leggans, E.K.; Duncan, K.K.; Barker, T.J.; Schleicher, K.D.; Boger, D.L. A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedented tubulin binding steric tolerance: C20’ urea derivatives. J. Med. Chem. 2013, 56, 628–639. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saba, N.; Seal, A. Identification of a less toxic vinca alkaloid derivative for use as a chemotherapeutic agent, based on in silico structural insights and metabolic interactions with CYP3A4 and CYP3A5. J. Mol. Model 2018, 24, 82. [Google Scholar] [CrossRef] [PubMed]
- Va, P.; Campbell, E.L.; Robertson, W.M.; Boger, D.L. Total synthesis and evaluation of a key series of C5-substituted vinblastine derivatives. J. Am. Chem. Soc. 2010, 132, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Bánóczi, Z.; Gorka-Kereskényi, A.; Reményi, J.; Orbán, E.; Hazai, L.; Tökési, N.; Oláh, J.; Ovádi, J.; Béni, Z.; Háda, V.; et al. Synthesis and in vitro antitumor effect of vinblastine derivative-oligoarginine conjugates. Bioconjugate Chem. 2010, 21, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; van Soest, R.; Matainaho, L.; Roberge, M.; Andersen, R.J. Ceratamines A and B, antimitotic heterocyclic alkaloids isolated from the marine Sponge Pseudoceratina sp. collected in Papua New Guinea. Org. Lett. 2003, 5, 4591–4594. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Pan, X.; Ji, M.; Chen, X.; Liu, Z. Efficient synthesis and cytotoxicity of novel microtubule-stabilizing agent ceratamine A analogues. Tetrahedron 2017, 73, 2159–2171. [Google Scholar] [CrossRef]
- Pan, X.; Tao, L.; Ji, M.; Chen, X.; Liu, Z. Synthesis and cytotoxicity of novel imidazo[4,5-d]azepine compounds derived from marine natural product ceratamine A. Bioorg. Med. Chem. Lett. 2018, 28, 866–868. [Google Scholar] [CrossRef]
- Nodwell, M.; Zimmerman, C.; Roberge, M.; Andersen, R.J. Synthetic analogues of the microtubule-stabilizing sponge alkaloid ceratamine A are more active than the natural product. J. Med. Chem. 2010, 53, 7843–7851. [Google Scholar] [CrossRef]
- Kowalski, R.J.; Giannakakou, P.; Gunasekera, S.P.; Longley, R.E.; Day, B.W.; Hamel, E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (Taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol. Pharmacol. 1997, 52, 613–622. [Google Scholar] [CrossRef]
- Paterson, I.; Florence, G.J. The chemical synthesis of discodermolide. In Tubulin-Binding Agents: Synthetic, Structural and Mechanistic Insights; Carlomagno, T., Ed.; Springer: Berlin, Germany, 2009; pp. 73–119. [Google Scholar]
- Pettit, G.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Bai, R.; Petit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal: Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem.Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Garteiz, D.A.; Madden, T.; Beck, D.E.; Huie, W.R.; McManus, K.T.; Abbruzzese, J.L.; Chen, W.; Newman, R.A. Quantitation of dolastatin-10 using HPLC/electrospray ionization mass spectrometry: Application in a phase I clinical trial. Cancer Chemother. Pharmacol. 1998, 41, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Pitot, H.C.; McElroy, E.A., Jr.; Reid, J.M.; Windebank, A.J.; Sloan, J.A.; Erlichman, C.; Bagniewski, P.G.; Walker, D.L.; Rubin, J.; Goldberg, R.M.; et al. Phase I trial of dolastatin-10 (NSC 376128) in patients with advanced solid tumors. Clin. Cancer Res. 1999, 5, 525–531. [Google Scholar] [PubMed]
- von Mehren, M.; Balcerzak, S.P.; Kraft, A.S.; Edmonson, J.H.; Okuno, S.H.; Davey, M.; McLaughlin, S.; Beard, M.T.; Rogatko, A. Phase II trial of dolastatin-10, a novel anti-tubulin agent, in metastatic soft tissue sarcomas. Sarcoma 2004, 8, 107–111. [Google Scholar] [CrossRef]
- Margolin, K.; Longmate, J.; Synold, T.W.; Gandara, D.R.; Weber, J.; Gonzalez, R.; Johansen, M.J.; Newman, R.; Baratta, T.; Doroshow, J.H. Dolastatin-10 in metastatic melanoma: A phase II and pharmokinetic trial of the california cancer consortium. Investig. New Drugs 2001, 19, 335–340. [Google Scholar] [CrossRef]
- Hoffman, M.A.; Blessing, J.A.; Lentz, S.S. A phase II trial of dolastatin-10 in recurrent platinum-sensitive ovarian carcinoma: A Gynecologic oncology group study. Gynecol. Oncol. 2003, 89, 95–98. [Google Scholar] [CrossRef]
- Kindler, H.L.; Tothy, P.K.; Wolff, R.; McCormack, R.A.; Abbruzzese, J.L.; Mani, S.; Wade-Oliver, K.T.; Vokes, E.E. Phase II trials of dolastatin-10 in advanced pancreaticobiliary cancers. Investig. New Drugs 2005, 23, 489–493. [Google Scholar] [CrossRef]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investig. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef]
- Maderna, A.; Doroski, M.; Subramanyam, C.; Porte, A.; Leverett, C.A.; Vetelino, B.C.; Chen, Z.; Risley, H.; Parris, K.; Pandit, J.; et al. Discovery of cytotoxic dolastatin 10 analogues with N-terminal modifications. J. Med. Chem. 2014, 57, 10527–10543. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- van de Donk, N.W.C.J.; Dhimolea, E. Brentuximab vedotin. MAbs 2012, 4, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.M.; Melchardt, T.; Egle, A.; Magnes, T.; Skrabs, C.; Staber, P.; Simonitsch-Klupp, I.; Panny, M.; Lehner, B.; Greil, R.; et al. Treatment with brentuximab vedotin plus bendamustine in unselected patients with CD30-positive aggressive lymphomas. Eur. J. Haem. 2020, 104, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bian, M.; Jiang, Q.; Zhang, C. Roles of aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 2007, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Magnaghi-Jaulin, L.; Eot-Houllier, G.; Gallaud, E.; Giet, R. Aurora A protein kinase: To the centrosome and beyond. Biomolecules 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.R.; Gergely, F. Aurora-A: The maker and breaker of spindle poles. J. Cell Sci. 2007, 120, 2987–2996. [Google Scholar] [CrossRef]
- Krenn, V.; Musacchio, A. The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front. Oncol. 2015, 5, 225. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.T.; Tang, C.J.C.; Tang, T.K. Possible role of Aurora-C in meiosis. Front. Oncol. 2015, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kaneko, S.; Yang, L.; Feldman, R.I.; Nicosia, S.V.; Chen, J.; Cheng, J.Q. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J. Biol. Chem. 2004, 279, 52175–52182. [Google Scholar] [CrossRef]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef]
- Sells, T.B.; Chau, R.; Ecsedy, J.A.; Gershman, R.E.; Hoar, K.; Huck, J.; Janowick, D.A.; Kadambi, V.J.; LeRoy, P.J.; Stirling, M.; et al. MLN8054 and alisertib (MLN8237): Discovery of selective oral aurora A inhibitors. ACS Med. Chem. Lett. 2015, 6, 630–634. [Google Scholar] [CrossRef]
- Li, J.P.; Yang, Y.X.; Liu, Q.L.; Pan, S.T.; He, Z.X.; Zhang, X.; Yang, T.; Chen, X.W.; Wang, D.; Qiu, J.X.; et al. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G(2)/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des. Devel. Ther. 2015, 9, 1627–1652. [Google Scholar] [PubMed]
- Fu, Y.; Zhang, Y.; Gao, M.; Quan, L.; Gui, R.; Liu, J. Alisertib induces apoptosis and autophagy through targeting the AKT/mTOR/AMPK/p38 pathway in leukemic cells. Mol. Med. Rep. 2016, 14, 394–398. [Google Scholar] [CrossRef][Green Version]
- Ren, B.J.; Zhou, Z.W.; Zhu, D.J.; Ju, Y.L.; Wu, J.H.; Ouyang, M.Z.; Chen, X.W.; Zhou, S.F. Alisertib induces cell cycle arrest, apoptosis, autophagy and suppresses EMT in HT29 and Caco-2 cells. Int. J. Mol. Sci. 2016, 17, 41. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Zhou, Z.W.; Xia, H.C.; Wang, X.Y.; Yang, Y.X.; He, Z.X.; Sun, T.; Zhou, S.F. Alisertib induces G(2)/M arrest, apoptosis, and autophagy via PI3K/Akt/mTOR- and p38 MAPK-mediated pathways in human glioblastoma cells. Am. J. Transl. Res. 2017, 9, 845–873. [Google Scholar] [PubMed]
- Shang, Y.Y.; Yao, M.; Zhou, Z.W.; Cui, J.; Xia, L.; Hu, R.Y.; Yu, Y.Y.; Gao, Q.; Yang, B.; Liu, Y.X.; et al. Alisertib promotes apoptosis and autophagy in melanoma through p38 MAPK-mediated aurora a signaling. Oncotarget 2017, 8, 107076–107088. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yu, X.; Zhou, Z.W.; Zhou, C.; Chen, X.W.; Zhou, S.F. Inhibition of aurora A kinase by alisertib induces autophagy and cell cycle arrest and increases chemosensitivity in human hepatocellular carcinoma HepG2 cells. Curr. Cancer Drug Targets 2017, 17, 386–401. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef]
- Niu, H.; Manfredi, M.; Ecsedy, J.A. Scientific rationale supporting the clinical development strategy for the investigational Aurora A kinase inhibitor alisertib in cancer. Front. Oncol. 2015, 5, 189. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J.; Huang, Y.; Gao, R.; Tang, B.; Li, S.; He, J.; Li, H. Domain-specific interactions between MLN8237 and human serum albumin estimated by STD and WaterLOGSY NMR, ITC, spectroscopic, and docking techniques. Sci. Rep. 2017, 7, 45514. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Özcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A.; et al. Randomized phase III study of alisertib or investigator’s choice (selected single agent) in patients with relapsed or refractory peripheral T-cell lymphoma. J. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef]
- Shah, H.A.; Fischer, J.H.; Venepalli, N.K.; Danciu, O.C.; Christian, S.; Russell, M.J.; Liu, L.C.; Zacny, J.P.; Dudek, A.Z. Phase I study of aurora A kinase inhibitor alisertib (MLN8237) in combination with selective VEGFR inhibitor pazopanib for therapy of advanced solid tumors. Am. J. Clin. Oncol. 2019, 42, 413–420. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Mosse, Y.P.; Fox, E.; Kudgus, R.A.; Reid, J.M.; McGovern, R.; Groshen, S.; Bagatell, R.; Maris, J.M.; Twist, C.J.; et al. Phase II trial of alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Clin. Cancer Res. 2018, 24, 6142–6149. [Google Scholar] [CrossRef] [PubMed]
- Currier, M.A.; Sprague, L.; Rizvi, T.A.; Nartker, B.; Chen, C.Y.; Wang, P.Y.; Hutzen, B.J.; Franczek, M.R.; Patel, A.V.; Chaney, K.E.; et al. Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget 2017, 8, 17412–17427. [Google Scholar] [CrossRef] [PubMed]
- Iankov, I.D.; Kurokawa, C.B.; D’Assoro, A.B.; Ingle, J.N.; Domingo-Musibay, E.; Allen, C.; Crosby, C.M.; Nair, A.A.; Liu, M.C.; Aderca, I.; et al. Inhibition of the aurora A kinase augments the anti-tumor efficacy of oncolytic measles virotherapy. Cancer Gene Ther. 2015, 22, 438–444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mortlock, A.A.; Foote, K.M.; Heron, N.M.; Jung, F.H.; Pasquet, G.; Lohmann, J.J.M.; Warin, N.; Renaud, F.; De Savi, C.; Roberts, N.J.; et al. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 2007, 50, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.W.; Odedra, R.; Heaton, S.P.; Wedge, S.R.; Keen, N.J.; Crafter, C.; Foster, J.R.; Brady, M.C.; Bigley, A.; Brown, E.; et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res. 2007, 13, 3682–3688. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.; Mesbahi, Y.; Ghanizadeh-Vesali, S.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Reactive oxygen species generation and increase in mitochondrial copy number: New insight into the potential mechanism of cytotoxicity induced by aurora kinase inhibitor, AZD1152-HQPA. Anticancer Drugs 2017, 28, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.; Mesbahi, Y.; Boustanipour, E.; Sadr, Z.; Ghaffari, S.H. The potential contribution of microRNAs in anti-cancer effects of aurora kinase inhibitor (AZD1152-HQPA). J. Mol. Neurosci. 2018, 65, 444–455. [Google Scholar] [CrossRef]
- Ashton, S.; Song, Y.S.; Nolan, J.; Cadogan, E.; Murray, J.; Odedra, R.; Foster, J.; Hall, P.A.; Low, S.; Taylor, P.; et al. Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo. Sci. Transl. Med. 2016, 8, 325ra17. [Google Scholar] [CrossRef]
- Palmisiano, N.D.; Kasner, M.T. Polo-like kinase and its inhibitors: Ready for the match to start? Am. J. Hematol. 2015, 90, 1071–1076. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Zhou, Z.; Wang, W.H.; Deng, A.; Andrisani, O.; Liu, X. Plk1-mediated phosphorylation of topors regulates p53 stability. J. Biol. Chem. 2009, 284, 18588–18592. [Google Scholar] [CrossRef] [PubMed]
- Goroshchuk, O.; Kolosenko, I.; Vidarsdottir, L.; Azimi, A.; Palm-Apergi, C. Polo-like kinases and acute leukemia. Oncogene 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- López-Sánchez, I.; Sanz-García, M.; Lazo, P.A. Plk3 interacts with and specifically phosphorylates VRK1 in Ser(342), a downstream target in a pathway that induces Golgi fragmentation. Mol. Cell. Biol. 2009, 29, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Ottmann, O.G.; Müller-Tidow, C.; Krämer, A.; Schlenk, R.F.; Lübbert, M.; Bug, G.; Krug, U.; Bochtler, T.; Voss, F.; Taube, T.; et al. Phase I dose-escalation trial investigating volasertib as monotherapy or in combination with cytarabine in patients with relapsed/refractory acute myeloid leukaemia. Br. J. Haematol. 2019, 184, 1018–1021. [Google Scholar] [CrossRef]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, A Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Deben, C.; de Pauw, I.; Lambrechts, H.; Hermans, C.; Deschoolmeester, V.; Jacobs, J.; Specenier, P.; Pauwels, P.; Vermorken, J.B.; et al. In vitro study of the Polo-like kinase 1 inhibitor volasertib in non-small-cell lung cancer reveals a role for the tumor suppressor p53. Mol. Oncol. 2019, 13, 1196–1213. [Google Scholar] [CrossRef]
- Solans, B.P.; Fleury, A.; Freiwald, M.; Fritsch, H.; Haug, K.; Trocóniz, I.F. Population pharmacokinetics of volasertib administered in patients with acute myeloid leukaemia as a single agent or in combination with cytarabine. Clin. Pharm. 2018, 57, 379–392. [Google Scholar] [CrossRef]
- Gumireddy, K.; Reddy, M.V.R.; Cosenza, S.C.; Boominathan, R.; Baker, S.J.; Papathi, N.; Jiang, J.; Holland, J.; Reddy, E.P. 1ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 2005, 7, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krssák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of Polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.W.; Messersmith, W.A.; Dy, G.K.; Weekes, C.D.; Whitworth, A.; Ren, C.; Maniar, M.; Wilhelm, F.; Eckhardt, S.G.; Adjei, A.A.; et al. Phase I study of rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin. Cancer Res. 2012, 18, 2048–2055. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Park, I.W.; Allen, H.; Zhang, X.; Reddy, M.V.R.; Boominathan, R.; Reddy, E.P.; Groopman, J.E. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene 2009, 28, 1518–1528. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer. 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Ritt, D.A.; Blanco, M.A.; Bindu, L.; Durrant, D.E.; Zhou, M.; Specht, S.I.; Stephen, A.G.; Holderfield, M.; Morrison, D.K. Inhibition of Ras/Raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol. Cell 2016, 64, 875–887. [Google Scholar] [CrossRef]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol. Cell 2017, 68, 210–223. [Google Scholar] [CrossRef]
- Baker, S.J.; Cosenza, S.C.; Athuluri-Divakar, S.; Reddy, M.V.R.; Vasquez-Del Carpio, R.; Jain, R.; Aggarwal, A.K.; Reddy, E.P. Mechanism of action of rigosertib does not involve tubulin binding. bioRxiv 2019. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.; Platzbecker, U.; et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): A randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 496–508. [Google Scholar] [CrossRef]
- Prasad, A.; Khudaynazar, N.; Tantravahi, R.V.; Gillum, A.M.; Hoffman, B.S. ON 01910.Na (rigosertib) inhibits PI3K/Akt pathway and activates oxidative stress signals in head and neck cancer cell lines. Oncotarget 2016, 7, 79388–79400. [Google Scholar] [CrossRef]
- Anderson, R.T.; Keysar, S.B.; Bowles, D.B.; Glogowska, M.J.; Astling, D.P.; Morton, J.P.; Le, P.; Umpierrez, A.; Eagles-Soukup, J.; Gan, G.N.; et al. The dual pathway inhibitor rigosertib is effective in direct-patient tumor xenografts of head and neck squamous cell carcinomas. Mol. Cancer Ther. 2013, 12, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.; Lin, A.W.; Safer, D.; Hart, C.L.; Naber, N.; Carragher, B.O.; Cain, S.M.; Pechatnikova, E.; Wilson-Kubalek, E.M.; Whittaker, M.; et al. A structural change in the kinesin motor protein that drives motility. Nature 1999, 402, 778–784. [Google Scholar] [CrossRef]
- Hepperla, A.J.; Willey, P.T.; Coombes, C.E.; Schuster, B.M.; Gerami-Nejad, M.; McClellan, M.; Mukherjee, S.; Fox, J.; Winey, M.; Odde, D.J.; et al. Minus-end-directed kinesin-14 motors align antiparallel microtubules to control metaphase spindle length. Dev. Cell 2014, 31, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hancock, W.O. Kinesin-5 is a microtubule polymerase. Nat. Commun. 2015, 6, 8160. [Google Scholar] [CrossRef] [PubMed]
- Trofimova, D.; Paydar, M.; Zara, A.; Talje, L.; Kwok, B.H.; Allingham, J.S. Ternary complex of Kif2A-bound tandem tubulin heterodimers represents a kinesin-13-mediated microtubule depolymerization reaction intermediate. Nat. Commun. 2018, 9, 2628. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef]
- Huszar, D.; Theoclitou, M.E.; Skolnik, J.; Herbst, R. Kinesin motor proteins as targets for cancer therapy. Cancer Metastasis Rev. 2009, 28, 197–208. [Google Scholar] [CrossRef]
- Mayer, T.U.; Kapoor, T.M.; Haggarty, S.J.; King, R.W.; Schreiber, S.L.; Mitchison, T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999, 286, 971–974. [Google Scholar] [CrossRef]
- Abnous, K.; Barati, B.; Mehri, S.; Reza, M.; Farimani, M.R.M.; Alibolandi, M.; Mohammadpour, F.; Ghandadi, M.; Hadizadeh, F. Synthesis and molecular modeling of six novel monastrol analogues: Evaluation of cytotoxicity and kinesin inhibitory activity against HeLa cell line. DARU 2013, 21, 70. [Google Scholar] [CrossRef]
- Kaan, H.Y.K.; Ulaganathan, V.; Rath, O.; Prokopcová, H.; Dallinger, D.; Kappe, C.O.; Kozielski, F. Structural basis for inhibition of Eg5 by dihydropyrimidines: Stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J. Med. Chem. 2010, 53, 5676–5683. [Google Scholar] [CrossRef]
- De Oliveira, F.S.; De Oliveira, P.M.; Farias, L.M.; Brinkerhoff, R.C.; Sobrinho, R.C.M.A.; Treptow, T.M.; D’Oca, C.R.M.; Marinho, M.A.G.; Hort, M.A.; Horn, A.P.; et al. Synthesis and antitumoral activity of novel analogues monastrol-fatty acids against glioma cells. Medchemcomm 2018, 9, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Bélanger, K.; Rao, S.C.; Petrella, T.M.; Tozer, R.G.; Wood, L.; Savage, K.J.; Eisenhauer, E.A.; Synold, T.W.; Wainman, N.; et al. A phase II study of ispinesib (SB-715992) in patients with metastatic or recurrent malignant melanoma: A National cancer institute of Canada clinical trials group trial. Investig. New Drugs 2008, 26, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.A.; Siu, L.L.; Chen, E.X.; Hotte, S.J.; Chia, S.; Schwarz, J.K.; Pond, G.R.; Johnson, C.; Colevas, A.D.; Synold, T.W.; et al. Phase II study of ispinesib in recurrent or metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 2008, 26, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Goldman, B.; Synold, T.W.; Ryan, C.W.; Vasist, L.S.; Van Veldhuizen, P.J., Jr.; Dakhil, S.R.; Lara, P.N., Jr.; Drelichman, A.; Hussain, M.H.A.; et al. Southwest oncology group phase II study of ispinesib in androgen-independent prostate cancer previously treated with taxanes. Clin. Genitourin. Cancer 2008, 6, 103–109. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Padmanabhan, S.; Stock, W.; Tallman, M.S.; Curt, G.A.; Li, J.; Osmukhina, A.; Wu, K.; Huszar, D.; Borthukar, G.; et al. Phase I/II multicenter study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of AZD4877 in patients with refractory acute myeloid leukemia. Investig. New Drugs 2012, 30, 1107–1115. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Goncalves, P.H.; Casetta, L.; Carter, J.A.; Litwiler, K.; Roseberry, D.; Rush, S.; Schreiber, J.; Simmons, H.M.; Ptaszynski, M.; et al. First-in-human phase 1 study of filanesib (ARRY-520), a kinesin spindle protein inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 440–449. [Google Scholar] [CrossRef]
- Holen, K.D.; Belani, C.P.; Wilding, G.; Ramalingam, S.; Volkman, J.L.; Ramanathan, R.K.; Vasist, L.S.; Bowen, C.J.; Hodge, J.P.; Dar, M.M.; et al. A first in human study of SB-743921, a kinesin spindle protein inhibitor, to determine pharmacokinetics, biologic effects and establish a recommended phase II dose. Cancer Chemother. Pharmacol. 2011, 67, 447–454. [Google Scholar] [CrossRef]
- Wakui, H.; Yamamoto, N.; Kitazono, S.; Mizugaki, H.; Nakamichi, S.; Fujiwara, Y.; Nokihara, H.; Yamada, Y.; Suzuki, K.; Kanda, H.; et al. A phase 1 and dose-finding study of LY2523355 (litronesib), an Eg5 inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 15–23. [Google Scholar] [CrossRef]
- Hollebecque, A.; Deutsch, E.; Massard, C.; Gomez-Roca, C.; Bahleda, R.; Ribrag, V.; Bourgier, C.; Lazar, V.; Lacroix, L.; Gazzah, A.; et al. A phase I, dose-escalation study of the Eg5-inhibitor EMD 534085 in patients with advanced solid tumors or lymphoma. Investig. New Drugs 2013, 31, 1530–1538. [Google Scholar] [CrossRef]
- Holen, K.; DiPaola, R.; Liu, G.; Tan, A.R.; Wilding, G.; Hsu, K.; Agrawal, N.; Chen, C.; Xue, L.; Rosenberg, E.; et al. A phase I trial of MK-0731, a Kinesin Spindle Protein (KSP) inhibitor, in patients with solid tumors. Investig. New Drugs 2012, 30, 1088–1095. [Google Scholar] [CrossRef]
- Wood, K.W.; Lad, L.; Luo, L.; Qian, X.; Knight, S.D.; Nevins, N.; Brejc, K.; Sutton, D.; Gilmartin, A.G.; Chua, P.R.; et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl. Acad. Sci. USA 2010, 107, 5839–5844. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Heath, E.I.; Schelman, W.R.; Johnson, B.M.; Kirby, L.C.; Lynch, K.M.; Botbyl, J.D.; Lampkin, T.A.; Holen, K.D. First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein E (CENP-E), in patients with refractory cancer. Cancer Chemother. Pharmacol. 2012, 69, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.P.; Martinez, R.; Zhu, Z.; Zager, M.; Blasina, A.; Rymer, I.; Hallin, J.; Xu, M.; Carroll, C.; Chionis, J.; et al. Chemogenetic evaluation of the mitotic kinesin CENP-E reveals a critical role in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, A.; Ohori, M.; Iwai, K.; Nambu, T.; Miyamoto, M.; Kawamoto, T.; Okaniwa, M. A novel time-dependent CENP-E inhibitor with potent antitumor activity. PLoS ONE 2015, 10, e0144675. [Google Scholar] [CrossRef]
- Yamane, M.; Sawada, J.I.; Ogo, N.; Ohba, M.; Ando, T.; Asai, A. Identification of benzo[d]pyrrolo[2,1-b]thiazole derivatives as CENP-E inhibitors. Biochem. Biophys. Res. Commun. 2019, 519, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/endoplasmic reticulum calcium ATPase inhibitors: Beyond anticancer perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef]
- Jurášek, M.; Rimpelová, S.; Kmoníčková, E.; Drašar, P.; Ruml, T. Tailor-made fluorescent trilobolide to study its biological relevance. J. Med. Chem. 2014, 57, 7947–7954. [Google Scholar] [CrossRef]
- Jurášek, M.; Černohorská, M.; Řehulka, J.; Spiwok, V.; Sulimenko, T.; Dráberová, E.; Darmostuk, M.; Gurská, S.; Frydrych, I.; Buriánová, R.; et al. Estradiol dimer inhibits tubulin polymerization and microtubule dynamics. J. Steroid. Biochem. Mol. Biol. 2018, 183, 68–79. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Number of Clinical Trials 1 |
|---|---|
| Colchicine | 171 |
| Paclitaxel | 3882 |
| Docetaxel | 2481 |
| Epothilones | 112 |
| Vinblastine | 208 |
| Vincristine | 1150 |
| Alisertib | 60 |
| Barasertib (AZD1152) | 10 |
| Rigosertib | 38 |
| Ispinesib | 16 |
| Compound | Condition | Status | Phase | Clinical Trial Identifier |
|---|---|---|---|---|
| Colchicine | Gout | Completed | IV | NCT01451645 NCT02060552 NCT01112982 NCT02500641 NCT02995512 NCT01310673 |
| Familiar Mediterranean fever | NCT02602028 | |||
| Hepatocellular carcinoma | Recruiting | II | NCT04264260 | |
| Paclitaxel | Non-small cell lung cancer | Completed | IV | NCT02151149 NCT00686322 NCT03092986 |
| Carcinoma, large cell Neuroendocrine tumors | NCT01317615 | |||
| Breast cancer | NCT01094184 NCT01301729 | |||
| Cancer | NCT00606515 | |||
| Docetaxel | Non-small cell lung cancer | Completed | IV | NCT01442909 NCT00883675 |
| Breast cancer | NCT02502864 NCT02445586 NCT01660542 NCT01094184 NCT01301729 | |||
| Prostatic neoplasm | NCT00280098 | |||
| Head and neck neoplasm | NCT00772681 | |||
| Squamous cell carcinoma | NCT02088515 | |||
| Ixabepilone | Breast cancer | Completed | III | NCT00082433 NCT00080301 |
| NCT00789581 | ||||
| Vinblastine | Non-small cell lung cancer | Completed | IV | NCT03092986 |
| Vincristine | Acute lymphoblastic leukemia | Completed | IV | NCT00411541 NCT00526305 NCT00526175 NCT00199069 NCT00526409 NCT00494897 NCT00853008 NCT02036489 |
| Acute lymphocytic leukemia | NCT00199069 NCT00199004 NCT00199095 NCT00199056 | |||
| Immune thrombocytopenia | NCT03229746 | |||
| Lymphoma | NCT00199017 | |||
| Vedotin/ Brentuximab | Hodgkin lymphoma | Completed | IV | NCT01990534 |
| Active | III | NCT01712490 | ||
| Anaplastic large-cell lymphoma | Recruiting | IV | NCT01909934 | |
| Active | III | NCT01777152 | ||
| Completed | NCT01578499 | |||
| Alisertib | Peripheral T-cell lymphoma | Completed | III | NCT01482962 |
| Barasertib | Acute myeloid leukemia | Completed | III | NCT00952588 |
| Volasertib | Acute myeloid leukemia | Active | III | NCT01721876 |
| Rigosertib | Metastatic Pancreatic Adenocarcinoma | Completed | III | NCT01360853 |
| Myelodysplastic syndrome | NCT01928537 NCT01241500 | |||
| Ispinesib | Renal cell cancer Breast cancer Head and neck cancer Ovarian cancer Prostate cancer Non-small cell lung cancer Melanoma (skin) Liver cancer Metastatic colorectal cancer | Completed | II | NCT00354250 NCT00089973 NCT00095628 NCT00097409 NCT00096499 NCT00085813 NCT00095953 NCT00095992 NCT00103311 |
| Cancer Cells | Normal Cells | ||||
|---|---|---|---|---|---|
| A549 | MCF-7 | LoVo | LoVo/DX | BALB/3T3 | |
| Comp. | IC50 (µM) | ||||
| 2a | 0.010 ± 0.0001 | 0.015 ± 0.002 | 0.014 ± 0.004 | 0.135 ± 0.012 | 0.103 ± 0.089 |
| 2b | 0.115 ± 0.007 | 0.178 ± 0.020 | 0.125 ± 0.044 | 0.700 ± 0.088 | 1.260 ± 0.796 |
| 2c | 0.074 ± 0.009 | 0.057 ± 0.011 | 0.074 ± 0.019 | 1.010 ± 0.020 | 0.104 ± 0.043 |
| 2d | 0.010 ± 0.0001 | 0.013 ± 0.002 | 0.007 ± 0.002 | 0.050 ± 0.010 | 0.066 ± 0.031 |
| 2e | 0.012 ± 0.004 | 0.018 ± 0.002 | 0.011 ± 0.004 | 0.071 ± 0.010 | 0.102 ± 0.063 |
| 2f | 0.030 ± 0.021 | 0.055 ± 0.026 | 0.018 ± 0.010 | 0.074 ± 0.007 | 0.138 ± 0.010 |
| 2g | 0.012 ± 0.004 | 0.027 ± 0.007 | 0.011 ± 0.0001 | 0.072 ± 0.011 | 0.116 ± 0.009 |
| 2h | 0.089 ± 0.020 | 0.132 ± 0.017 | 0.054 ± 0.017 | 0.089 ± 0.026 | 0.173 ± 0.108 |
| 2i | 0.095 ± 0.005 | 0.125 ± 0.014 | 0.062 ± 0.013 | 0.091 ± 0.009 | 0.146 ± 0.014 |
| 2j | 0.093 ± 0.014 | 0.125 ± 0.015 | 0.281 ± 0.185 | 4.240 ± 1.330 | 0.135 ± 0.015 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škubník, J.; Jurášek, M.; Ruml, T.; Rimpelová, S. Mitotic Poisons in Research and Medicine. Molecules 2020, 25, 4632. https://doi.org/10.3390/molecules25204632
Škubník J, Jurášek M, Ruml T, Rimpelová S. Mitotic Poisons in Research and Medicine. Molecules. 2020; 25(20):4632. https://doi.org/10.3390/molecules25204632
Chicago/Turabian StyleŠkubník, Jan, Michal Jurášek, Tomáš Ruml, and Silvie Rimpelová. 2020. "Mitotic Poisons in Research and Medicine" Molecules 25, no. 20: 4632. https://doi.org/10.3390/molecules25204632
APA StyleŠkubník, J., Jurášek, M., Ruml, T., & Rimpelová, S. (2020). Mitotic Poisons in Research and Medicine. Molecules, 25(20), 4632. https://doi.org/10.3390/molecules25204632