Identification and Repurposing of Trisubstituted Harmine Derivatives as Novel Inhibitors of Mycobacterium tuberculosis Phosphoserine Phosphatase

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
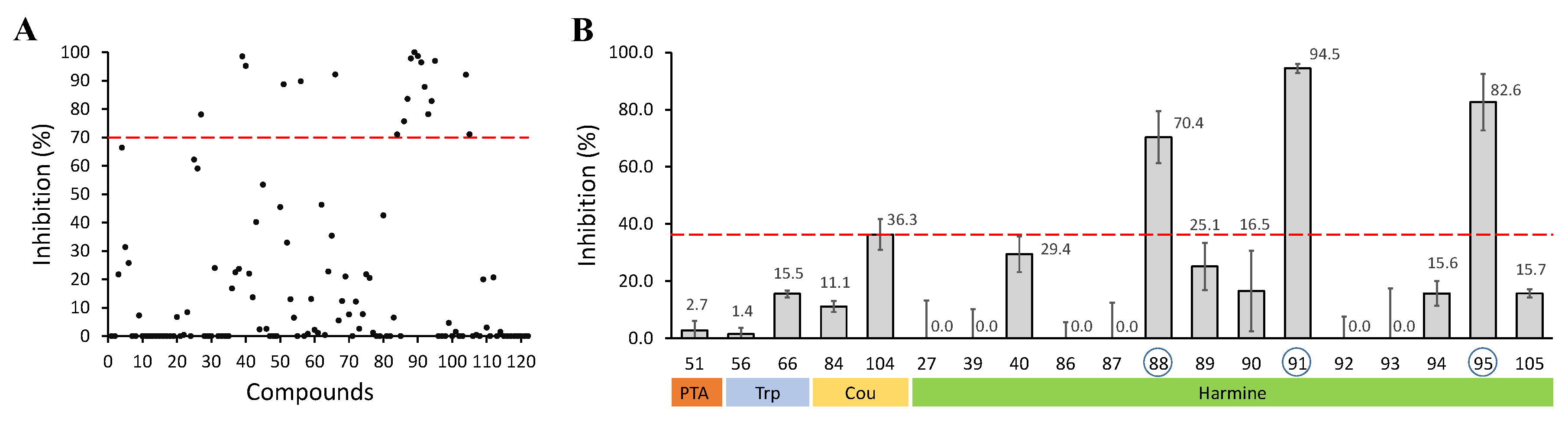
2.1. Screening and Identification of Inhibitors
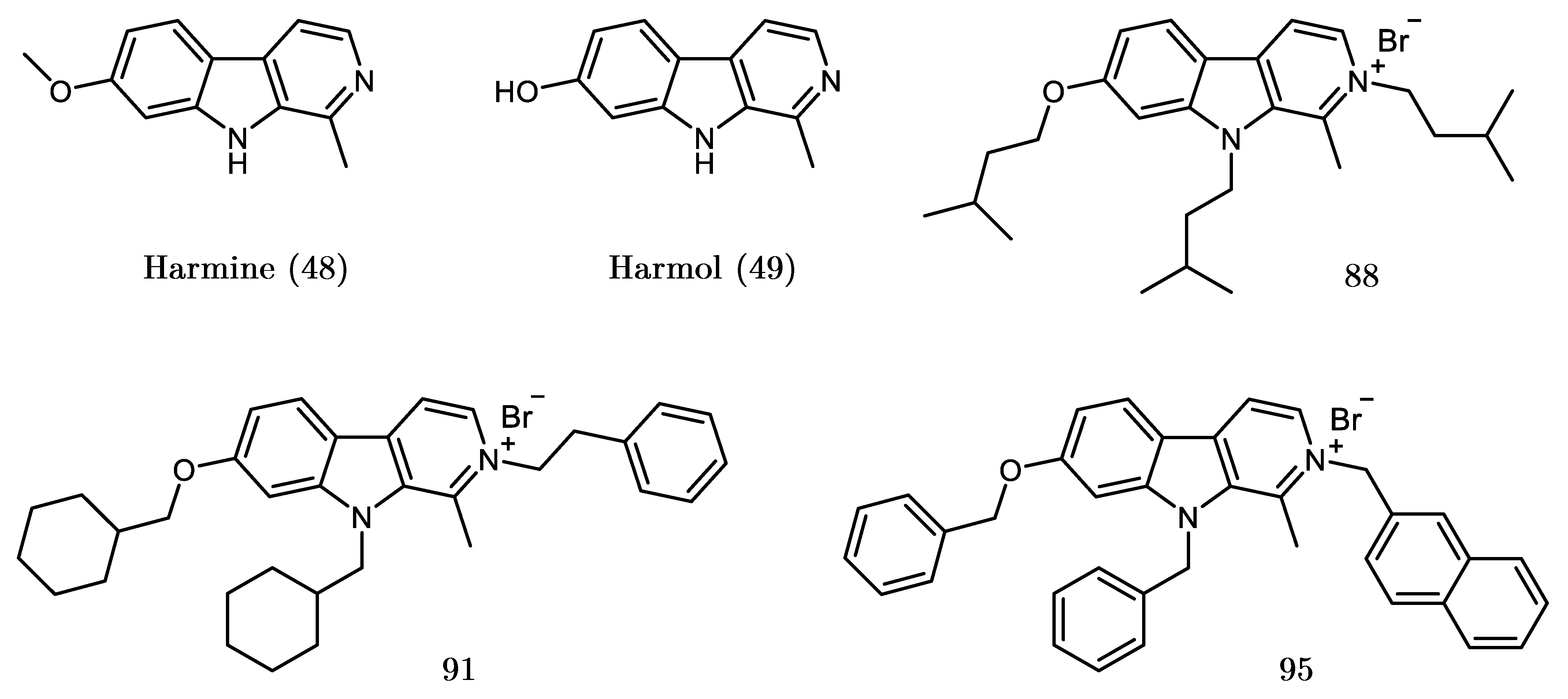
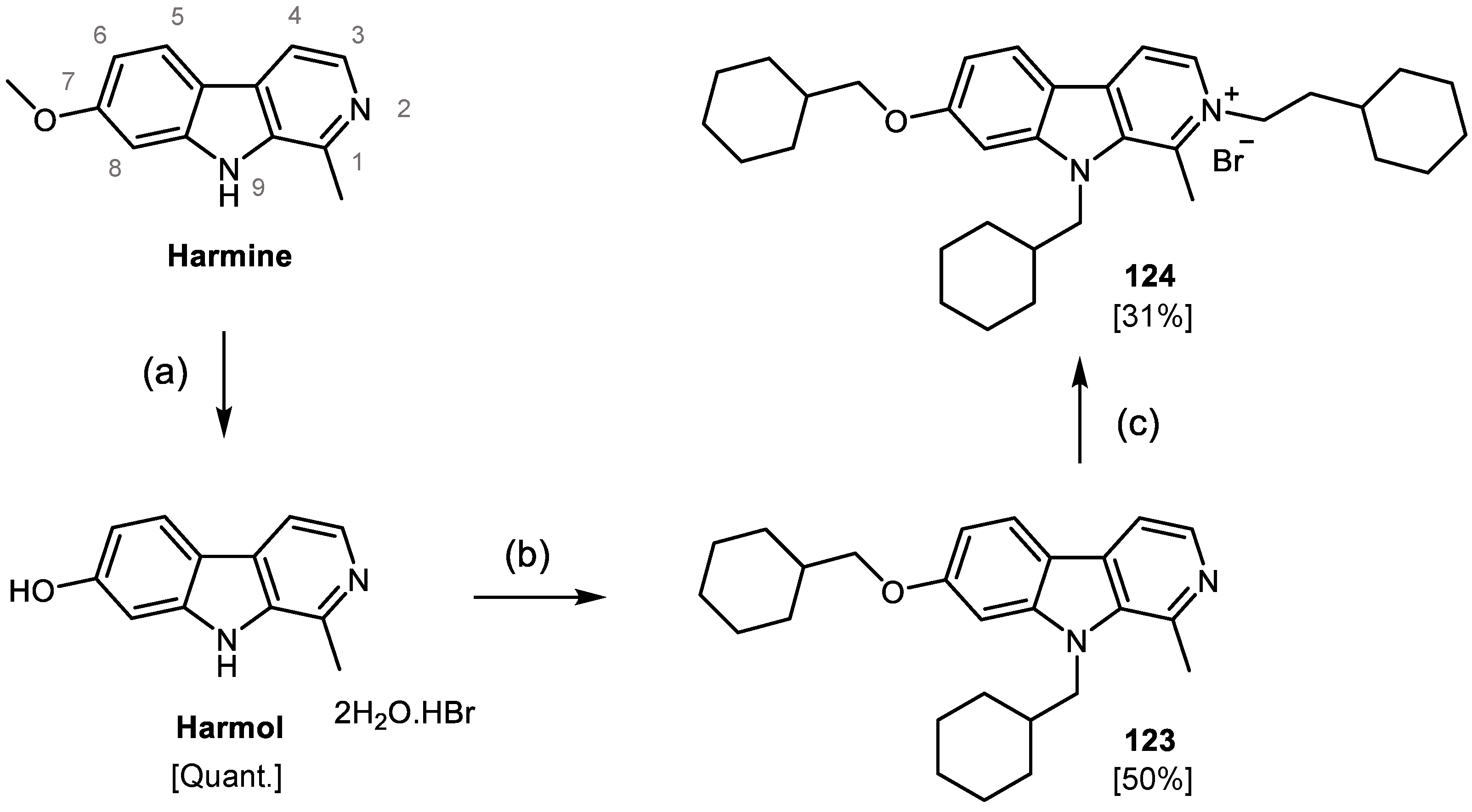
2.2. Design, Synthesis and Evaluation of a New Derivative
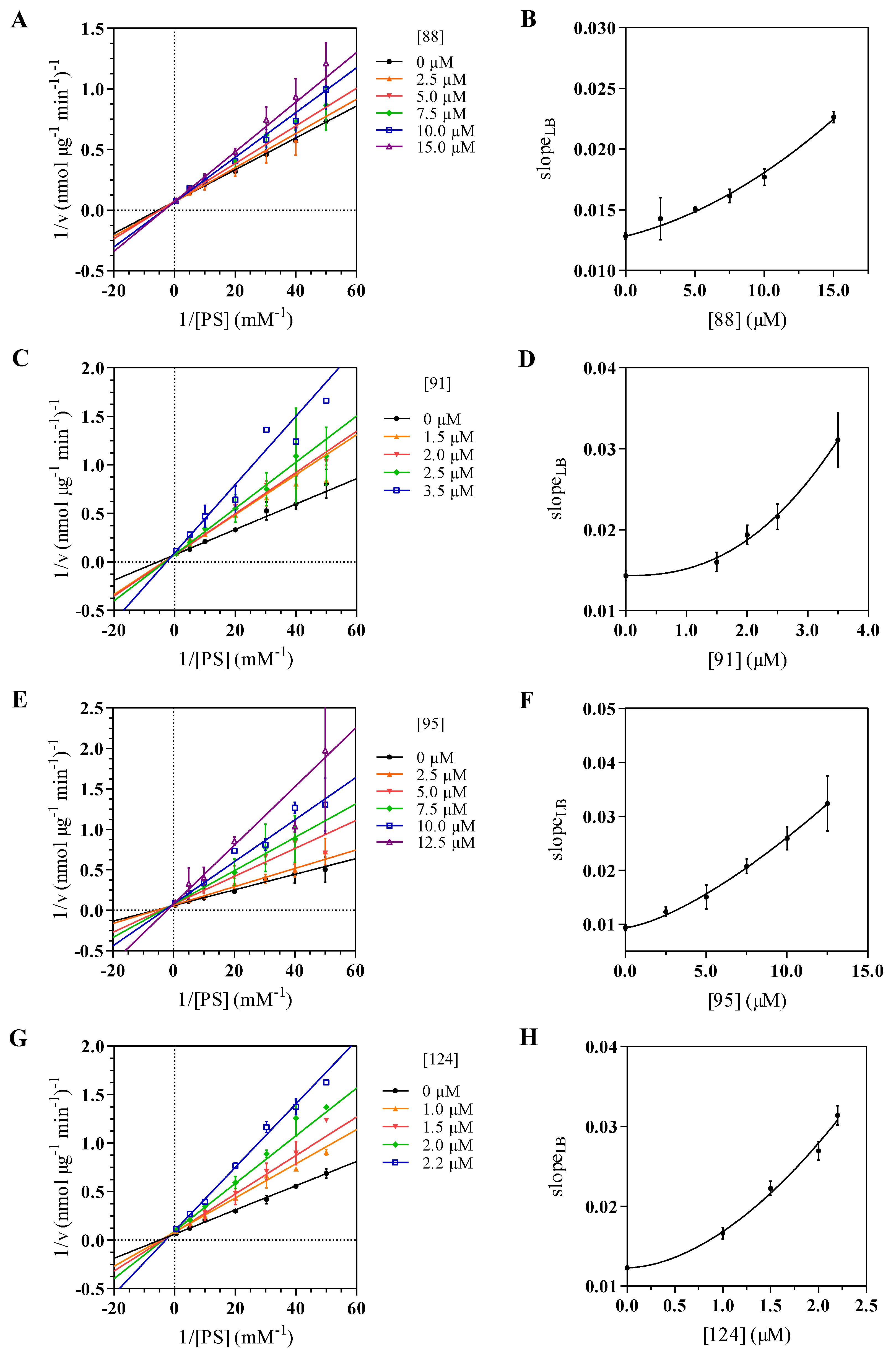
2.3. Kinetics Assay and Determination of the Inhibition Mode
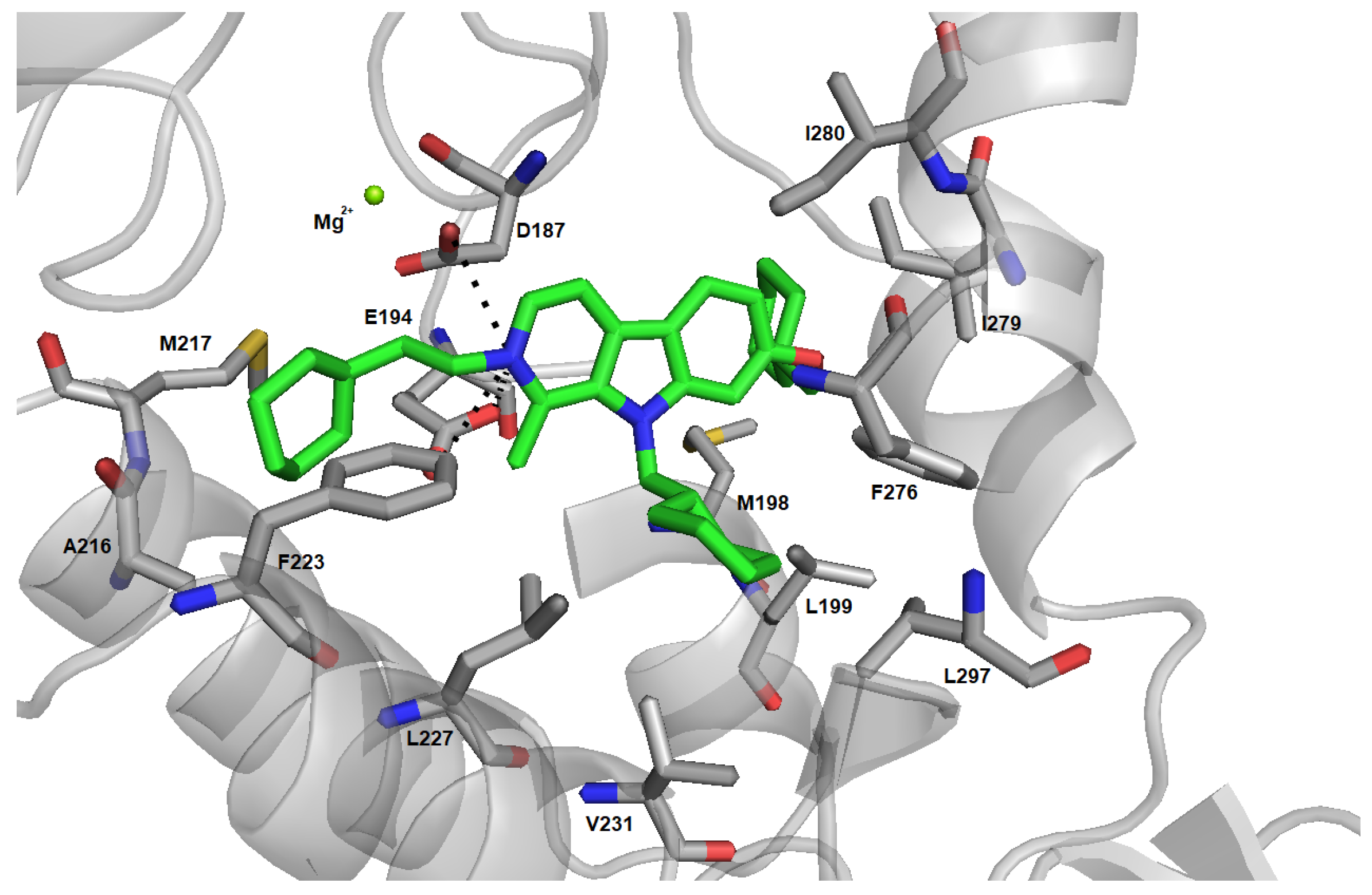
2.4. Induced Fit Docking and Binding Free Energy Calculation
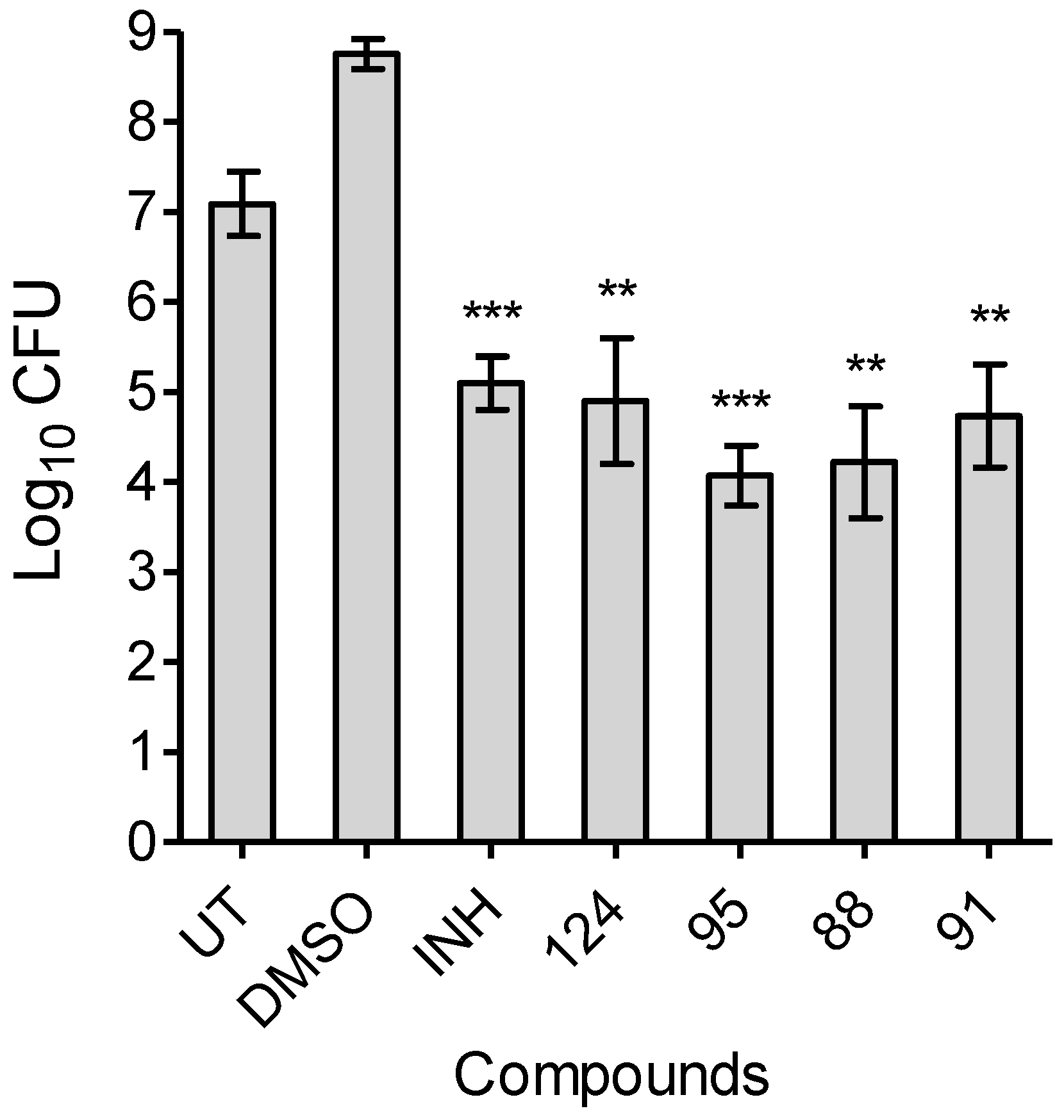
2.5. MIC Determination Assays and Time-Dependent Bactericidal/bacteriostatic Activity
3. Conclusions
4. Materials and Methods
4.1. Expression and Purification of SerB2
4.2. Enzymatic Assay, Screening of Inhibitors and Inhibition Kinetics Studies
4.3. Evaluation of Kinetic Parameters
4.4. Evaluation of Hydrophobic Parameters
4.5. Induced Fit Docking
4.6. MIC Determination Assays
4.7. Time-Dependent Bactericidal/Bacteriostatic Activity of Harmine Derivatives
4.8. Synthesis
4.8.1. General
4.8.2. Synthesis of 1-methyl-9H-pyrido[3,4-b]indol-7-ol hydrochloride dihydrate (Harmol)
4.8.3. 7-(cyclohexylmethoxy)-9(cyclohexylmethyl)-1-methyl--carboline (123)
4.8.4. 7-(cyclohexylmethoxy)-9-(cyclohexylmethyl)-1-methyl-2-(2-cyclohexylethyl)--carboline -2-ium bromide (124)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CFU | Colony Forming Unit |
| DCM | Dichloromethane |
| DMF | N,N-Dimethylformamide |
| HRV 3C | Human rhinovirus 3C |
| IMAC | Immobilized metal affinity chromatography |
| INH | isoniazid |
| inhibition constant | |
| MIC | minimal inhibitory concentration |
| Mtb | Mycobacterium tuberculosis |
| PDB | Protein Data Bank |
| Pi | inorganic phosphate |
| PS | O-phospho-L-serine |
| TB | Tuberculosis |
| THF | Tetrahydrofuran |
References
- World Health Organization. Global Tuberculosis Report 2018. Available online: https://www.who.int/ (accessed on 24 September 2019).
- The Global Alliance for TB Drug Development. Available online: https://www.tballiance.org/ (accessed on 2 October 2019).
- Lytras, T.; Kalkouni, O. The global tuberculosis epidemic: Turning political will into concrete action. J. Thorac. Dis. 2018, 10, S3149–S3152. [Google Scholar] [CrossRef]
- Floyd, K.; Glaziou, P.; Zumla, A.; Raviglione, M. The global tuberculosis epidemic and progress in care, prevention, and research: An overview in year 3 of the End TB era. Lancet Respir. Med. 2018, 6, 299–314. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.; Eiglmeier, K.; Gas, S.; Barry, C., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- Singh, V.; Donini, S.; Pacitto, A.; Sala, C.; Hartkoorn, R.C.; Dhar, N.; Keri, G.; Ascher, D.B.; Mondesert, G.; Vocat, A.; et al. The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect. Dis. 2016, 3, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.U.; Singh, V.; Ferraris, D.M.; Rizzi, M.; Kharkar, P.S. Hit discovery of Mycobacterium tuberculosis inosine 5-monophosphate dehydrogenase, GuaB2, inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1714–1718. [Google Scholar] [CrossRef]
- Burke, C.; Abrahams, K.A.; Richardson, E.J.; Loman, N.J.; Alemparte, C.; Lelievre, J.; Besra, G.S. Development of a whole-cell high-throughput phenotypic screen to identify inhibitors of mycobacterial amino acid biosynthesis. FASEB BioAdv. 2019, 1, 246–254. [Google Scholar] [CrossRef]
- Berney, M.; Berney-Meyer, L.; Wong, K.W.; Chen, B.; Chen, M.; Kim, J.; Wang, J.; Harris, D.; Parkhill, J.; Chan, J.; et al. Essential roles of methionine and S-adenosylmethionine in the autarkic lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 10008–10013. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, V.; Warner, D.F. Death of Mycobacterium tuberculosis by L-arginine starvation. Proc. Natl. Acad. Sci. USA 2018, 115, 9658–9660. [Google Scholar] [CrossRef] [PubMed]
- Haufroid, M.; Wouters, J. Targeting the Serine Pathway: A Promising Approach against Tuberculosis? Pharmaceuticals 2019, 12, 66. [Google Scholar] [CrossRef]
- Lowry, M.; Hall, D.E.; Hall, M.S.; Brosnan, J.T. Renal metabolism of amino acids in vivo: Studies on serine and glycine fluxes. Am. J. Physiol.-Ren. Physiol. 1987, 252, F304–F309. [Google Scholar] [CrossRef] [PubMed]
- Snell, K. The duality of pathways for serine biosynthesis is a fallacy. Trends Biochem. Sci. 1986, 11, 241–243. [Google Scholar] [CrossRef]
- Ros, R.; Muñoz-Bertomeu, J.; Krueger, S. Serine in plants: Biosynthesis, metabolism, and functions. Trends Plant Sci. 2014, 19, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Schaak, D.D.; Smith, E.A.; McDonough, K.A. Dysregulation of serine biosynthesis contributes to the growth defect of a Mycobacterium tuberculosis crp mutant. Mol. Microbiol. 2011, 82, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Hu, Z.; Xu, X.L.; Sacchettini, J.C.; Grant, G.A. D-3-Phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 2005, 280, 14884–14891. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, F.; Lassalle, E.; Baker, H.M.; Baker, E.N. Structure of phosphoserine aminotransferase from Mycobacterium tuberculosis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 553–563. [Google Scholar] [CrossRef]
- Yadav, G.P.; Shree, S.; Maurya, R.; Rai, N.; Singh, D.K.; Srivastava, K.K.; Ramachandran, R. Characterization of M. tuberculosis SerB2, an essential HAD-family phosphatase, reveals novel properties. PLoS ONE 2014, 9, e115409. [Google Scholar] [CrossRef]
- Shree, S.; Singh, A.K.; Saxena, R.; Kumar, H.; Agarwal, A.; Sharma, V.K.; Srivastava, K.; Srivastava, K.K.; Sanyal, S.; Ramachandran, R. The M. tuberculosis HAD phosphatase (Rv3042c) interacts with host proteins and is inhibited by Clofazimine. Cell. Mol. Life Sci. 2016, 73, 3401–3417. [Google Scholar] [CrossRef]
- Anishetty, S.; Pulimi, M.; Pennathur, G. Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput. Biol. Chem. 2005, 29, 368–378. [Google Scholar] [CrossRef]
- Arora, G.; Tiwari, P.; Mandal, R.S.; Gupta, A.; Sharma, D.; Saha, S.; Singh, R. High throughput screen identifies small molecule inhibitors specific for Mycobacterium tuberculosis phosphoserine phosphatase. J. Biol. Chem. 2014, 289, 25149–25165. [Google Scholar] [CrossRef]
- Marx, S.; Van Gysel, M.; Breuer, A.; Dal Maso, T.; Michiels, C.; Wouters, J.; Le Calvé, B. Potentialization of anticancer agents by identification of new chemosensitizers active under hypoxia. Biochem. Pharmacol. 2019, 162, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Axelsson-Robertson, R.; Rao, M.; Singh, N.; Master, I.; Lutckii, A.; Keshavjee, S.; Andersson, J.; Zumla, A.; Maeurer, M. Totally drug-resistant tuberculosis and adjunct therapies. J. Intern. Med. 2015, 277, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Bates, S.; Kolvekar, T.; Devarajan, P.V.; Guzman, J.D.; Bhakta, S. Repurposing—A ray of hope in tackling extensively drug resistance in tuberculosis. Int. J. Infect. Dis. 2015, 32, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Bates, S.; Shaik, M.; Evangelopoulos, D.; Abubakar, I.; McHugh, T.D.; Lipman, M.; Bhakta, S. Repurposing drugs for treatment of tuberculosis: A role for non-steroidal anti-inflammatory drugs. Br. Med. Bull. 2016, 118, 138. [Google Scholar] [CrossRef] [PubMed]
- Harausz, E.P.; Garcia-Prats, A.J.; Seddon, J.A.; Schaaf, H.S.; Hesseling, A.C.; Achar, J.; Bernheimer, J.; Cruz, A.T.; D’Ambrosio, L.; Detjen, A.; et al. New and repurposed drugs for pediatric multidrug-resistant tuberculosis. Practice-based recommendations. Am. J. Respir. Crit. Care Med. 2017, 195, 1300–1310. [Google Scholar] [CrossRef]
- Gopal, M.; Padayatchi, N.; Metcalfe, J.; O’Donnell, M. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2013, 17, 1001–1007. [Google Scholar] [CrossRef]
- Falzon, D.; Schünemann, H.J.; Harausz, E.; González-Angulo, L.; Lienhardt, C.; Jaramillo, E.; Weyer, K. World Health Organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur. Respir. J. 2017, 49, 1602308. [Google Scholar] [CrossRef]
- Silva, D.R.; Dalcolmo, M.; Tiberi, S.; Arbex, M.A.; Munoz-Torrico, M.; Duarte, R.; D’Ambrosio, L.; Visca, D.; Rendon, A.; Gaga, M.; et al. New and repurposed drugs to treat multidrug-and extensively drug-resistant tuberculosis. J. Bras. Pneumol. 2018, 44, 153–160. [Google Scholar] [CrossRef]
- Tonby, K.; Wergeland, I.; Lieske, N.V.; Kvale, D.; Tasken, K.; Dyrhol-Riise, A.M. The COX-inhibitor indomethacin reduces Th1 effector and T regulatory cells in vitro in Mycobacterium tuberculosis infection. BMC Infect. Dis. 2016, 16, 599. [Google Scholar] [CrossRef]
- Kroesen, V.M.; Gröschel, M.I.; Martinson, N.; Zumla, A.; Maeurer, M.; van der Werf, T.S.; Vilaplana, C. Non-steroidal anti-inflammatory drugs as host-directed therapy for tuberculosis: A systematic review. Front. Immunol. 2017, 8, 772. [Google Scholar] [CrossRef]
- Gold, B.; Pingle, M.; Brickner, S.J.; Shah, N.; Roberts, J.; Rundell, M.; Bracken, W.C.; Warrier, T.; Somersan, S.; Venugopal, A.; et al. Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proc. Natl. Acad. Sci. USA 2012, 109, 16004–16011. [Google Scholar] [CrossRef] [PubMed]
- Ivanyi, J.; Zumla, A. Nonsteroidal Antiinflammatory Drugs for Adjunctive Tuberculosis Treatment. J. Infect. Dis. 2013, 208, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Chen, Q.; Hou, X.; Chen, H.; Guan, H.; Ma, Y.; Peng, W.; Xu, A. Synthesis, acute toxicities, and antitumor effects of novel 9-substituted β-carboline derivatives. Bioorg. Med. Chem. 2004, 12, 4613–4623. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Gadewar, M.; Tripathi, R.; Prasad, S.; Patel, D.K. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid “Harmine”. Asian Pac. J. Trop. Biomed. 2012, 2, 660–664. [Google Scholar] [CrossRef]
- Song, H.; Liu, Y.; Liu, Y.; Wang, L.; Wang, Q. Synthesis and antiviral and fungicidal activity evaluation of β-carboline, dihydro-β-carboline, tetrahydro-β-carboline alkaloids, and their derivatives. J. Agric. Food Chem. 2014, 62, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. β-Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef]
- Begum, S.; Ali, S.N.; Siddiqui, B.S. Antitubercular Alkaloid. U.S. Patent 8420660 B2, 16 April 2013. [Google Scholar]
- Davoodi, H.; Ghaemi, E.; Mazandarani, M.; Shakeri, F.; Javid, S.; Klishadi, M. Anti-mycobacterial and anti-inflammatory activity of Peganum harmala. Chem. Pharm. Res. 2015, 7, 1611–1616. [Google Scholar]
- Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Synthesis and evaluation of β-carboline derivatives as potential monoamine oxidase inhibitors. Bioorg. Med. Chem. 2011, 19, 134–144. [Google Scholar] [CrossRef]
- Frédédrick, R.; Bruyere, C.; Vancraeynest, C.; Reniers, J.; Meinguet, C.; Pochet, L.; Backlund, A.; Masereel, B.; Kiss, R.; Wouters, J. Novel trisubstituted harmine derivatives with original in vitro anticancer activity. J. Med. Chem. 2012, 55, 6489–6501. [Google Scholar] [CrossRef]
- Meinguet, C.; Bruyère, C.; Frédérick, R.; Mathieu, V.; Vancraeynest, C.; Pochet, L.; Laloy, J.; Mortier, J.; Wolber, G.; Kiss, R.; et al. 3D-QSAR, design, synthesis and characterization of trisubstituted harmine derivatives with in vitro antiproliferative properties. Eur. J. Med. Chem. 2015, 94, 45–55. [Google Scholar] [CrossRef]
- Meinguet, C.; Masereel, B.; Wouters, J. Preparation and characterization of a new harmine-based antiproliferative compound in complex with cyclodextrin: Increasing solubility while maintaining biological activity. Eur. J. Pharmaceut. Sci. 2015, 77, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Chu, J.; Meinguet, C.; Kiss, R.; Vandenbussche, G.; Masereel, B.; Wouters, J.; Kornienko, A.; Pelletier, J.; Mathieu, V. A harmine-derived beta-carboline displays anti-cancer effects in vitro by targeting protein synthesis. Eur. J. Pharmacol. 2017, 805, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.; Bodart, L.; Tumanov, N.; Wouters, J. Design and Synthesis of a New Soluble Natural β-Carboline Derivative for Preclinical Study by Intravenous Injection. Int. J. Mol. Sci. 2019, 20, 1491. [Google Scholar] [CrossRef] [PubMed]
- Itaya, K.; Ui, M. A new micromethod for the colorimetric determination of inorganic phosphate. Clin. Chim. Acta 1966, 14, 361–366. [Google Scholar] [CrossRef]
- Baykov, A.; Evtushenko, O.; Avaeva, S. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 1988, 171, 266–270. [Google Scholar] [CrossRef]
- Fujita, T.; Iwasa, J.; Hansch, C. A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar] [CrossRef]
- Leskovac, V. Hyperbolic and parabolic inhibition. In Comprehensive Enzyme Kinetics; Kluwer: New York, NY, USA, 2003; pp. 95–110. [Google Scholar]
- Segel, I. Simple inhibition systems. In Enzyme Kinetics: Behaviour and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; John Wiley & Sons: Hoboken, NJ, USA, 1993; pp. 100–160. [Google Scholar]
- Cao, R.; Zeidan, A.A.; Rådström, P.; van Niel, E.W. Inhibition kinetics of catabolic dehydrogenases by elevated moieties of ATP and ADP—Implication for a new regulation mechanism in Lactococcus lactis. FEBS J. 2010, 277, 1843–1852. [Google Scholar] [CrossRef]
- Pala, D.; Rivara, S.; Mor, M.; Milazzo, F.M.; Roscilli, G.; Pavoni, E.; Giannini, G. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology 2016, 26, 640–654. [Google Scholar] [CrossRef]
- Hammond, E.; Handley, P.; Dredge, K.; Bytheway, I. Mechanisms of heparanase inhibition by the heparan sulfate mimetic PG545 and three structural analogues. FEBS Open Bio 2013, 3, 346–351. [Google Scholar] [CrossRef]
- Abendroth, J.; Gardberg, A.S.; Robinson, J.I.; Christensen, J.S.; Staker, B.L.; Myler, P.J.; Stewart, L.J.; Edwards, T.E. SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genomics 2011, 12, 83–95. [Google Scholar] [CrossRef]
- Haufroid, M.; Mirgaux, M.; Leherte, L.; Wouters, J. Crystal structures and snapshots along the reaction pathway of human phosphoserine phosphatase. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Heo, Y.S.; Kim, J.H.; Park, M.H.; Moon, J.; Kim, E.; Kwon, D.; Yoon, J.; Shin, D.; Jeong, E.J.; et al. Molecular basis for the local conformational rearrangement of human phosphoserine phosphatase. J. Biol. Chem. 2002, 277, 46651–46658. [Google Scholar] [CrossRef] [PubMed]
- Peeraer, Y.; Rabijns, A.; Verboven, C.; Collet, J.F.; Van Schaftingen, E.; De Ranter, C. High-resolution structure of human phosphoserine phosphatase in open conformation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.; Singh, R. Recent advances for identification of new scaffolds and drug targets for Mycobacterium tuberculosis. IUBMB Life 2018, 70, 905–916. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2015, 12, 281–296. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein- ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.C.; Wolf, R.M. MM/GBSA binding energy prediction on the PDBbind data set: Successes, failures, and directions for further improvement. J. Chem. Inf. Model. 2012, 53, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef] [PubMed]
- Sirin, S.; Kumar, R.; Martinez, C.; Karmilowicz, M.J.; Ghosh, P.; Abramov, Y.A.; Martin, V.; Sherman, W. A computational approach to enzyme design: Predicting ω-aminotransferase catalytic activity using docking and MM-GBSA scoring. J. Chem. Inf. Model. 2014, 54, 2334–2346. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Set | Compound | R/R | R | (R) | Inhibition at 10 μM (%) |
|---|---|---|---|---|---|
| A | 39 |  π(R7)/π(R9) 2.13/1.67 |  | −0.02 | 0 ± 10.2 |
| 86 |  | −2.25 | 0 ± 5.6 | ||
| 92 |  | 0.03 | 0 ± 7.6 | ||
| 94 |  | 0.19 | 15.6 ± 4.3 | ||
| 95 |  | 1.17 | 82.6 ± 9.9 | ||
| B | 87 |  π(R7)/π(R9) 2.19/1.72 |  | −2.25 | 0 ± 12.4 |
| 88 |  | 0.03 | 70.4 ± 9.1 | ||
| 93 | - | 2.80 | 0 ± 17.4 | ||
| C | 89 |  π(R7)/π(R9) 2.81/2.36 |  | −2.25 | 25.1 ± 8.2 |
| 90 |  | 0.03 | 16.5 ± 13.3 | ||
| 91 |  | 0.19 | 94.5 ± 1.4 |
| Compound | (μM) | n | R | (kcal/mol) | MIC(μM) |
|---|---|---|---|---|---|
| 88 | 18.3 ± 2.3 | 1.45 ± 0.38 | 0.985 | 6.3 | |
| 91 | 3.27 ± 0.12 | 2.39 ± 0.45 | 0.996 | 1.5 | |
| 95 | 6.66 ± 0.39 | 1.42 ± 0.16 | 0.998 | 3.6 | |
| 124 | 1.75 ± 0.11 | 1.78 ± 0.53 | 0.993 | 0.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierson, E.; Haufroid, M.; Gosain, T.P.; Chopra, P.; Singh, R.; Wouters, J. Identification and Repurposing of Trisubstituted Harmine Derivatives as Novel Inhibitors of Mycobacterium tuberculosis Phosphoserine Phosphatase. Molecules 2020, 25, 415. https://doi.org/10.3390/molecules25020415
Pierson E, Haufroid M, Gosain TP, Chopra P, Singh R, Wouters J. Identification and Repurposing of Trisubstituted Harmine Derivatives as Novel Inhibitors of Mycobacterium tuberculosis Phosphoserine Phosphatase. Molecules. 2020; 25(2):415. https://doi.org/10.3390/molecules25020415
Chicago/Turabian StylePierson, Elise, Marie Haufroid, Tannu Priya Gosain, Pankaj Chopra, Ramandeep Singh, and Johan Wouters. 2020. "Identification and Repurposing of Trisubstituted Harmine Derivatives as Novel Inhibitors of Mycobacterium tuberculosis Phosphoserine Phosphatase" Molecules 25, no. 2: 415. https://doi.org/10.3390/molecules25020415
APA StylePierson, E., Haufroid, M., Gosain, T. P., Chopra, P., Singh, R., & Wouters, J. (2020). Identification and Repurposing of Trisubstituted Harmine Derivatives as Novel Inhibitors of Mycobacterium tuberculosis Phosphoserine Phosphatase. Molecules, 25(2), 415. https://doi.org/10.3390/molecules25020415