3. Materials and Methods
All reactions were carried out in oven-dried glassware under an argon atmosphere. Flash column chromatography was performed with silica gel 60 (230–400 mesh ASTM, Merck, Darmstadt, Germany). TLC analysis was performed on Merck 0.25 mm Silicagel 60 F254 plates. Melting points were determined on a hot stage microscope apparatus (ATM-01, AS ONE, Osaka, Japan) and were uncorrected. NMR spectra were recorded on a JEOLRESONANCE EXC-400 or ECX-500 spectrometer (JEOL, Akishima, Japan) operating at 400 MHz or 500 MHz for 1H-NMR, and 100 MHz and 125 MHz for 13C NMR. Chemical shifts (δ ppm) in CDCl3 were reported downfield from TMS (=0) for 1H-NMR. For 13C-NMR, chemical shifts were reported in the scale relative to CDCl3 (77.00 ppm) as an internal reference. Mass spectra were measured on a JMS-T100LC spectrometer (JEOL, Akishima, Japan). HPLC data were obtained on a SHIMADZU (Kyoto, Japan) HPLC system (consisting of the following: LC-20AT, CMB20A, CTO-20AC, and detector SPD-20A measured at 254 nm) using Chiracel AD-H or Ad-3 column (Daicel, Himeji, Japan, 25 cm) at 25 °C. Optical rotations were measured on a JASCO DIP-370 (Na lamp, 589 nm).
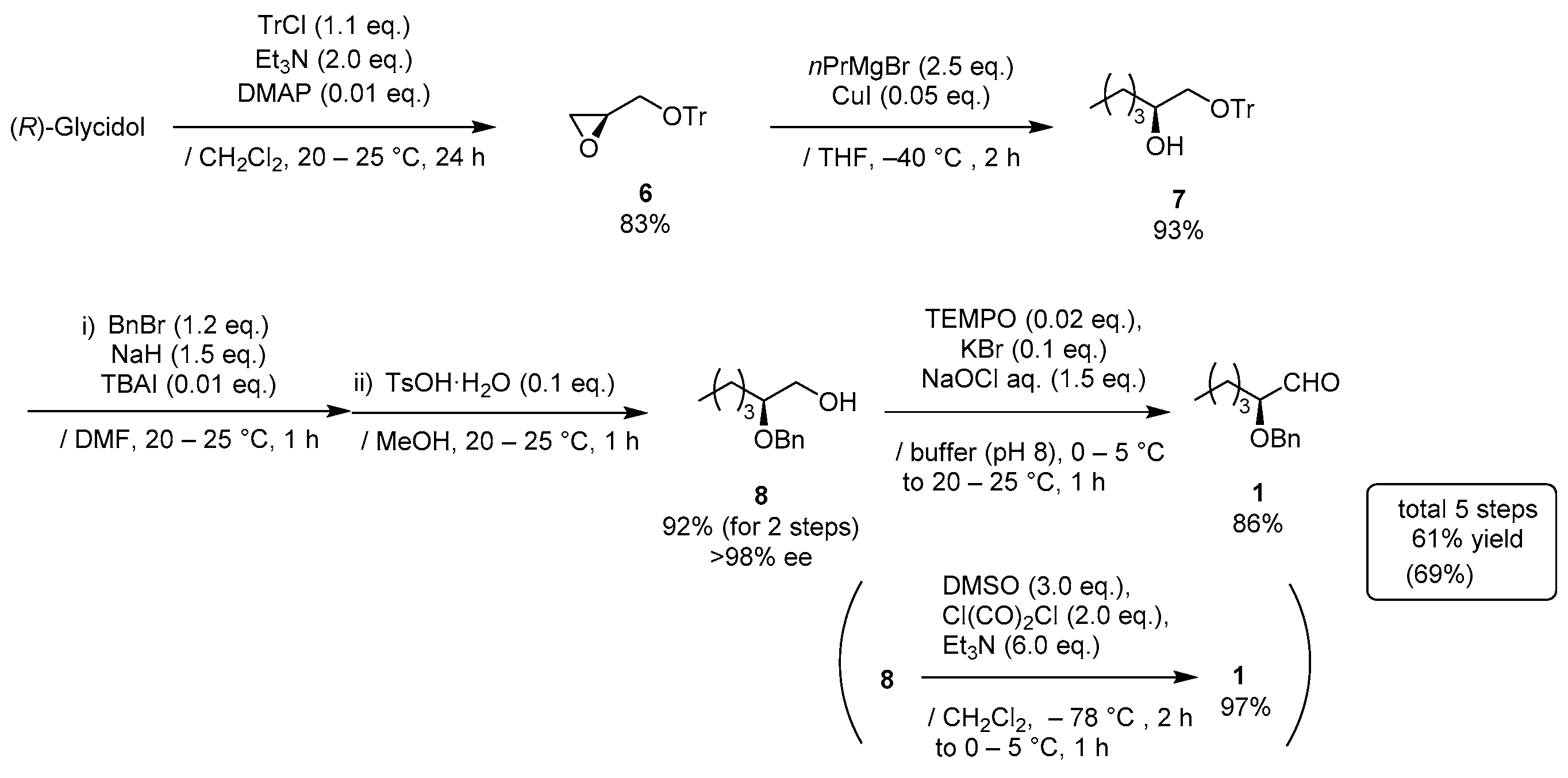
(R)-2-((trityloxy)methyl)oxirane (6)
TrCl (15.3 g, 55 mmol) in CH2Cl2 (35 mL) was added to a stirred solution of (R)-(+)-glycidol (3.70 g, 50 mmol) and Et3N (13.9 mL, 100 mmol) and DMAP (61 mg, 0.5 mmol) in CH2Cl2 (15 mL) at 0–5 °C under an Ar atmosphere, followed by stirring at 20–25 °C for 24 h. The mixture was quenched with sat. NH4Cl aq., which was extracted three times with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude solid was purified by recrystallization from MeOH (100 mL) to give the desired product 6 (13.1 g, 83%).
Colorless crystals, mp 99–100 °C [lit. [
25], 100 °C (EtOH)];
1H-NMR (400 MHz, CDCl
3): δ = 2.63 (dd,
J = 2.3 Hz, 5.0 Hz, 1H), 2.78 (dd,
J = 4.6, 1H), 3.09–3.18 (m, 2H), 3.32 (dd,
J = 2.3 Hz, 10.0 Hz, 1H), 7.20–7.35 (m, 10H), 7.42–7.50 (m, 5H);
13C-NMR (100 MHz, CDCl
3): δ = 44.6, 51.0, 64.7, 86.6, 127.0 (3C), 127.8 (6C), 128.6 (6C), 143.8.
(S)-1-(Trityloxy)hexan-2-ol (7)
1-Bromopropane (8.60 mL, 95 mmol) was gradually added to a stirred Mg granular (2.31 g, 95 mmol) and a small amounts of I2 in THF (60 mL) at 20–25 °C under an Ar atmosphere, and the mixture was stirred for 0.5 h at 20–25 °C. CuI (143 mg, 0.80 mmol) was added, the mixture was cooled down to −40 °C and (S)-oxirane 6 (12.1 g, 38 mmol) in THF (100 mL) was added to the mixture at the same temperature, followed by stirring for 2 h. The mixture was quenched with sat. NH4Cl aq., which was extracted three times with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2–column chromatography (hexane/AcOEt = 15/1) to give the desired alcohol 7 (12.7 g, 93%).
Pale yellow oil; 1H-NMR (400 MHz, CDCl3): δ = 0.86 (t, J = 6.9 Hz, 3H), 1.16–1.46 (m, 6H), 2.30 (d, J = 3.7 Hz, 1H), 3.02 (dd, J = 7.8 Hz, 9.2 Hz, 1H), 3.18 (dd, J = 3.2 Hz, 9.2 Hz, 1H), 3.72–3.80 (m, 1H), 7.19–7.35 (m, 10H), 7.40–7.47 (m, 5H); 13C-NMR (100 MHz, CDCl3): δ = 13.9, 22.6, 27.6, 33.0, 67.7, 70.9, 86.6, 127.0 (3C), 127.8 (6C), 128.6 (6C), 143.8.
(
S)-2-(Benzyloxy)hexan-1-ol (
8) [
15]
A mixture of benzyl bromide (4.85 mL, 41 mmol) and (S)-alcohol 7 (12.4 g, 34 mmol) in DMF (25 mL) were added to a stirred suspension of NaH (60%; 2.04 mg, 51 mmol) in DMF (10 mL) at 0–5 °C under an Ar atmosphere. TBAI (126 mg, 0.3 mmol) was added to the mixture and the mixture was allowed to warm up to 20–25 °C, followed by stirring for 1 h. The mixture was quenched with MeOH and K2CO3, which was extracted three times with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude oil (15.6 g) was used for the next step without purification.
TsOH·H2O (647 mg, 3.4 mmol) was added to a solution of the oil (15.6 g) in MeOH (70 mL) at 20–25 °C under an Ar atmosphere, and the mixture was stirred for 1 h at the same temperature. The mixture was quenched with sat. NaHCO3 aq. and concentrated, which was extracted three times with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2–column chromatography (hexane/AcOEt = 15:1–3:1) to give 8 (6.52 g, 92% for 2 steps, >98% ee).
Yellow oil;
+21.4 (
c 1.16, CHCl
3) [lit. [
15], [α
+22.3 (
c 1.13, CHCl
3)];
1H-NMR (400 MHz, CDCl
3): δ = 0.90 (t,
J = 6.9 Hz, 3H), 1.23–1.40 (m, 4H), 1.44–1.71 (m, 2H), 1.93 (brs, 1H), 3.47–3.58 (m, 2H), 3.65–3.75 (m, 1H), 4.54 (d,
J = 11.5 Hz, 1H), 4.63 (d,
J = 11.5 Hz, 1H), 7.27–7.39 (m, 5H);
13C NMR (500 MHz, CDCl
3): δ = 13.8, 22.6, 27.3, 30.3, 63.9, 71.3, 79.7, 127.4, 127.6 (2C), 128.2 (2C), 138.3. HPLC analysis (AD-H, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 30/1) t
R(racemic) = 9.33 min and 10.27 min. t
R[(
S)-form] = 8.95 min.
(
S)-2-(Benzyloxy)hexanal (
1) [
15]
TEMPO (106 mg, 0.68 mmol) and KBr (407 mg, 3.4 mmol) was added to a stirred solution of alcohol 8 (7.08 g, 34 mmol) in CH2Cl2 (34 mL) at 0–5 °C under an Ar atmosphere. A mixture of NaOCl aq. (1.5 M, 34 mL, 51 mmol), NaHCO3 (6.7 g, 80 mmol), and Na2CO3 (318 mg, 3 mmol) in water (220 mL), was added to the solution at same temperature. The mixture was allowed to warm to 20–25 °C, followed by stirring at the same temperature for 1 h. The mixture was quenched with water, which was extracted twice with CH2Cl2. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by Florisil® column chromatography (hexane/AcOEt = 5:1) to give the desired product 1 (6.04 g, 86%).
Yellow oil; [α
−81.2 (
c 1.08, CHCl
3) [lit. [
15], [α
−86.1 (
c 0.98, CHCl
3)];
1H-NMR (500 MHz, CDCl
3): δ = 0.90 (t,
J = 7.5 Hz, 3H), 1.24–1.49 (m, 4H), 1.69 (q,
J = 6.9 Hz, 13.8 Hz, 2H), 3.76 (t,
J = 6.3 Hz, 1H), 4.54 (d,
J = 11.5 Hz, 1H), 4.68 (d,
J = 11.5 Hz, 1H), 7,27–7.41 (m, 5H), 9.66 (s, 1H);
13C NMR (125 MHz, CDCl
3): δ = 13.7, 22.3, 26.7, 29.6, 72.3, 83.3, 127.8, 127.9, 128.4, 137.3, 203.6.
An alternative method is following:
DMSO (4.26 mL, 60 mmol) in CH2Cl2 (20 mL) was added slowly to a stirred solution of oxalyl dichloride (3.43 mL, 40 mmol) in CH2Cl2 (60 mL) at −78 °C under an Ar atmosphere. After the mixture was stirred for 5 min, 8 (4.22 g, 20 mmol) in CH2Cl2 (20 mL) was added and the mixture was stirred for 0.5 h at the same temperature. Et3N (16.6 mL, 120 mmol) was added to the mixture and the mixture was allowed to warm up to 0–5 °C over a period of 1 h, followed by stirring for 1 h at 0–5 °C. The mixture was quenched with water, which was extracted three times with Et2O. The combined organic phase was washed with a large amounts of water, brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2–column chromatography (hexane/AcOEt = 25/1) to give the desired product 1 (3.99 g, 97%).
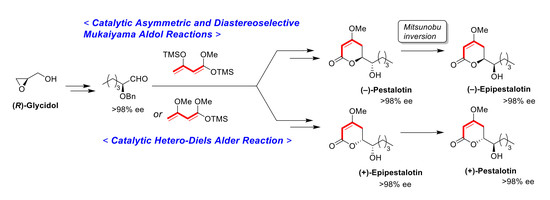
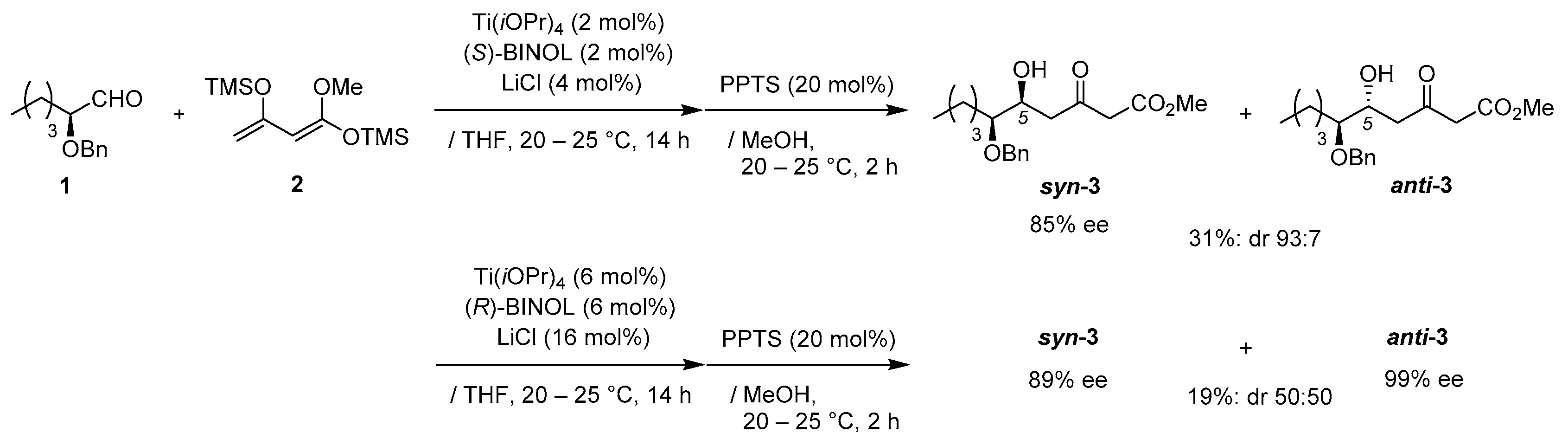
Methyl (5
S,6
S)-6-(benzyloxy)-5-hydroxy-3-oxodecanoate (
syn-
3) [
15]
Preparation for Ti-BINOL solution: A suspension of Ti(OiPr)4 (2.9 mg, 10 μmol), and (S)-BINOL (2.8 mg, 10 μmol) in THF (0.4 mL) was stirred stirred at 20–25 °C under an Ar atmosphere for 1 h.
Asymmetric Mukaiyama aldol reaction: Ti-BINOL solution was added to a stirred suspension of aldehyde 1 (103 mg, 0.50 mmol) and LiCl (0.85 mg, 20 μmol) in THF (0.5 mL) at 20–25 °C under an Ar atmosphere, followed by stirring at the same temperature for 0.5 h. Chan’s diene 2 (260 mg, 1.0 mmol) in THF (0.3 mL) was added slowly to the mixture, which was stirred for 14 h. PPTS (25 mg, 0.10 mmol) in MeOH (1.0 mL) was added to the mixture, followed by stirring at the same temperature for 2 h. The resulting mixture was quenched with sat. NaHCO3 aq., which was extracted three times with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2–column chromatography (hexane/AcOEt = 8/1) to give the desired product syn-3 (85% ee, dr 93:7, 51 mg, 31%).
Pale yellow oil; [α]
+1.0 (
c 1.0, CHCl
3) [lit. [
15], [α
+1.2 (
c 1.00, CHCl
3)]; 85% ee; HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 30:1) t
R(racemic) = 13.51 min, 14.13 min, 18.89 min and 19.82 min. t
R[(5
S,6
S)-form] = 18.69 min.
1H-NMR (500 MHz, CDCl
3): δ = 0.91 (t,
J = 6.9 Hz, 3H), 1.24–1.70 (m, 6H), 2.62–2.64 (m, 1H), 2.71-2.74 (m, 1H), 3.34–3.37 (m, 1H), 3.477 (s, 1H), 3.480 (s, 1H), 3.73 (s, 3H), 4.13–4.18 (m, 1H), 4.49 (d,
J = 11.5 Hz, 1H), 4.63 (d,
J = 11.5 Hz, 1H), 7.28–7.37 (m, 5H);
13C-NMR (125 MHz, CDCl
3): δ = 14.0, 22.8, 27.6, 29.3, 46.0, 49.6, 52.3, 68.3, 72.2, 80.8, 127.8, 127.9, 128.4, 138.2, 167.4, 202.7.
(E)-((1,3-dimethoxybuta-1,3-dien-1-yl)oxy)trimethylsilane (4) (Brassard’s diene)
Concentrated H2SO4 (0.27 mL, 5.0 mmol) was added to a stirred mixture of methyl acetoacetate (11.6g, 100 mmol) and trimethyl orthoformate (26.5 g, 250 mmol) at 0–5 °C under an Ar atmosphere, followed by stirring at 20–25 °C for 24 h. K2CO3 (5.0 g) was added to the mixture, which was filtered through a glass filter. The filtrate was concentrated under reduced pressure. The obtained crude oil was purified by distillation (bp 72–75 °C/3.2 kPa) to give the desired (E)-methyl-3-methoxybut-2-enoate (9.08 g, 70%).
nBuLi (1.63 M in hexane, 13.6 mL, 22 mmol) was added to stirred solution of iPr2NH (3.11 mL, 22 mmol) in THF (10 mL) at 0–5 °C under an Ar atmosphere, followed by stirring for 10 min. The mixture was cooled down to −78 °C and (E)-methyl-3-methoxybut-2-enoate (2.22 g, 17 mmol) in THF (4.0 mL) was added to the mixture, followed by stirring at the same temperature for 0.5 h. TMSCl (2.58 mL, 20 mmol) in THF (3.0 mL) was added to the mixture at the same temperature and the mixture was allowed to warm up to 0–5 °C over a period of 1 h. The mixture was concentrated and filtered through Celite® (No.503) using a glass filter, and washing with hexane (10 mL × 3). The filtrate was concentrated under reduced pressure and the obtained crude oil was purified by distillation to give the desired product 4 (2.62 g, 76%).
Colorless oil; bp 40–43 °C/50 Pa; 1H-NMR (400 MHz, CDCl3): δ = 0.26 (s, 9H), 3.56 (s, 3H), 3.57 (s, 3H), 3.99 (t, J = 1.4 Hz, 1H), 4.03 (d, J = 1.4 Hz, 1H), 4.34 (d, J = 1.8, 1H); 13C NMR (100 MHz, CDCl3): δ = 0.3, 54.0, 55.0, 75.5, 78.6, 158.7
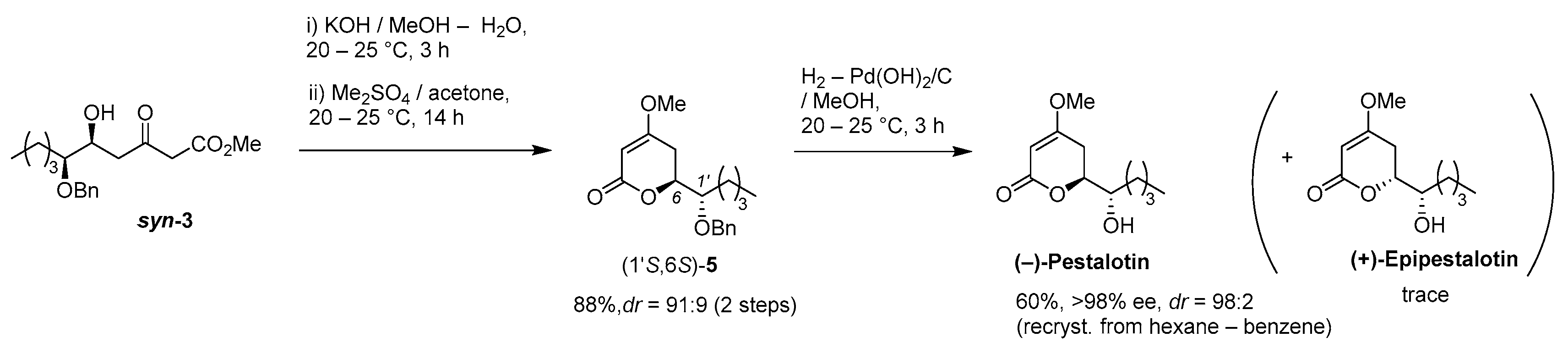
(
S)-6-[(
S)-1-(Benzyloxy)pentyl]-4-methoxy-5,6-dihydro-2
H-pyran-2-one [(1′
S,6
S)-
5] [
15]
(1) 1M-KOH aq. (0.37 mL) was added to a stirred solution of (5S,6S)-aldol adduct syn-3 (108 mg, 0.33 mmol) in MeOH (0.37 mL) at 20–25 °C under an Ar atmosphere, followed by stirring at the same temperature for 3 h. The mixture was quenched with 1M-HCl aq., which was extracted twice with AcOEt. The combined organic phase was washed with brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2–gel column chromatography (hexane/AcOEt = 5/1–2/1) to give the desired 4-hydroxy-5,6-dihydro-2H-pyran-2-one precursor (dr 93:7, 94 mg, 98%).
1H-NMR (500 MHz, CDCl3): δ = 0.92 (t, J = 6.9 Hz, 3H), 1.28–1.40 (m, 4H), 1.71–1.77 (m, 2H), 2.58 (dd, J = 5.2 Hz, 17.2 Hz, 1H), 2.76 (dd, J = 5.2 Hz, 17.2 Hz, 1H), 3.30 (d, J = 20.1 Hz, 1H), 3.41 (d, J = 20.1 Hz, 1H), 3.42–3.45 (m, 1H), 4.44 (d, J = 10.9 Hz, 1H), 4.59 (d, J = 10.9 Hz, 1H), 4.71–4.74 (m, 1H), 7.26–7.37 (m, 1H); 13C-NMR (125 MHz, CDCl3): δ = 13.9, 22.7, 27.5, 29.3, 40.6, 46.2, 72.3, 75.9, 80.0, 128.1, 128.2, 128.5, 136.9, 167.7, 199.4
K2CO3 (80 mg, 0.58 mmol) was added to a stirred suspension of the precursor (85 mg, 029 mmol) and Me2SO4 (55 mg, 0.44 mmol) in acetone (1.5 mL) at 20–25 °C under an Ar atmosphere, followed by stirring at the same temperature for 14 h. The mixture was quenched with water, which was extracted three times with Et2O. The combined organic phase was washed with brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2–gel column chromatography (hexane/AcOEt = 6/1–4/1) to give the desired product (1′S,6S)-5 (dr 91:9, 79 mg, 89%).
Pale yellow oil; [α
−93.7 (
c 0.72, CHCl
3)]. [lit. [
15], [α
−99.1 (
c 0.93, CHCl
3)].
(2) TiCl4 (0.02 mL, 0.2 mmol) was added to a solution of aldehyde 1 (206 mg, 1.0 m mol) in CH2Cl2 (3.0 mL) at 0–5 °C under an Ar atmosphere, followed by stirring at the same temperature for 10 min. Chan’s diene (61 % purity, 520 mg, 1.2 mmol) was added to the mixture, which was stirred at 0–5 °C for 5 min and at 20–25 °C for 1 h. MeOH (2 mL) and PPTS (125 mg, 0.5 mmol) was successively added to the mixture, followed by stirring at the same temperature for 2 h. The mixture was quenched with sat. NaHCO3 aq., which was filtered through Celite®. The filtrate was extracted twice with AcOEt, and the combined organic phase was washed with water, brine dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2-column chromatography (hexane/AcOEt = 4:1) to give the desired product (1’S,6S)-5 [165 mg, 49%, 91% ee, dr = 87:13].
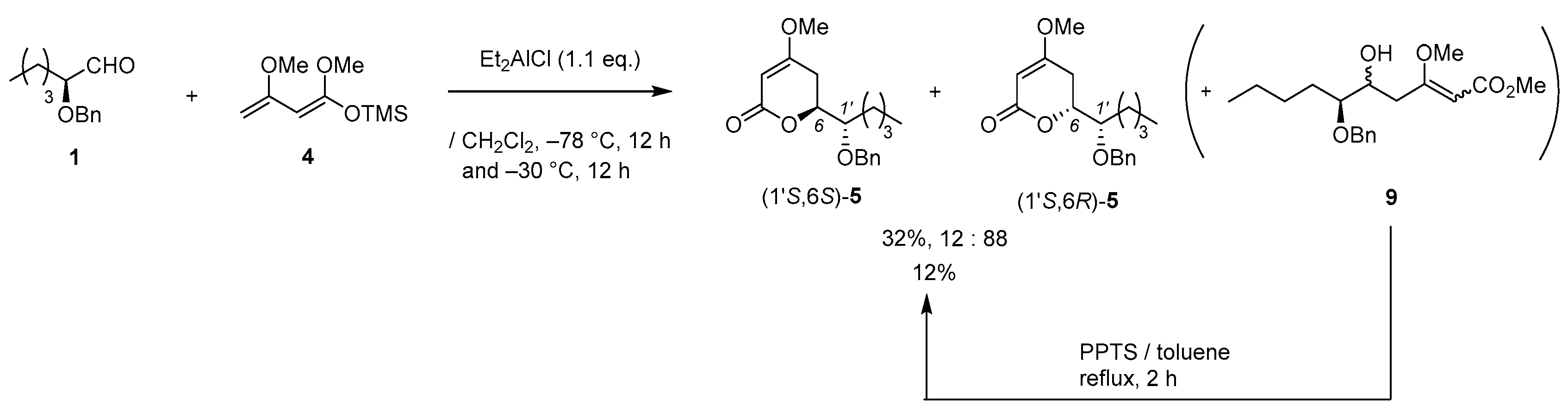
(3) Aldehyde 1 (413 mg, 2.0 mmol) in CH2Cl2 (1.0 mL) was added to a stirred suspension of Eu(fod)3 (104 mg, 0.1 mmol) in CH2Cl2 (1.0 mL) at 0–5 °C under an Ar atmosphere, followed by stirring at the same temperature for 5 min. Brassard’s diene 4 (607 mg, 3.0 mmol) in CH2Cl2 (2.0 mL) was added to the mixture at the same temperature, followed by stirring for 2 h. The mixture was quenched with water, which was extracted three times with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2–column chromatography (hexane/AcOEt = 3/1) to give the desired product [(1’S,6S)-5] (370 mg, 67%, >98% ee, dr = 98:2). HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 30:1) tR(racemic) = 23.25 min and 24.77 min. tR[(1S,6S)-form] = 25.53 min.; 1H-NMR (500 MHz, CDCl3): δ = 0.89 (t, J = 6.9 Hz, 3H), 1.25–1.72 (m, 6H), 2.26 (dd, J = 4.0 Hz, 17.2 Hz, 1H), 2.70 (ddd, J = 1.7 Hz, 13.2 Hz, 17.2 Hz, 1H), 3.58–3.61 (m, 1H), 3.74 (s, 3H), 4.52 (dt, J = 4.0 Hz, 13.2 Hz, 1H), 4.62 (d, J = 11.5 Hz, 1H), 4.66 (d, J = 11.5 Hz, 1H), 5.13 (d, J = 1.7 Hz, 1H), 7.27–7.36 (m, 5H); 13C NMR (125 MHz, CDCl3): δ = 14.0, 22.7, 27.9, 28.4, 29.3, 56.1, 72.9, 76.3, 79.0, 90.2, 127.8, 127.9, 128.4, 138.1, 167.0, 173.3.
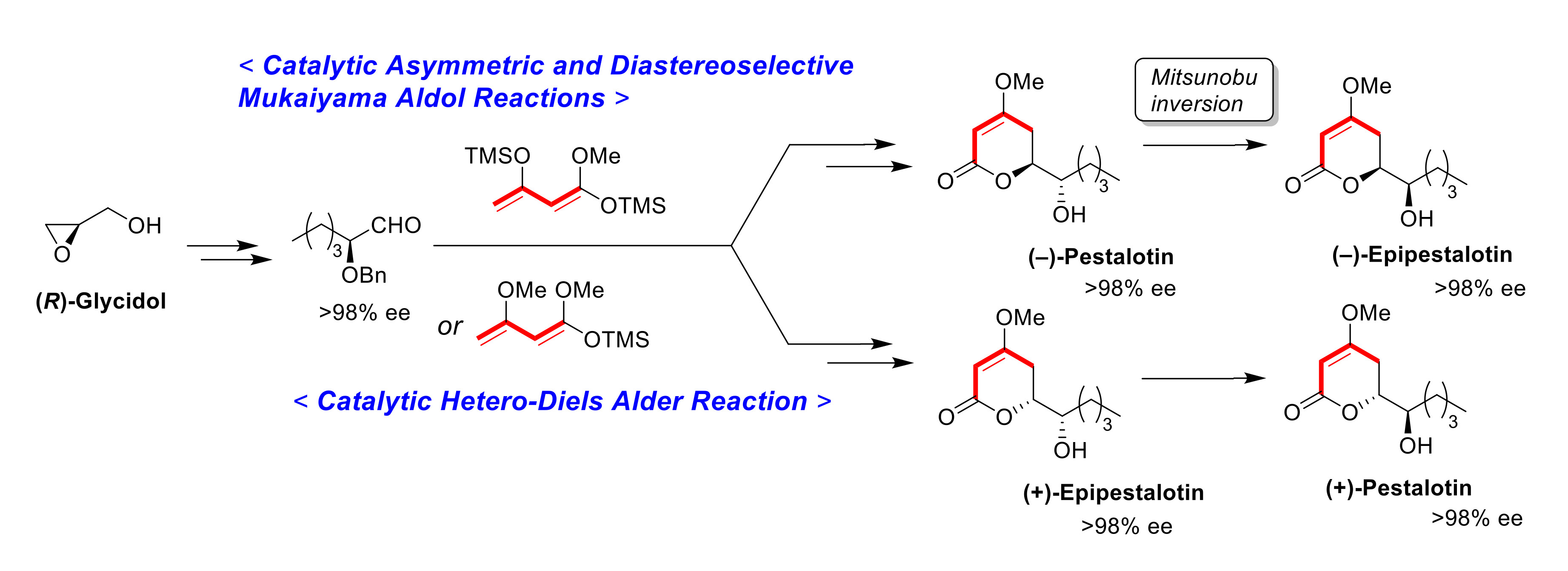
(−)-Pestalotin; (
S)-6-[(
S)-1-Hydroxypentyl]-4-methoxy-5,6-dihydro-2
H-pyran-2-one [
9]
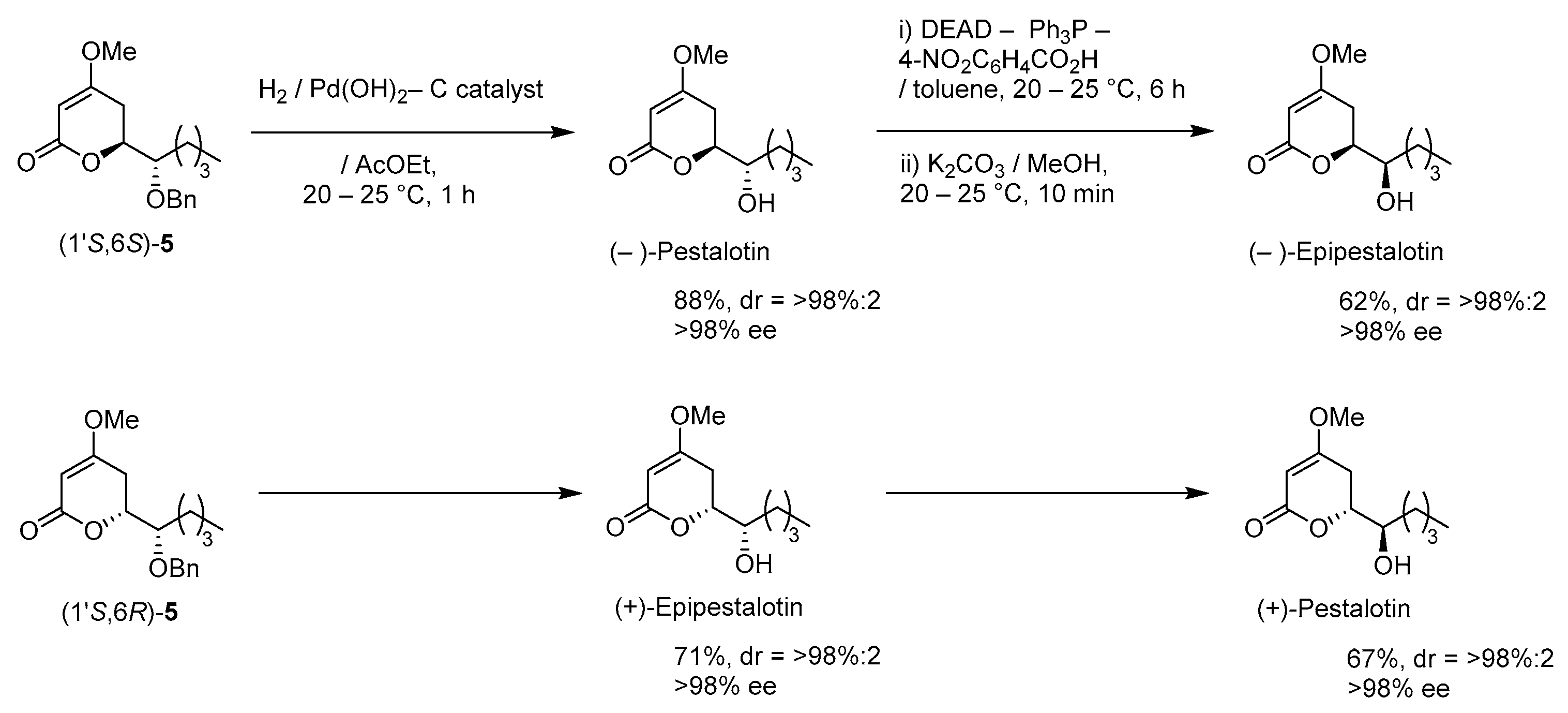
A suspension of benzyl ether [(1S,6S)-5] (448 mg, 1.5 mmol) and 20% Pd(OH)2/C (53 mg, 0.08 mmol) in AcOEt (15 mL), equipped with a H2 balloon, was stirred at 20–25 °C for 1 h. The mixture was filtered through Celite® (No.503) using glass filter and the filtrate was concentrated under reduced pressure. The obtained crude solid (384 mg) was purified by SiO2–column chromatography (hexane/AcOEt = 3:2) to give the desired (−)-pestalotin (283 mg, 88%, >98% ee, dr = >98:2).
Colorless crystals; mp 84–86 °C (lit. [
9], 85.8–86.0 °C); [α
−91.9 (
c 0.44, MeOH) [lit. [
9], [α
−90.2 (
c 1.17, MeOH)]; HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 30:1) t
R(racemic) = 45.23 min and 48.13 min. t
R[(1
S,6
S)-form] = 45.85 min.;
1H-NMR (500 MHz, CDCl
3): δ = 0.92 (t,
J = 6.9 Hz, 3H), 1.30–1.67 (m, 6H), 2.07 (brs, 1H), 2.25 (dd,
J = 4.0 Hz, 17.2 Hz, 1H), 2.80 (ddd,
J = 1.7 Hz, 12.6 Hz, 17.2 Hz, 1H), 3.61–3.64 (m, 1H), 3.76 (s, 3H), 4.30 (dt,
J = 4.0 Hz, 12.6 Hz, 1H), 5.15 (d,
J = 1.7 Hz, 1H);
13C-NMR (125 MHz, CDCl
3): δ = 13.9, 22.6, 27.6, 29.6, 32.4, 56.1, 72.4, 78.4, 90.0, 166.7, 173.1.
(R)-6-[(S)-1-(Benzyloxy)pentyl]-4-methoxy-5,6-dihydro-2H-pyran-2-one [(1’S,6R)-5]
(1) Aldehyde 1 (206 mg, 1.0 mmol) was added to a stirred suspension of ZrCl4 (47 mg, 0.2 mmol) in CH2Cl2 (0.9 mL) at 0–5 °C under an Ar atmosphere. After 10 min, Chan’s diene (ca. 60% purity; 520 mg, 1.2 mmol) was added to the mixture, which was allowed to warm up to 20–25 °C, followed by stirring for 1 h. MeOH (2.0 mL) and PPTS (125 mg 0.5 mmol) was successively added to the solution, followed by stirring at 40–45 °C for 14 h. Sat. NaHCO3 aq. solution was added to the mixture, which was filtered through Cerite®. The filtrate was extracted twice with AcOEt, and the combined organic phase was washed with water, brine dried (Na2SO4), and concentrated. The obtained crude oil was purified by SiO2-column chromatography (hexane/AcOEt = 4:1) to give the desired product (1′S,6R)-5 (126 mg, 41%, >98% ee, dr = 35:65).
Colorless oil. HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 30:1) tR(racemic) = 17.72 min and 19.60 min. tR[(1S,6R)-form] = 18.10 min.; 1H-NMR (500 MHz, CDCl3): δ = 0.89 (t, J = 6.9 Hz, 3H), 1.29-1.64 (m, 6H), 2.35 (dd, J = 4.0 Hz, 17.2 Hz, 1H), 2.81 (ddd, J = 1.7 Hz, 12.6 Hz, 17.2 Hz, 1H), 3.74 (s, 3H), 3.73-3.78 (m, 1H), 4.39 (dt, J = 4.0, 12.6, 1H), 4.63 (d, J = 11.5, 1H), 4.74 (d, J = 11.5, 1H), 5.14 (d, J = 1.7, 1H), 7.28-7.35 (m, 5H ); 13C-NMR (125 MHz, CDCl3): δ = 13.9, 22.6, 27.4, 27.9, 30.7, 56.0, 73.3, 78.4, 79.1, 90.0, 127.6, 127.8, 128.3, 138.3, 167.0, 173.4.
(2) Et2AlCl (1.0 M, 0.6 mL, 0.6 mmol) was added to a stirred solution of aldehyde 1 (103 mg, 0.5 mmol) in CH2Cl2 (0.5 mL) at −78 °C under an Ar atmosphere. After 5 min, diene 4 (202 mg, 0.6 mmol) in CH2Cl2 (0.5 mL) was added to the mixture, which was stirred for 14 h at the same temperature. The mixture was allowed to warm up to −30 °C, followed by stirring for 14 h. The mixture was quenched by MeOH, which was extracted three times with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2-column chromatography (hexane/AcOEt = 5/1) to give a mixture of aldol adduct 9 and (1′S,6R)-5 (45:55, 48 mg, 30%). The mixture (48 mg) and PPTS (2 mg, 0.007 mmol) in toluene (1.4 mL), was added at 80–85 °C for 1 h under an Ar atmosphere. After cooling to room temperature, water was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude solid purified by SiO2-column chromatography (hexane/AcOEt = 5/1) to give the desired product (1’S,6R)-5 (23 mg, 2 steps 15%, ca. 30% of (1’S,6S)-5 was contained).
(+)-Epipestalotin; (
R)-6-[(
S)-1-Hydroxypentyl]-4-methoxy-5,6-dihydro-2
H-pyran-2-one [
9]
A suspension of benzyl ether [(1S,6R)-5] (365 mg, 1.2 mmol) and 20% Pd(OH)2/C (42 mg, 0.06 mmol) in AcOEt (12 mL), equipped with a H2 balloon, was stirred stirred at 20–25 °C for 1 h. The mixture was filtered through Celite® (No.503) using glass filter and the filtrate was concentrated under reduced pressure. The obtained crude solid was purified SiO2–column chromatography (hexane/AcOEt = 3/2) to give the desired (+)-epipestalotin (187 mg, 71%, >98% ee, dr = 98:2).
Colorless crystals; mp 92–94 °C (lit. [
9], 93.0–94.0 °C); [α
+ 75.3 (
c 0.39, MeOH) [lit. [
9], [α
+ 75.9 (
c 0.39, MeOH)]; >99% ee; HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 25:1) t
R(racemic) = 33.00 min and 35.46 min. t
R[(1
S,6
R)-form] = 34.81 min.;
1H NMR (500 MHz, CDCl
3): δ = 0.92 (t,
J = 6.9 Hz, 3H), 1.30–1.56 (m, 6H), 2.04 (brs, 1H), 2.24 (dd,
J = 4.0 Hz, 17.2 Hz, 1H), 2.84 (ddd,
J = 1.7 Hz, 12.6 Hz, 17.2 Hz, 1H), 3.76 (s, 3H), 3.94–3.97 (m, 1H), 4.34 (dt,
J = 3.4 Hz, 12.6 Hz, 1H), 5.14 (d, 1.7 Hz, 1H);
13C NMR (125 MHz, CDCl
3): δ = 13.8, 22.4, 26.8, 27.7, 31.4, 56.0, 71.3, 78.7, 89.7, 167.1, 173.5.
(−)-Epipestalotin; (
S)-6-[(
R)-1-Hydroxypentyl]-4-methoxy-5,6-dihydro-2
H-pyran-2-one [
9]
DEAD (40% in toluene, 0.91 mL, 2.0 mmol) was added slowly to a stirred mixture of (−)-pestalotin (214 mg, 1.0 mmol) and 4-nitrobenzoic acid (334 mg, 2.0 mmol) and PPh3 (525 mg, 2.0 mmol) in toluene (10 mL) at 0–5 °C under an Ar atmosphere, followed by stirring at 20–25 °C for 6 h. The mixture was quenched with water, which was extracted three times with AcOEt. The combined organic phase was washed with sat. NaHCO3 aq., brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2–column chromatography (hexane/AcOEt = 3:1) to give a mixture of the desired (−)-Epipestalotin and diethyl hydrazodicarboxylate, which was used in the next step without further purification.
A suspension of the mixture and K2CO3 (138 mg, 1.0 mmol) in MeOH (10 mL) was stirred at 20–25 °C under an Ar atmosphere for 10 min. The mixture was filtered through Celite® (No.503) using a glass filter washing with AcOEt (5 mL × 3). The filtrate was concentrated under reduced pressure and the obtained crude oil, which was purified by SiO2–column chromatography (hexane/AcOEt = 2:1) to give the desired (−)-epipestalotin (133 mg, 62% for 2 steps, >98% ee, dr = >98:2).
Colorless crystals; mp 89–91 °C (lit. [
9], 90.7–91.2 °C); [α
−75.8 (
c 0.58, MeOH) [lit. [
9], [α
−75.6 (
c 0.58, MeOH)]; HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 25:1) t
R(racemic) = 33.00 min and 35.46 min. t
R[(1
R,6
S)-form] = 32.31 min.;
1H-NMR (500 MHz, CDCl
3): δ = 0.92 (t,
J = 6.9 Hz, 3H), 1.30–1.55 (m, 6H), 2.04 (brs, 1H), 2.24 (dd,
J = 4.0 Hz, 17.2 Hz, 1H), 2.84 (ddd,
J = 1.7 Hz, 12.6 Hz, 17.2 Hz, 1H), 3.76 (s, 3H), 3.94–3.97 (m, 1H), 4.34 (dt,
J = 3.4 Hz, 12.6 Hz, 1H), 5.14 (d,
J = 1.7 Hz, 1H);
13C-NMR (125 MHz, CDCl
3): δ = 13.8, 22.4, 26.8, 27.7, 31.4, 56.0, 71.3, 78.7, 89.7, 167.1, 173.5.
(+)-Pestalotin; (
R)-6-[(
R)-1-Hydroxypentyl]-4-methoxy-5,6-dihydro-2
H-pyran-2-one [
9]
Following the procedure for the preparation of (−)-epipestalotin, the reaction of (+)-epipestalotin (107 mg, 0.5 mmol) using DEAD (40% in toluene, 0.45 mL, 1.0 mmol), 4-nitrobenzoic acid (167 mg, 1.0 mmol), PPh3 (262 mg, 1.0 mmol), and K2CO3 (69 mg, 0.5 mmol) give the desired (+)-pestalotin (72 mg, 67% for 2 steps, >98% ee, dr > 98:2,).
Colorless crystals; mp 82–84 °C (lit. [
9], 83.0–84.5 °C); [α
+97.5 (
c 0.65, MeOH) [lit. [
9], [α
+88.7 (
c 0.65, MeOH)]; HPLC analysis (AD-3, flow rate 1.00 mL/min, solvent: hexane/2-propanol = 30:1) t
R(racemic) = 45.23 min and 48.13 min. t
R[(1
R,6
R)-form] = 49.57 min;
1H-NMR (500 MHz, CDCl
3): δ = 0.92 (t,
J = 6.9 Hz, 3H), 1.30–1.67 (m, 6H), 2.07 (brs, 1H), 2.25 (dd,
J = 4.0 Hz, 17.2 Hz, 1H), 2.80 (ddd,
J = 1.7 Hz, 12.6 Hz, 17.2 Hz, 1H), 3.61–3.64 (m, 1H), 3.76 (s, 3H), 4.30 (dt,
J = 4.0 Hz, 12.6 Hz, 1H), 5.15 (d,
J = 1.7 Hz, 1H);
13C-NMR (125 MHz, CDCl
3): δ = 13.9, 22.6, 27.6, 29.6, 32.4, 56.1, 72.4, 78.4, 90.0, 166.7, 173.1.
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

