Synergistic Approach of Ultrafast Spectroscopy and Molecular Simulations in the Characterization of Intramolecular Charge Transfer in Push-Pull Molecules

 ,
, 
Abstract

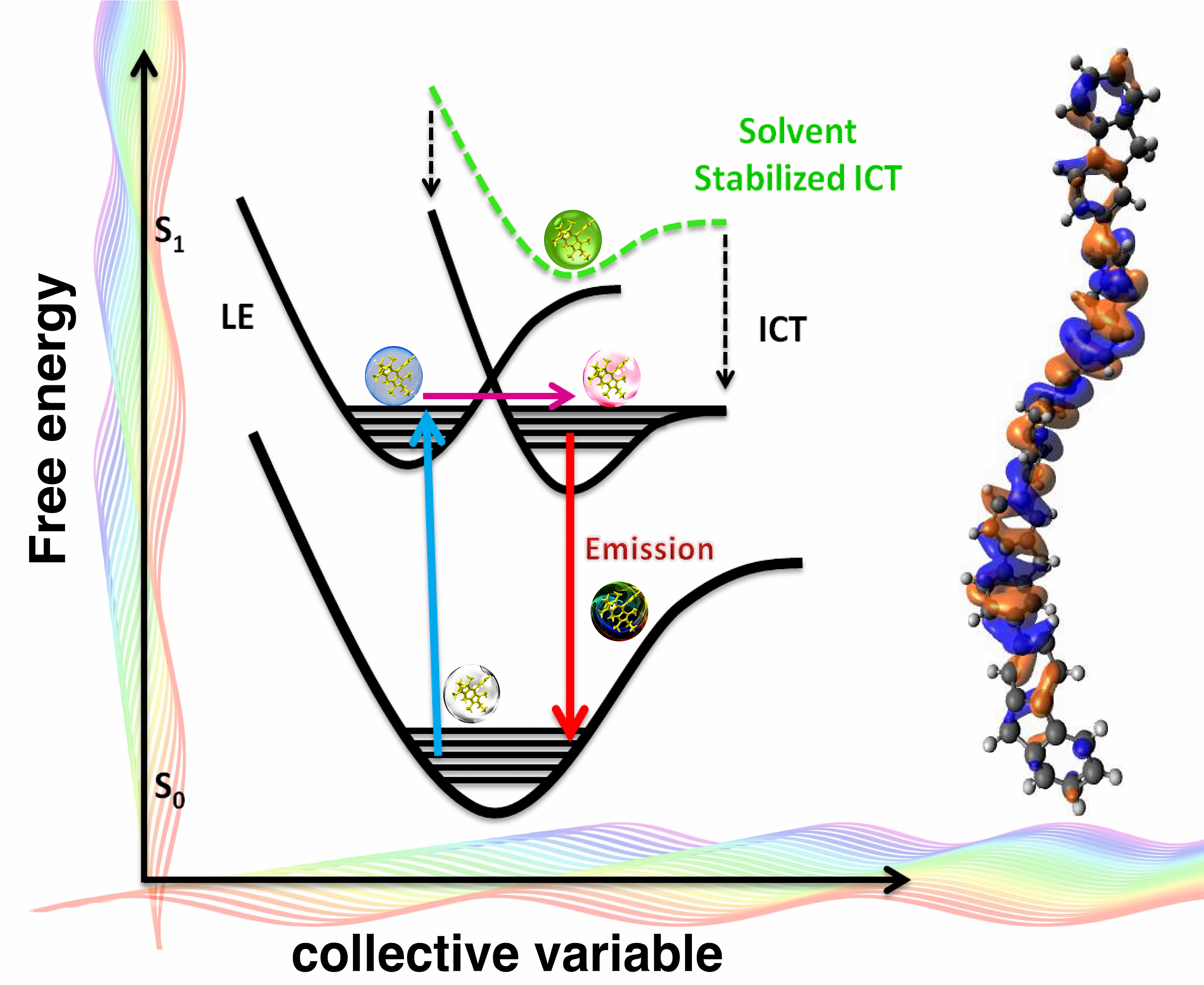{kind=link}
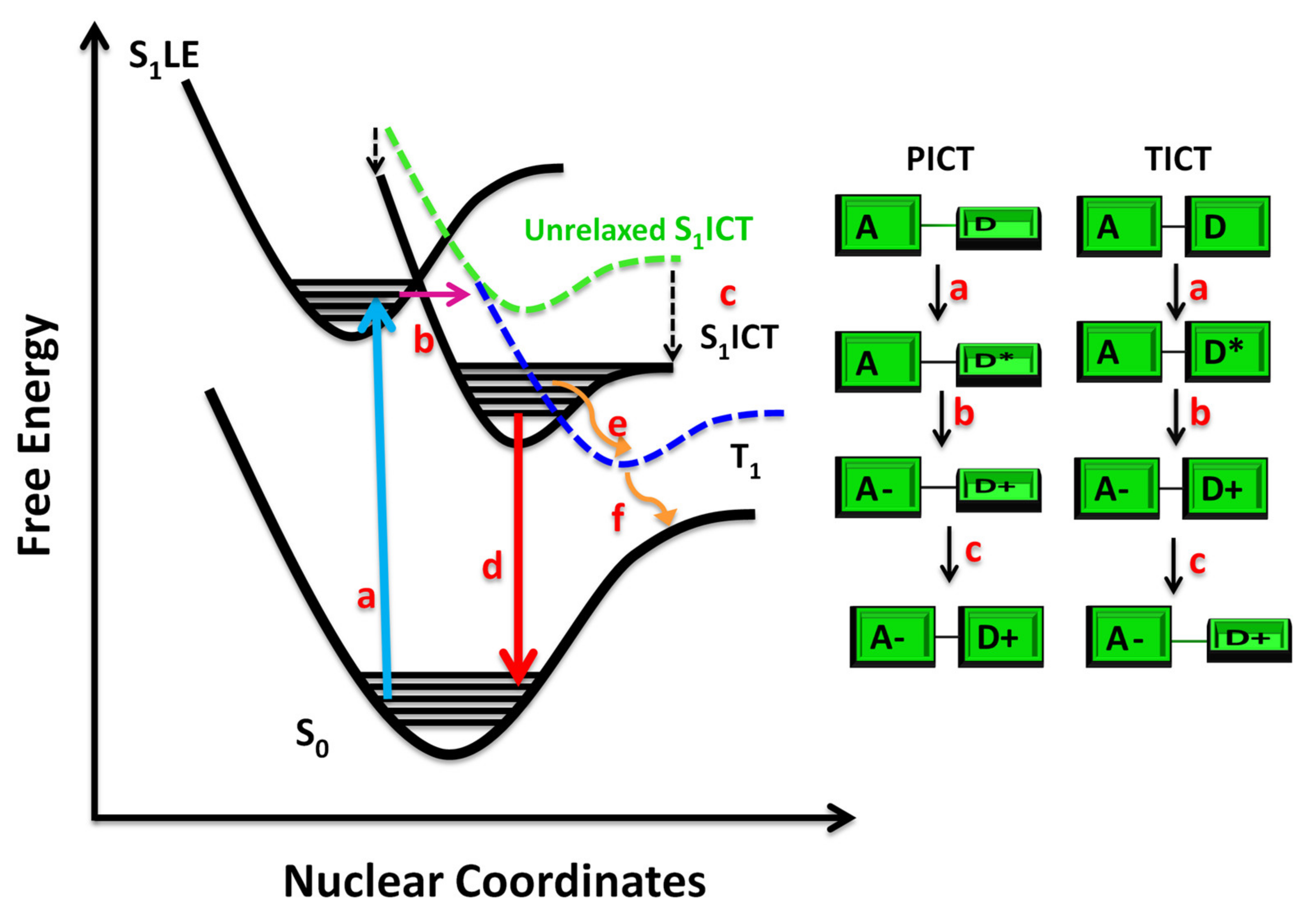{kind=link}
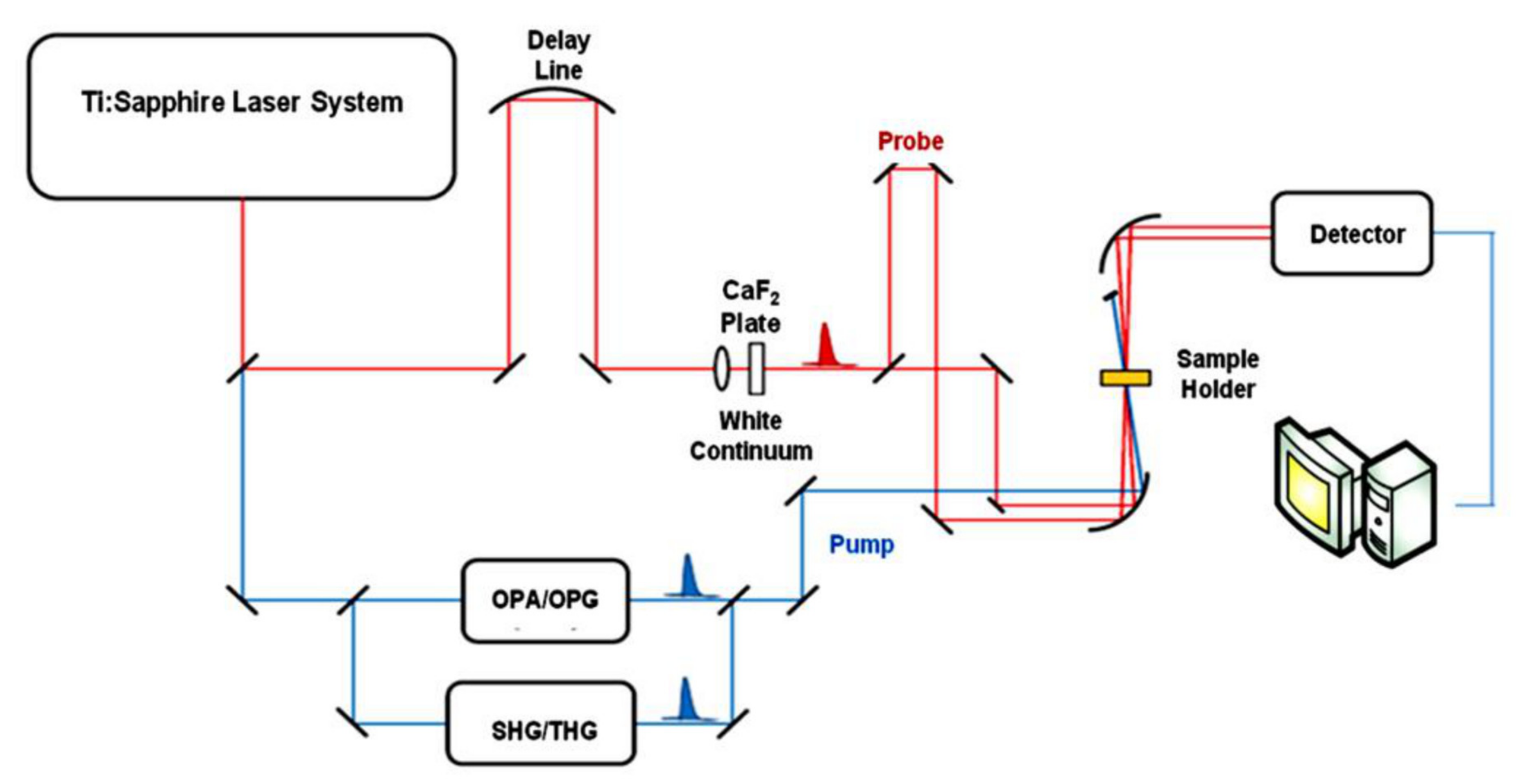{kind=link}
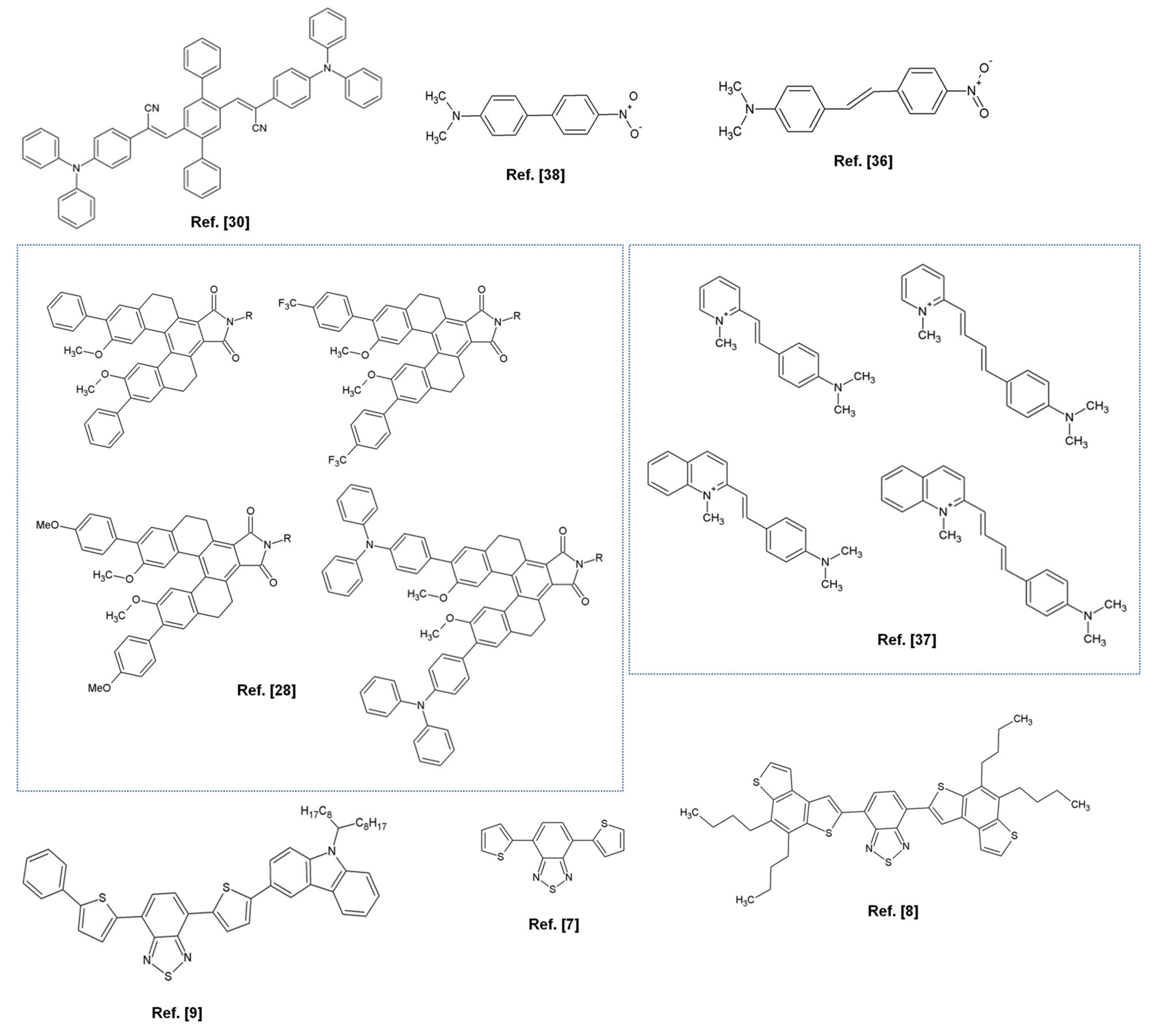{kind=link}
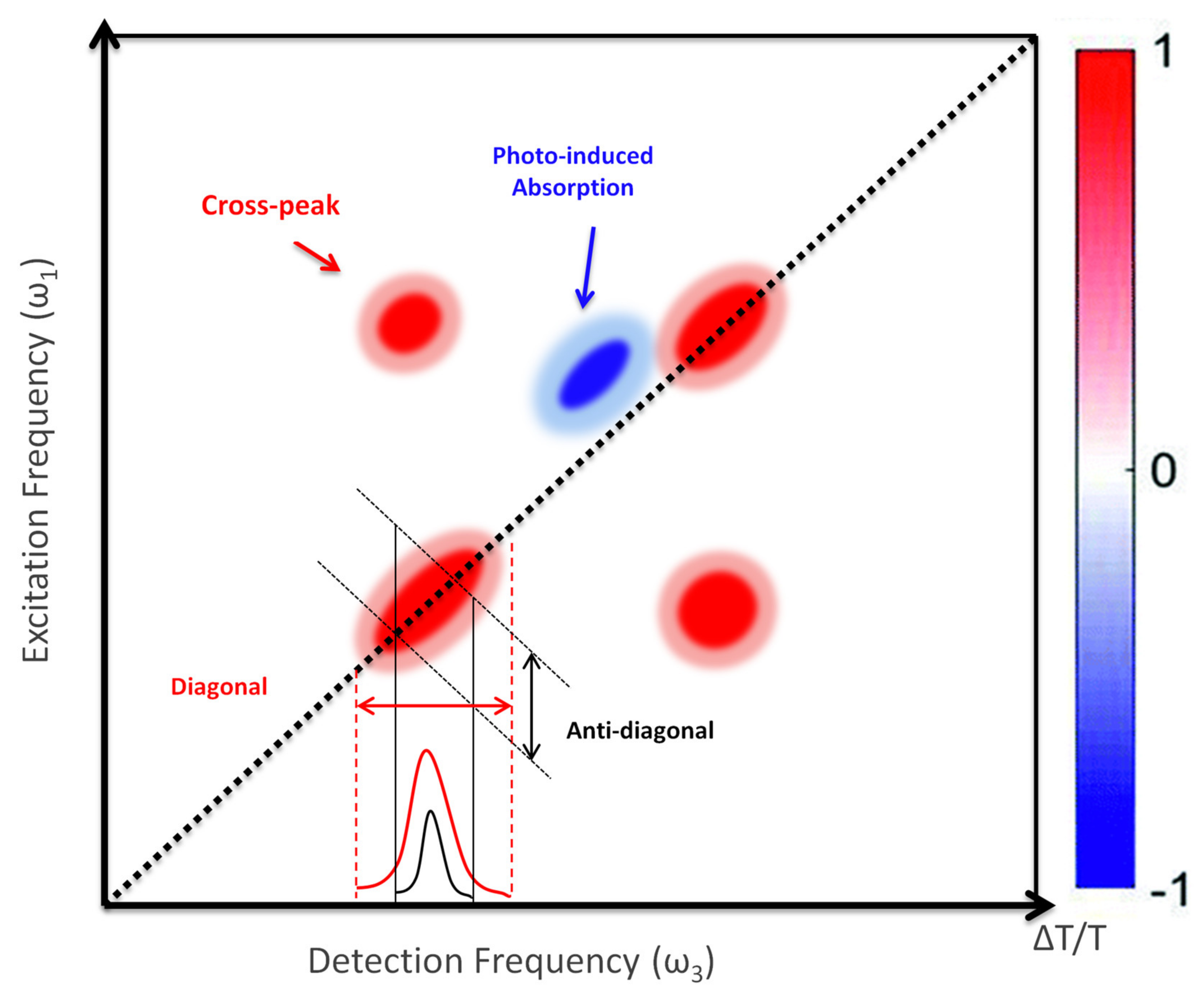{kind=link}
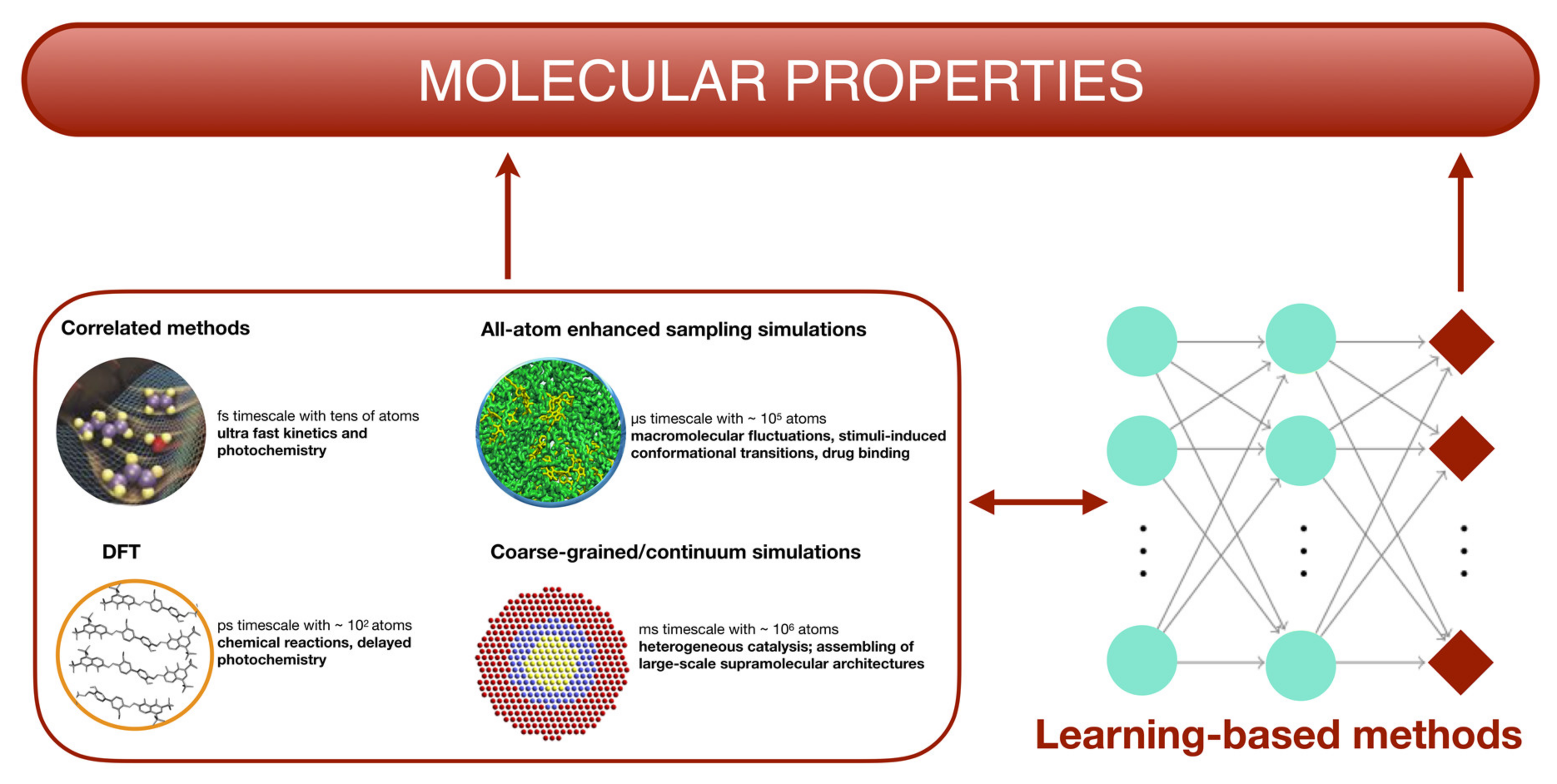{kind=link}
1. Introduction
2. Basic principles of Transient Absorption Spectroscopy (TAS) and Its Application to Study Push-Pull Molecules
Study of the Photophysical Properties and Excited State Dynamics of Push-Pull Molecules by UV-vis TAS from the Recent Literature
3. Recent Advances in Multidimensional Ultrafast Spectroscopy and Its Application in Unraveling the Intramolecular Charge Transfer (ICT) Nature of Push-Pull Molecular Systems
4. Ab-Initio Approaches to Predict Charge Transfer Properties in Push-Pull Systems
Selected Applications of Ab-Initio Predictions in Push-Pull Frameworks
5. Outlooks on Machine Learning-Based Methods and Large-Scale Simulations of Light-Induced π-Delocalized Frameworks
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sawaya, N.P.D.; Rappoport, D.; Tabor, D.P.; Aspuru-Guzik, A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12, 6410–6420. [Google Scholar] [CrossRef] [PubMed]
- Angiolini, L.; Benelli, T.; Giorgini, L.; Mauriello, F.; Salatelli, E. Chiroptical and optical thermoplastic acid sensors based on chiral methacrylic polymers containing azoaromatic moieties. Sensors Actuators B Chem. 2007, 126, 56–61. [Google Scholar] [CrossRef]
- Angiolini, L.; Benelli, T.; Giorgini, L.; Mauriello, F.; Salatelli, E.; Bozio, R.; Daurù, A.; Pedron, D. Synthesis, chiroptical properties and photoinduced birefringence of optically active methacrylic copolymers bearing side-chain bisazoaromatic moieties. Eur. Polym. J. 2007, 43, 3550–3561. [Google Scholar] [CrossRef]
- Fehrentz, T.; Huber, F.M.E.; Hartrampf, N.; Bruegmann, T.; Frank, J.A.; Fine, N.H.F.; Malan, D.; Danzl, J.G.; Tikhonov, D.B.; Sumser, M.; et al. Optical control of L-type Ca2+ channels using a diltiazem photoswitch. Nat. Chem. Biol. 2018, 14, 764–767. [Google Scholar] [CrossRef]
- Shao, B.; Baroncini, M.; Qian, H.; Bussotti, L.; Di Donato, M.; Credi, A.; Aprahamian, I. Solution and Solid-State Emission Toggling of a Photochromic Hydrazone. J. Am. Chem. Soc. 2018, 140, 12323–12327. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, W. Organic sensitizers from D–π–A to D–A–π–A: effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances. Chem. Soc. Rev. 2013, 42, 2039–2058. [Google Scholar] [CrossRef]
- Iagatti, A.; Patrizi, B.; Basagni, A.; Marcelli, A.; Alessi, A.; Zanardi, S.; Fusco, R.; Salvalaggio, M.; Bussotti, L.; Foggi, P. Photophysical properties and excited state dynamics of 4,7-dithien-2-yl-2,1,3-benzothiadiazole. Phys. Chem. Chem. Phys. 2017, 19, 13604–13613. [Google Scholar] [CrossRef]
- Patrizi, B.; Iagatti, A.; Abbondanza, L.; Bussotti, L.; Zanardi, S.; Salvalaggio, M.; Fusco, R.; Foggi, P. Ultrafast Intramolecular and Solvation Dynamics in 4,7-Bis (4,5-dibutylbenzo[1,2-b:4,3-b′]bisthiophene[1,2-b:4,3-b′]bisthiophen-2-yl)-2,1,3-benzothiadiazole. J. Phys. Chem. C 2019, 123, 5840–5852. [Google Scholar] [CrossRef]
- Scarongella, M.; Laktionov, A.; Rothlisberger, U.; Banerji, N. Charge transfer relaxation in donor–acceptor type conjugated materials. J. Mater. Chem. C 2013, 1, 2308–2319. [Google Scholar] [CrossRef]
- Allard, S.; Forster, M.; Souharce, B.; Thiem, H.; Scherf, U. Organic Semiconductors for Solution-Processable Field-Effect Transistors (OFETs). Angew. Chem. Int. Ed. 2008, 47, 4070–4098. [Google Scholar] [CrossRef]
- Ohmori, Y. Development of organic light-emitting diodes for electro-optical integrated devices. Laser Photon. Rev. 2010, 4, 300–310. [Google Scholar] [CrossRef]
- García, G.; Adamo, C.; Ciofini, I. Evaluating push–pull dye efficiency using TD-DFT and charge transfer indices. Phys. Chem. Chem. Phys. 2013, 15, 20210–20219. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Cho, Y.-J.; Lee, A.-R.; Son, H.; Han, W.-S.; Cho, D.W.; Kang, S.O. Influence of π-conjugation structural changes on intramolecular charge transfer and photoinduced electron transfer in donor–π–acceptor dyads. Phys. Chem. Chem. Phys. 2017, 19, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Buck, J.T.; Wilson, R.W.; Mani, T. Intramolecular Long-Range Charge-Transfer Emission in Donor–Bridge–Acceptor Systems. J. Phys. Chem. Lett. 2019, 10, 3080–3086. [Google Scholar] [CrossRef] [PubMed]
- Nazim, M.; Ameen, S.; Seo, H.-K.; Shin, H.S. Effective D-A-D type chromophore of fumaronitrile-core and terminal alkylated bithiophene for solution-processed small molecule organic solar cells. Sci. Rep. 2015, 5, 11143. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Eilers-König, N.; Senyushkina, T.A.; Ernsting, N.P. Charge Transfer and Solvation of Betaine-30 in Polar SolventsA Femtosecond Broadband Transient Absorption Study. J. Phys. Chem. A 2001, 105, 4834–4843. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef]
- Sasaki, S.; Drummen, G.P.C.; Konishi, G. Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef]
- Glasbeek, M.; Zhang, H. Femtosecond Studies of Solvation and Intramolecular Configurational Dynamics of Fluorophores in Liquid Solution. Chem. Rev. 2004, 104, 1929–1954. [Google Scholar] [CrossRef]
- Haberhauer, G.; Gleiter, R.; Burkhart, C. Planarized Intramolecular Charge Transfer: A Concept for Fluorophores with both Large Stokes Shifts and High Fluorescence Quantum Yields. Chem. J. 2016, 22, 971–978. [Google Scholar] [CrossRef]
- Boubeta, F.M.; Boechi, L.; Estrin, D.; Patrizi, B.; Di Donato, M.; Iagatti, A.; Giordano, D.; Verde, C.; Bruno, S.; Abbruzzetti, S.; et al. Cold-Adaptation Signatures in the Ligand Rebinding Kinetics to the Truncated Hemoglobin of the Antarctic Bacterium Pseudoalteromonas haloplanktis TAC125. J. Phys. Chem. B 2018, 122, 11649–11661. [Google Scholar] [CrossRef]
- Delcanale, P.; Pennacchietti, F.; Maestrini, G.; Rodríguez-Amigo, B.; Bianchini, P.; Diaspro, A.; Iagatti, A.; Patrizi, B.; Foggi, P.; Agut, M.; et al. Subdiffraction localization of a nanostructured photosensitizer in bacterial cells. Sci. Rep. 2015, 5, 15564. [Google Scholar] [CrossRef]
- Hussain, M.; Taddei, M.; Bussotti, L.; Foggi, P.; Zhao, J.; Liu, Q.; Di Donato, M. Intersystem Crossing in Naphthalenediimide–Oxoverdazyl Dyads: Synthesis and Study of the Photophysical Properties. Chem. J. 2019, 25, 15615–15627. [Google Scholar] [CrossRef]
- Lapini, A.; Di Donato, M.; Patrizi, B.; Marcelli, A.; Lima, M.; Righini, R.; Foggi, P.; Sciamanna, N.; Boffi, A. Carbon monoxide recombination dynamics in truncated hemoglobins studied with visible-pump midir-probe spectroscopy. J. Phys. Chem. B 2012, 116, 8753–8761. [Google Scholar] [CrossRef]
- Patrizi, B.; Lapini, A.; Di Donato, M.; Marcelli, A.; Lima, M.; Righini, R.; Foggi, P.; Baiocco, P.; Bonamore, A.; Boffi, A. Role of local structure and dynamics of small ligand migration in proteins: A study of a mutated truncated hemoprotein from Thermobifida fusca by time resolved MIR spectroscopy. J. Phys. Chem. B 2014, 118, 9209–9217. [Google Scholar] [CrossRef]
- Berera, R.; van Grondelle, R.; Kennis, J.T.M. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef]
- Moroni, L.; Gellini, C.; Salvi, P.R.; Marcelli, A.; Foggi, P. Excited States of Porphyrin Macrocycles. J. Phys. Chem. A 2008, 112, 11044–11051. [Google Scholar] [CrossRef]
- Zhu, H.; Li, M.; Hu, J.; Wang, X.; Jie, J.; Guo, Q.; Chen, C.; Xia, A. Ultrafast Investigation of Intramolecular Charge Transfer and Solvation Dynamics of Tetrahydro[5]-helicene-Based Imide Derivatives. Sci. Rep. 2016, 6, 24313. [Google Scholar] [CrossRef]
- Niu, X.; Gautam, P.; Kuang, Z.; Yu, C.P.; Guo, Y.; Song, H.; Guo, Q.; Chan, J.M.W.; Xia, A. Intramolecular charge transfer and solvation dynamics of push–pull dyes with different π-conjugated linkers. Phys. Chem. Chem. Phys. 2019, 21, 17323–17331. [Google Scholar] [CrossRef]
- Song, H.; Wang, K.; Kuang, Z.; Zhao, Y.S.; Guo, Q.; Xia, A. Solvent modulated excited state processes of push–pull molecule with hybridized local excitation and intramolecular charge transfer character. Phys. Chem. Chem. Phys. 2019, 21, 3894–3902. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, M.; Niu, Y.; Guo, Q.; Xia, A. Solvent-dependent intramolecular charge transfer delocalization/localization in multibranched push-pull chromophores. J. Chem. Phys. 2015, 143, 34309. [Google Scholar] [CrossRef]
- Gong, Y.; Guo, X.; Wang, S.; Su, H.; Xia, A.; He, Q.; Bai, F. Photophysical Properties of Photoactive Molecules with Conjugated Push–Pull Structures. J. Phys. Chem. A 2007, 111, 5806–5812. [Google Scholar] [CrossRef]
- Campioli, E.; Sanyal, S.; Marcelli, A.; Di Donato, M.; Blanchard-Desce, M.; Mongin, O.; Painelli, A.; Terenziani, F. Addressing Charge-Transfer and Locally-Excited States in a Twisted Biphenyl Push-Pull Chromophore. ChemPhysChem 2019, 20, 2860–2873. [Google Scholar] [CrossRef]
- Kühne, T.D. Second generation Car–Parrinello molecular dynamics. WIREs Comput. Mol. Sci. 2014, 4, 391–406. [Google Scholar] [CrossRef]
- Tuckerman, M.E. Ab initiomolecular dynamics: basic concepts, current trends and novel applications. J. Phys. Condens. Matter 2002, 14, R1297–R1355. [Google Scholar] [CrossRef]
- Singh, C.; Ghosh, R.; Mondal, J.A.; Palit, D.K. Excited state dynamics of a push–pull stilbene: A femtosecond transient absorption spectroscopic study. J. Photochem. Photobiol. A Chem. 2013, 263, 50–60. [Google Scholar] [CrossRef]
- Carlotti, B.; Benassi, E.; Barone, V.; Consiglio, G.; Elisei, F.; Mazzoli, A.; Spalletti, A. Effect of the π Bridge and Acceptor on Intramolecular Charge Transfer in Push–Pull Cationic Chromophores: An Ultrafast Spectroscopic and TD-DFT Computational Study. ChemPhysChem 2015, 16, 1440–1450. [Google Scholar] [CrossRef]
- Ghosh, R.; Nandi, A.; Palit, D.K. Solvent sensitive intramolecular charge transfer dynamics in the excited states of 4-N,N-dimethylamino-4′-nitrobiphenyl. Phys. Chem. Chem. Phys. 2016, 18, 7661–7671. [Google Scholar] [CrossRef]
- Shen, P.; Zhuang, Z.; Jiang, X.-F.; Li, J.; Yao, S.; Zhao, Z.; Tang, B.Z. Through-Space Conjugation: An Effective Strategy for Stabilizing Intramolecular Charge-Transfer States. J. Phys. Chem. Lett. 2019, 10, 2648–2656. [Google Scholar] [CrossRef]
- Bredenbeck, J.; Helbing, J.; Kolano, C.; Hamm, P. Ultrafast 2D–IR Spectroscopy of Transient Species. ChemPhysChem 2007, 8, 1747–1756. [Google Scholar] [CrossRef]
- Lima, M.; Candelaresi, M.; Foggi, P. 2D-IR spectroscopy: an additional dimension to investigate ultrafast structural dynamics. J. Raman Spectrosc. 2013, 44, 1470–1477. [Google Scholar] [CrossRef]
- Scholes, G.D.; Fleming, G.R.; Chen, L.X.; Aspuru-Guzik, A.; Buchleitner, A.; Coker, D.F.; Engel, G.S.; van Grondelle, R.; Ishizaki, A.; Jonas, D.M.; et al. Using coherence to enhance function in chemical and biophysical systems. Nature 2017, 543, 647–656. [Google Scholar] [CrossRef]
- Borrego-Varillas, R.; Nenov, A.; Ganzer, L.; Oriana, A.; Manzoni, C.; Tolomelli, A.; Rivalta, I.; Mukamel, S.; Garavelli, M.; Cerullo, G. Two-dimensional UV spectroscopy: a new insight into the structure and dynamics of biomolecules. Chem. Sci. 2019, 10, 9907–9921. [Google Scholar] [CrossRef]
- De Sio, A.; Troiani, F.; Maiuri, M.; Réhault, J.; Sommer, E.; Lim, J.; Huelga, S.F.; Plenio, M.B.; Rozzi, C.A.; Cerullo, G.; et al. Tracking the coherent generation of polaron pairs in conjugated polymers. Nat. Commun. 2016, 7, 13742. [Google Scholar] [CrossRef]
- Brixner, T.; Mančal, T.; Stiopkin, I.V.; Fleming, G.R. Phase-stabilized two-dimensional electronic spectroscopy. J. Chem. Phys. 2004, 121, 4221–4236. [Google Scholar] [CrossRef]
- Ruetzel, S.; Diekmann, M.; Nuernberger, P.; Walter, C.; Engels, B.; Brixner, T. Multidimensional spectroscopy of photoreactivity. Proc. Natl. Acad. Sci. USA 2014, 111, 4764–4769. [Google Scholar] [CrossRef]
- Oliver, T.A.A. Recent advances in multidimensional ultrafast spectroscopy. R. Soc. Open Sci. 2019, 5, 171425. [Google Scholar] [CrossRef]
- Corsi, C.; Liontos, I.; Bellini, M.; Cavalieri, S.; Cancio Pastor, P.; Siciliani de Cumis, M.; Eramo, R. Ultimate Limit in the Spectral Resolution of Extreme Ultraviolet Frequency Combs. Phys. Rev. Lett. 2017, 118, 143201. [Google Scholar] [CrossRef]
- Shim, S.-H.; Strasfeld, D.B.; Ling, Y.L.; Zanni, M.T. Automated 2D IR spectroscopy using a mid-IR pulse shaper and application of this technology to the human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 2007, 104, 14197–14202. [Google Scholar] [CrossRef]
- Cowan, M.L.; Ogilvie, J.P.; Miller, R.J.D. Two-dimensional spectroscopy using diffractive optics based phased-locked photon echoes. Chem. Phys. Lett. 2004, 386, 184–189. [Google Scholar] [CrossRef]
- Mukamel, S.; Tanimura, Y.; Hamm, P. Coherent Multidimensional Optical Spectroscopy. Acc. Chem. Res. 2009, 42, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Cundiff, S.T.; Bristow, A.D.; Siemens, M.; Li, H.; Moody, G.; Karaiskaj, D.; Dai, X.; Zhang, T. Optical 2-D Fourier Transform Spectroscopy of Excitons in Semiconductor Nanostructures. IEEE J. Sel. Top. Quantum Electron. 2012, 18, 318–328. [Google Scholar] [CrossRef]
- Lewis, K.L.M.; Ogilvie, J.P. Probing Photosynthetic Energy and Charge Transfer with Two-Dimensional Electronic Spectroscopy. J. Phys. Chem. Lett. 2012, 3, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Doria, S.; Lapini, A.; Di Donato, M.; Righini, R.; Azzaroli, N.; Iagatti, A.; Caram, J.R.; Sinclair, T.S.; Cupellini, L.; Jurinovich, S.; et al. Understanding the influence of disorder on the exciton dynamics and energy transfer in Zn-phthalocyanine H-aggregates. Phys. Chem. Chem. Phys. 2018, 20, 22331–22341. [Google Scholar] [CrossRef]
- Bolzonello, L.; Polo, A.; Volpato, A.; Meneghin, E.; Cordaro, M.; Trapani, M.; Fortino, M.; Pedone, A.; Castriciano, M.A.; Collini, E. Two-Dimensional Electronic Spectroscopy Reveals Dynamics and Mechanisms of Solvent-Driven Inertial Relaxation in Polar BODIPY Dyes. J. Phys. Chem. Lett. 2018, 9, 1079–1085. [Google Scholar] [CrossRef]
- Lee, Y.; Das, S.; Malamakal, R.M.; Meloni, S.; Chenoweth, D.M.; Anna, J.M. Ultrafast Solvation Dynamics and Vibrational Coherences of Halogenated Boron-Dipyrromethene Derivatives Revealed through Two-Dimensional Electronic Spectroscopy. J. Am. Chem. Soc. 2017, 139, 14733–14742. [Google Scholar] [CrossRef]
- Maurer, R.J.; Freysoldt, C.; Reilly, A.M.; Brandenburg, J.G.; Hofmann, O.T.; Björkman, T.; Lebègue, S.; Tkatchenko, A. Advances in Density-Functional Calculations for Materials Modeling. Annu. Rev. Mater. Res. 2019, 49, 1–30. [Google Scholar] [CrossRef]
- Pershin, A.; Hall, D.; Lemaur, V.; Sancho-Garcia, J.-C.; Muccioli, L.; Zysman-Colman, E.; Beljonne, D.; Olivier, Y. Highly emissive excitons with reduced exchange energy in thermally activated delayed fluorescent molecules. Nat. Commun. 2019, 10, 597. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: The Zincbacteriochlorin–Bacteriochlorin and Bacteriochlorophyll–Spheroidene Complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Benfield, P.; Helgaker, T.; Tozer, D.J. Excitation energies in density functional theory: An evaluation and a diagnostic test. J. Chem. Phys. 2008, 128, 44118. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Tozer, D.J. Illustration of a TDDFT spatial overlap diagnostic by basis function exponent scaling. J. Mol. Struct. THEOCHEM 2009, 914, 110–114. [Google Scholar] [CrossRef]
- Guido, C.A.; Cortona, P.; Mennucci, B.; Adamo, C. On the Metric of Charge Transfer Molecular Excitations: A Simple Chemical Descriptor. J. Chem. Theory Comput. 2013, 9, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.A.; Cortona, P.; Adamo, C. Effective electron displacements: A tool for time-dependent density functional theory computational spectroscopy. J. Chem. Phys. 2014, 140, 104101. [Google Scholar] [CrossRef] [PubMed]
- Olivier, Y.; Sancho-Garcia, J.-C.; Muccioli, L.; D’Avino, G.; Beljonne, D. Computational Design of Thermally Activated Delayed Fluorescence Materials: The Challenges Ahead. J. Phys. Chem. Lett. 2018, 9, 6149–6163. [Google Scholar] [CrossRef]
- Le Bahers, T.; Adamo, C.; Ciofini, I. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef]
- Etienne, T.; Assfeld, X.; Monari, A. Toward a Quantitative Assessment of Electronic Transitions’ Charge-Transfer Character. J. Chem. Theory Comput. 2014, 10, 3896–3905. [Google Scholar] [CrossRef] [PubMed]
- Campetella, M.; Maschietto, F.; Frisch, M.J.; Scalmani, G.; Ciofini, I.; Adamo, C. Charge transfer excitations in TDDFT: A ghost-hunter index. J. Comput. Chem. 2017, 38, 2151–2156. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Manna, A.K.; Balamurugan, D.; Cheung, M.S.; Dunietz, B.D. Unraveling the Mechanism of Photoinduced Charge Transfer in Carotenoid–Porphyrin–C60 Molecular Triad. J. Phys. Chem. Lett. 2015, 6, 1231–1237. [Google Scholar] [CrossRef]
- Zhang, C.-R.; Sears, J.S.; Yang, B.; Aziz, S.G.; Coropceanu, V.; Brédas, J.-L. Theoretical Study of the Local and Charge-Transfer Excitations in Model Complexes of Pentacene-C60 Using Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2014, 10, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Manna, A.K.; Lee, M.H.; McMahon, K.L.; Dunietz, B.D. Calculating High Energy Charge Transfer States Using Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2015, 11, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Eisenberg, H.; Kronik, L.; Baer, R. Fundamental Gaps in Finite Systems from Eigenvalues of a Generalized Kohn-Sham Method. Phys. Rev. Lett. 2010, 105, 266802. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, S.; Cheung, M.S.; Geva, E.; Kronik, L.; Dunietz, B.D. Fundamental Gaps of Condensed-Phase Organic Semiconductors from Single-Molecule Calculations using Polarization-Consistent Optimally Tuned Screened Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2018, 14, 6287–6294. [Google Scholar] [CrossRef]
- Zheng, Z.; Egger, D.A.; Brédas, J.-L.; Kronik, L.; Coropceanu, V. Effect of Solid-State Polarization on Charge-Transfer Excitations and Transport Levels at Organic Interfaces from a Screened Range-Separated Hybrid Functional. J. Phys. Chem. Lett. 2017, 8, 3277–3283. [Google Scholar] [CrossRef]
- Refaely-Abramson, S.; Sharifzadeh, S.; Jain, M.; Baer, R.; Neaton, J.B.; Kronik, L. Gap renormalization of molecular crystals from density-functional theory. Phys. Rev. B 2013, 88, 81204. [Google Scholar] [CrossRef]
- Bhandari, S.; Dunietz, B.D. Quantitative Accuracy in Calculating Charge Transfer State Energies in Solvated Molecular Complexes Using a Screened Range Separated Hybrid Functional within a Polarized Continuum Model. J. Chem. Theory Comput. 2019, 15, 4305–4311. [Google Scholar] [CrossRef]
- Wang, L.; Beljonne, D. Charge transport in organic semiconductors: Assessment of the mean field theory in the hopping regime. J. Chem. Phys. 2013, 139, 64316. [Google Scholar] [CrossRef]
- Filatov, M.; Martínez, T.J.; Kim, K.S. Description of ground and excited electronic states by ensemble density functional method with extended active space. J. Chem. Phys. 2017, 147, 64104. [Google Scholar] [CrossRef] [PubMed]
- Menger, M.F.S.J.; Plasser, F.; Mennucci, B.; González, L. Surface Hopping within an Exciton Picture. An Electrostatic Embedding Scheme. J. Chem. Theory Comput. 2018, 14, 6139–6148. [Google Scholar] [CrossRef] [PubMed]
- Vuong, V.Q.; Akkarapattiakal Kuriappan, J.; Kubillus, M.; Kranz, J.J.; Mast, T.; Niehaus, T.A.; Irle, S.; Elstner, M. Parametrization and Benchmark of Long-Range Corrected DFTB2 for Organic Molecules. J. Chem. Theory Comput. 2018, 14, 115–125. [Google Scholar] [CrossRef] [PubMed]
- De Wergifosse, M.; Grimme, S. Nonlinear-response properties in a simplified time-dependent density functional theory (sTD-DFT) framework: Evaluation of the first hyperpolarizability. J. Chem. Phys. 2018, 149, 24108. [Google Scholar] [CrossRef]
- Sifain, A.E.; Bjorgaard, J.A.; Nelson, T.R.; Nebgen, B.T.; White, A.J.; Gifford, B.J.; Gao, D.W.; Prezhdo, O.V.; Fernandez-Alberti, S.; Roitberg, A.E.; et al. Photoexcited Nonadiabatic Dynamics of Solvated Push–Pull π-Conjugated Oligomers with the NEXMD Software. J. Chem. Theory Comput. 2018, 14, 3955–3966. [Google Scholar] [CrossRef]
- Alam, M.M.; Chattopadhyaya, M.; Chakrabarti, S.; Ruud, K. Chemical Control of Channel Interference in Two-Photon Absorption Processes. Acc. Chem. Res. 2014, 47, 1604–1612. [Google Scholar] [CrossRef]
- Tonnelé, C.; Champagne, B.; Muccioli, L.; Castet, F. Second-order nonlinear optical properties of Stenhouse photoswitches: insights from density functional theory. Phys. Chem. Chem. Phys. 2018, 20, 27658–27667. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Cozza, C.; Zhang, Z.; Nakano, T. Temperature-dependent UV absorption of biphenyl based on intra-molecular rotation investigated within a combined experimental and TD-DFT approach. Liq. Cryst. 2018, 45, 2048–2053. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Nakano, T. Molecular Mechanism of Polyacrylate Helix Sense Switching across Its Free Energy Landscape. J. Am. Chem. Soc. 2013, 135, 5509–5512. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Wang, Y.; Nakano, T. Predicting the Switchable Screw Sense in Fluorene-Based Polymers. Angew. Chemie Int. Ed. 2015, 54, 2688–2692. [Google Scholar] [CrossRef]
- Nénon, S.; Champagne, B. SCC-DFTB calculation of the static first hyperpolarizability: From gas phase molecules to functionalized surfaces. J. Chem. Phys. 2013, 138, 204107. [Google Scholar] [CrossRef] [PubMed]
- Suhina, T.; Amirjalayer, S.; Mennucci, B.; Woutersen, S.; Hilbers, M.; Bonn, D.; Brouwer, A.M. Excited-State Decay Pathways of Molecular Rotors: Twisted Intermediate or Conical Intersection? J. Phys. Chem. Lett. 2016, 7, 4285–4290. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Prado, L.R.; Bortolini, G.; Rego, L.G.C. Charge Transfer Driven Structural Relaxation in a Push–Pull Azobenzene Dye–Semiconductor Complex. J. Phys. Chem. Lett. 2018, 9, 5926–5933. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 84106. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunning, T.H. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H+H2→H2+H reaction. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- Wilson, A.K.; van Mourik, T.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct. THEOCHEM 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Benassi, E.; Carlotti, B.; Segado, M.; Cesaretti, A.; Spalletti, A.; Elisei, F.; Barone, V. Presence of Two Emissive Minima in the Lowest Excited State of a Push–Pull Cationic Dye Unequivocally Proved by Femtosecond Up-Conversion Spectroscopy and Vibronic Quantum-Mechanical Computations. J. Phys. Chem. B 2015, 119, 6035–6040. [Google Scholar] [CrossRef] [PubMed]
- Muniz-Miranda, F.; Pedone, A.; Muniz-Miranda, M. Spectroscopic and DFT investigation on the photo-chemical properties of a push-pull chromophore: 4-Dimethylamino-4′-nitrostilbene. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 33–39. [Google Scholar] [CrossRef]
- Sutton, J.; Barnsley, J.; Mapley, J.; Wagner, P.; Officer, D.; Gordon, K. Modulation of Donor-Acceptor Distance in a Series of Carbazole Push-Pull Dyes; A Spectroscopic and Computational Study. Molecules 2018, 23, 421. [Google Scholar] [CrossRef]
- Dettori, R.; Ceriotti, M.; Hunger, J.; Colombo, L.; Donadio, D. Energy Relaxation and Thermal Diffusion in Infrared Pump–Probe Spectroscopy of Hydrogen-Bonded Liquids. J. Phys. Chem. Lett. 2019, 10, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Segarra-Martí, J.; Mukamel, S.; Garavelli, M.; Nenov, A.; Rivalta, I. Towards Accurate Simulation of Two-Dimensional Electronic Spectroscopy. Top. Curr. Chem. 2018, 376, 24. [Google Scholar] [CrossRef] [PubMed]
- Provazza, J.; Segatta, F.; Garavelli, M.; Coker, D.F. Semiclassical Path Integral Calculation of Nonlinear Optical Spectroscopy. J. Chem. Theory Comput. 2018, 14, 856–866. [Google Scholar] [CrossRef]
- Segatta, F.; Cupellini, L.; Garavelli, M.; Mennucci, B. Quantum Chemical Modeling of the Photoinduced Activity of Multichromophoric Biosystems. Chem. Rev. 2019, 119, 9361–9380. [Google Scholar] [CrossRef]
- Duan, C.; Janet, J.P.; Liu, F.; Nandy, A.; Kulik, H.J. Learning from Failure: Predicting Electronic Structure Calculation Outcomes with Machine Learning Models. J. Chem. Theory Comput. 2019, 15, 2331–2345. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Cheng, L.; Welborn, M.; Christensen, A.S.; Miller, T.F. A universal density matrix functional from molecular orbital-based machine learning: Transferability across organic molecules. J. Chem. Phys. 2019, 150, 131103. [Google Scholar] [CrossRef]
- Welborn, M.; Cheng, L.; Miller, T.F. Transferability in Machine Learning for Electronic Structure via the Molecular Orbital Basis. J. Chem. Theory Comput. 2018, 14, 4772–4779. [Google Scholar] [CrossRef] [PubMed]
- Brockherde, F.; Vogt, L.; Li, L.; Tuckerman, M.E.; Burke, K.; Müller, K.-R. Bypassing the Kohn-Sham equations with machine learning. Nat. Commun. 2017, 8, 872. [Google Scholar] [CrossRef] [PubMed]
- Häse, F.; Kreisbeck, C.; Aspuru-Guzik, A. Machine learning for quantum dynamics: deep learning of excitation energy transfer properties. Chem. Sci. 2017, 8, 8419–8426. [Google Scholar] [CrossRef] [PubMed]
- Häse, F.; Valleau, S.; Pyzer-Knapp, E.; Aspuru-Guzik, A. Machine learning exciton dynamics. Chem. Sci. 2016, 7, 5139–5147. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, B.G.; Noid, D.W. Potential energy surfaces for macromolecules. A neural network technique. Chem. Phys. Lett. 1992, 192, 455–462. [Google Scholar] [CrossRef]
- Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401. [Google Scholar] [CrossRef]
- Handley, C.M.; Popelier, P.L.A. Potential Energy Surfaces Fitted by Artificial Neural Networks. J. Phys. Chem. A 2010, 114, 3371–3383. [Google Scholar] [CrossRef]
- Grisafi, A.; Fabrizio, A.; Meyer, B.; Wilkins, D.M.; Corminboeuf, C.; Ceriotti, M. Transferable Machine-Learning Model of the Electron Density. ACS Cent. Sci. 2019, 5, 57–64. [Google Scholar] [CrossRef]
- Wilkins, D.M.; Grisafi, A.; Yang, Y.; Lao, K.U.; DiStasio, R.A.; Ceriotti, M. Accurate molecular polarizabilities with coupled cluster theory and machine learning. Proc. Natl. Acad. Sci. USA 2019, 116, 3401–3406. [Google Scholar] [CrossRef]
- Nebgen, B.; Lubbers, N.; Smith, J.S.; Sifain, A.E.; Lokhov, A.; Isayev, O.; Roitberg, A.E.; Barros, K.; Tretiak, S. Transferable Dynamic Molecular Charge Assignment Using Deep Neural Networks. J. Chem. Theory Comput. 2018, 14, 4687–4698. [Google Scholar] [CrossRef]
- Dral, P.O.; Barbatti, M.; Thiel, W. Nonadiabatic Excited-State Dynamics with Machine Learning. J. Phys. Chem. Lett. 2018, 9, 5660–5663. [Google Scholar] [CrossRef] [PubMed]
- Westermayr, J.; Gastegger, M.; Menger, M.F.S.J.; Mai, S.; González, L.; Marquetand, P. Machine learning enables long time scale molecular photodynamics simulations. Chem. Sci. 2019, 10, 8100–8107. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, S.; Sauceda, H.E.; Müller, K.-R.; Tkatchenko, A. Towards exact molecular dynamics simulations with machine-learned force fields. Nat. Commun. 2018, 9, 3887. [Google Scholar] [CrossRef] [PubMed]
- Bonati, L.; Zhang, Y.-Y.; Parrinello, M. Neural networks-based variationally enhanced sampling. Proc. Natl. Acad. Sci. USA 2019, 116, 17641–17647. [Google Scholar] [CrossRef]
- Ribeiro, J.M.L.; Bravo, P.; Wang, Y.; Tiwary, P. Reweighted autoencoded variational Bayes for enhanced sampling (RAVE). J. Chem. Phys. 2018, 149, 72301. [Google Scholar] [CrossRef]
- Trapl, D.; Horvacanin, I.; Mareska, V.; Ozcelik, F.; Unal, G.; Spiwok, V. Anncolvar: Approximation of Complex Collective Variables by Artificial Neural Networks for Analysis and Biasing of Molecular Simulations. Front. Mol. Biosci. 2019, 6, 25. [Google Scholar] [CrossRef]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef]
- Bonomi, M.; Bussi, G.; Camilloni, C.; Tribello, G.A.; Banáš, P.; Barducci, A.; Bernetti, M.; Bolhuis, P.G.; Bottaro, S.; Branduardi, D.; et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar]
- Pfaendtner, J.; Bonomi, M. Efficient Sampling of High-Dimensional Free-Energy Landscapes with Parallel Bias Metadynamics. J. Chem. Theory Comput. 2015, 11, 5062–5067. [Google Scholar] [CrossRef]
- Pfaendtner, J. Metadynamics to Enhance Sampling in Biomolecular Simulations. In Biomolecular Simulations: Methods and Protocols; Bonomi, M., Camilloni, C., Eds.; Springer: New York, NY, USA, 2019; pp. 179–200. [Google Scholar]
- Hovan, L.; Comitani, F.; Gervasio, F.L. Defining an Optimal Metric for the Path Collective Variables. J. Chem. Theory Comput. 2019, 15, 25–32. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Tang, S.; Raymo, F.M. Free-energy predictions and absorption spectra calculations for supramolecular nanocarriers and their photoactive cargo. Nanoscale 2017, 9, 4989–4994. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, A.; Branduardi, D.; Bonomi, M.; Parrinello, M. A chirality-based metrics for free-energy calculations in biomolecular systems. J. Comput. Chem. 2011, 32, 2627–2637. [Google Scholar] [CrossRef] [PubMed]
- Sjöqvist, J.; González-Cano, R.C.; López Navarrete, J.T.; Casado, J.; Ruiz Delgado, M.C.; Linares, M.; Norman, P. A combined MD/QM and experimental exploration of conformational richness in branched oligothiophenes. Phys. Chem. Chem. Phys. 2014, 16, 24841–24852. [Google Scholar] [CrossRef] [PubMed]
- Cacelli, I.; Ferretti, A.; Prampolini, G. Predicting light absorption properties of anthocyanidins in solution: A multi-level computational approach. Theor. Chem. Acc. 2016, 135, 156. [Google Scholar] [CrossRef]
- Northey, T.; Stacey, J.; Penfold, T.J. The role of solid state solvation on the charge transfer state of a thermally activated delayed fluorescence emitter. J. Mater. Chem. C 2017, 5, 11001–11009. [Google Scholar] [CrossRef]
- Cozza, C.; Bonomi, M.; Pietropaolo, A. A Versatile Computational Strategy To Characterize the Free-Energy Landscape of Excited States in Oligofluorenes. J. Chem. Theory Comput. 2018, 14, 5441–5445. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 20603. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patrizi, B.; Cozza, C.; Pietropaolo, A.; Foggi, P.; Siciliani de Cumis, M. Synergistic Approach of Ultrafast Spectroscopy and Molecular Simulations in the Characterization of Intramolecular Charge Transfer in Push-Pull Molecules. Molecules 2020, 25, 430. https://doi.org/10.3390/molecules25020430
Patrizi B, Cozza C, Pietropaolo A, Foggi P, Siciliani de Cumis M. Synergistic Approach of Ultrafast Spectroscopy and Molecular Simulations in the Characterization of Intramolecular Charge Transfer in Push-Pull Molecules. Molecules. 2020; 25(2):430. https://doi.org/10.3390/molecules25020430
Chicago/Turabian StylePatrizi, Barbara, Concetta Cozza, Adriana Pietropaolo, Paolo Foggi, and Mario Siciliani de Cumis. 2020. "Synergistic Approach of Ultrafast Spectroscopy and Molecular Simulations in the Characterization of Intramolecular Charge Transfer in Push-Pull Molecules" Molecules 25, no. 2: 430. https://doi.org/10.3390/molecules25020430
APA StylePatrizi, B., Cozza, C., Pietropaolo, A., Foggi, P., & Siciliani de Cumis, M. (2020). Synergistic Approach of Ultrafast Spectroscopy and Molecular Simulations in the Characterization of Intramolecular Charge Transfer in Push-Pull Molecules. Molecules, 25(2), 430. https://doi.org/10.3390/molecules25020430