
Preparative Method for Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid

 , , ,
, , ,
Abstract
1. Introduction
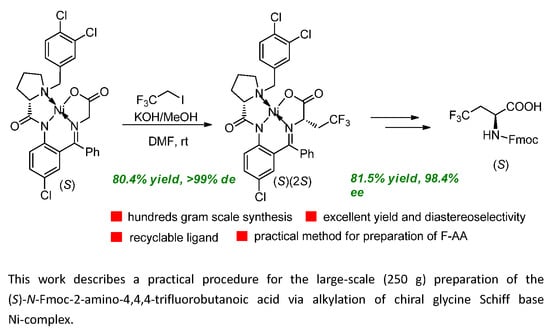
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Alkylation of (S)-6 with ICH2CF3
3.2.1. 80 g Scale Reaction
3.2.2. 250 g Scale Reaction
3.3. Disassembly of Major Diastereomer (S)(2S)-7 and Preparation of (S)-12
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S. Unnatural amino acids in drug discovery. Chim. Oggi 2003, 21, 65–68. [Google Scholar]
- Hodgson, D.R.W.; Sanderson, J.M. The synthesis of peptides and proteins containing non-natural amino acids. Chem. Soc. Rev. 2004, 33, 422–430. [Google Scholar] [CrossRef]
- Sato, T.; Izawa, K.; Aceña, J.L.; Liu, H.; Soloshonok, V.A. Tailor-Made α-Amino Acids in Pharmaceutical Industry: Synthetic Approaches to (1R,2S)-1-Amino-2-vinylcyclopropane-1-carboxylic Acid (Vinyl-ACCA). Eur. J. Org. Chem. 2016, 16, 2757–2774. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Wang, J.; Sato, T.; Izawa, K.; Soloshonok, V.A.; Liu, H. The second-generation of highly potent hepatitis C virus (HCV) NS3/4A protease inhibitors: Evolutionary design based on tailor-made amino acids, synthesis and major features of bio-activity. Curr. Pharm. Des. 2017, 23, 4493–4554. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Izawa, K. (Eds.) Asymmetric Synthesis and Application of alpha-Amino Acids; ACS Symposium Series #1009; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluor. Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.L.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Tao, J.Y.; Song, W.T.; Sun, L.; Li, Z.Z.; Fu, B.; Cai, Z.C.; Xu, H.J. Synthesis, Photo-physical Properties and Cell Imaging of Meso-2,6-dichlorophenyl Boron-dipyrromethene Derivatives. Chin. J. Struct. Chem. 2019, 38, 1503–1510. [Google Scholar]
- Yang, J.; Fan, Y.; Cai, F.; Xu, X.; Fu, B.; Wang, S.; Shen, Z.; Tian, J.; Xu, H. BODIPY derivatives bearing borneol moieties: Enhancing cell membrane permeability for living cell imaging. Dyes Pigment. 2019, 164, 105–111. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Smits, R.; Cadicamo, C.D.; Burger, K.; Koksch, B. Synthetic strategies to α-trifluoromethyl and α-difluoromethyl substituted α-amino acids. Chem. Soc. Rev. 2008, 37, 1727–1739. [Google Scholar] [CrossRef]
- Kukhar, V.P.; Sorochinsky, A.E.; Soloshonok, V.A. Practical synthesis of fluorine-containing α-and β-amino acids: Recipes from Kiev, Ukraine. Future Med. Chem. 2009, 1, 793–819. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of fluorine-containing amines, amino alcohols, α- and β-amino acids mediated by chiral sulfinyl group. J. Fluor. Chem. 2010, 131, 127–139. [Google Scholar] [CrossRef]
- Tarui, A.; Sato, K.; Omote, M.; Kumadaki, I.; Ando, A. Stereoselective synthesis of α-fluorinated amino acid derivatives. Adv. Synth. Catal. 2010, 352, 2733–2744. [Google Scholar] [CrossRef]
- Czekelius, C.; Tzschucke, C.C. Synthesis of halogenated carboxylic acids and amino acids. Synthesis 2010, 4, 543–566. [Google Scholar] [CrossRef][Green Version]
- Qiu, X.L.; Qing, F.L. Recent advances in the synthesis of fluorinated amino acids. Eur. J. Org. Chem. 2011, 18, 3261–3278. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Roeschenthaler, G.V.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Recent advances in asymmetric synthesis of α-(trifluoromethyl)-containing α-amino acids. Synthesis 2012, 44, 1591–1602. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of fluorine-containing α-amino acids in enantiomerically pure form via homologation of Ni(II) complexes of glycine and alanine Schiff bases. J. Fluor. Chem. 2013, 155, 21–38. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sánchez-Roselló, M.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Synthesis of Fluorine Containing β-Amino Acids. Synthesis 2011, 19, 3045–3079. [Google Scholar]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic Transamination—A Metal-Free Alternative to the Reductive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth. 2011, 8, 281–294. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Kirilenko, A.G.; Fokina, N.A.; Shishkina, I.P.; Galushko, S.V.; Kukhar, V.P.; Svedas, V.K.; Kozlova, E.V. Biocatalytic Resolution of β-Fluoroalkyl-β-Amino Acids. Tetrahedron 1994, 5, 1119–1126. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Fluorine-Containing Amino Acids: Synthesis and Properties; Kukhar, V.P., Soloshonok, V.A., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1994. [Google Scholar]
- Hao, J.; Milcent, T.; Retailleau, P.; Soloshonok, V.A.; Ongeri, S.; Crousse, B. Asymmetric Synthesis of Cyclic Fluorinated Amino Acids. Eur. J. Org. Chem. 2018, 27, 3688–3692. [Google Scholar] [CrossRef]
- Peng, Y.Y.; Liu, P.; Liu, Z.J.; Liu, J.T.; Mao, H.F.; Yao, Y.L. Regio- and diastereoselective Reformatsky reaction of chiral fluoroalkyl α,β-unsaturated N-tert-butanesulfinyl ketimines: Efficient asymmetric synthesis of β-fluoroalkyl β-vinyl β-amino esters. Tetrahedron 2018, 74, 3074–3080. [Google Scholar] [CrossRef]
- Kondratov, I.S.; Bugera, M.Y.; Tolmachova, N.A.; Daniliuc, C.G.; Haufe, G. Straightforward synthesis of fluorinated amino acids by Michael addition of ethyl bromodifluoroacetate to α,β-unsaturated α-amino acid derivatives. J. Fluor. Chem. 2018, 211, 100–108. [Google Scholar] [CrossRef]
- Oliver, M.; Gadais, C.; Garcia-Pindado, J.; Teixido, M.; Lensen, N.; Chaume, G.; Brigaud, T. Trifluoromethylated proline analogues as efficient tools to enhance the hydrophobicity and to promote passive diffusion transport of the L-prolyl-L-leucyl glycinamide (PLG) tripeptide. RSC Adv. 2018, 8, 14597–14602. [Google Scholar] [CrossRef]
- Makki, M.S.T.; Reda, M.; Alharbi, A.S. Synthetic Approach for Novel Fluorine Substituted α-Aminophosphonic Acids Containing 1,2,4-Triazin-5-One Moiety as Antioxidant Agents. Int. J. Org. Chem. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Zhao, J.B.; Ren, X.; Zheng, B.Q.; Ji, J.; Qiu, Z.B.; Li, Y. A diastereoselective Mannich reaction of α-fluoroketones with ketimines: Construction of β-fluoroamine motifs with vicinal tetrasubstituted stereocenters. Tetrahedron Lett. 2018, 59, 2091–2094. [Google Scholar] [CrossRef]
- Bucci, R.; Das, P.; Iannuzzi, F.; Feligioni, M.; Gandolfi, R.; Gelmi, M.L.; Reches, M.; Pellegrino, S. Self-assembly of an amphipathic ααβ-tripeptide into cationic spherical particles for intracellular delivery. Org. Biomol. Chem. 2017, 15, 6773–6779. [Google Scholar] [CrossRef] [PubMed]
- Betts, H.M.; Milicevic Sephton, S.; Tong, C.; Awais, R.O.; Hill, P.J.; Perkins, A.C.; Aigbirhio, F.I. Synthesis, in vitro evaluation, and radiolabeling of fluorinated puromycin analogues: Potential candidates for PET imaging of protein synthesis. J. Med. Chem. 2016, 59, 9422–9430. [Google Scholar] [CrossRef]
- Bouhlel, A.; Alyami, W.; Li, A.; Yuan, L.; Rich, K.; McConathy, J. Effect of α-Methyl versus α-Hydrogen Substitution on Brain Availability and Tumor Imaging Properties of Heptanoic [F-18] Fluoroalkyl Amino Acids for Positron Emission Tomography (PET). J. Med. Chem. 2016, 59, 3515–3531. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Cordes, D.B.; Slawin, A.M.Z.; O’Hagan, D. Fluorine containing amino acids: Synthesis and peptide coupling of amino acids containing the all-cis tetrafluorocyclohexyl motif. Org. Biomol. Chem. 2015, 13, 5621–5624. [Google Scholar] [CrossRef]
- Bandak, D.; Babii, O.; Vasiuta, R.; Komarov, I.V.; Mykhailiuk, P.K. Design and synthesis of novel 19F-amino acid: A promising 19F NMR label for peptide studies. Org. Lett. 2015, 17, 226–229. [Google Scholar] [CrossRef]
- Usuki, Y.; Wakamatsu, Y.; Yabu, M.; Iio, H. A New Access to Fluorine-containing Asparagine and Glutamine Analogues via Pd-catalyzed Formate Reduction. Asian J. Org. Chem. 2014, 3, 1270–1272. [Google Scholar] [CrossRef]
- Tkachenko, A.N.; Mykhailiuk, P.K.; Radchenko, D.S.; Babii, O.; Afonin, S.; Ulrich, A.S.; Komarov, I.V. Design and Synthesis of a Monofluoro-Substituted Aromatic Amino Acid as a Conformationally Restricted 19F NMR Label for Membrane-Bound Peptides. Eur. J. Org. Chem. 2014, 17, 3584–3591. [Google Scholar] [CrossRef]
- Shibata, N.; Nishimine, T.; Shibata, N.; Tokunaga, E.; Kawada, K.; Kagawa, T.; Sorochinsky, A.E.; Soloshonok, V.A. Organic base-catalyzed stereodivergent synthesis of (R)- and (S)-3-amino-4,4,4-trifluorobutanoic acids. Chem. Commun. 2012, 48, 4124–4126. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nishimine, T.; Shibata, N.; Tokunaga, E.; Kawada, K.; Kagawa, T.; Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric Mannich reaction between (S)-N-(tertbutanesulfinyl)-3,3,3-trifluoroacetaldimine and malonic acid derivatives. Stereodivergent synthesis of (R)- and (S)-3-amino-4,4,4-trifluorobutanoic acids. Org. Biomol. Chem. 2014, 12, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Fueloep, F. Selective Synthesis of Fluorine-Containing Cyclic β-Amino Acid Scaffolds. Chem. Rec. 2018, 18, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Milcent, T.; Hao, J.; Kawada, K.; Soloshonok, V.A.; Ongeri, S.; Crousse, B. Highly Stereoselective aza-Baylis–Hillman Reactions of CF3-Sulfinylimines: Straightforward Access to α-Methylene β-CF3 β-Amino Acids. Eur. J. Org. Chem. 2014, 15, 3072–3075. [Google Scholar] [CrossRef]
- Drouet, F.; Noisier, A.F.M.; Harris, C.S.; Furkert, D.P.; Brimble, M.A. A Convenient Method for the Asymmetric Synthesis of Fluorinated α-Amino Acids from Alcohols. Eur. J. Org. Chem. 2014, 6, 1195–1201. [Google Scholar] [CrossRef]
- Mei, H.; Hiramatsu, T.; Takeda, R.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Expedient Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid via Alkylation of Chiral Nucleophilic Glycine Equivalent. Org. Process Res. Dev. 2019, 23, 629–634. [Google Scholar] [CrossRef]
- Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-trifluoro-Norleucine. Symmetry 2019, 11, 578. [Google Scholar] [CrossRef]
- Yin, Z.; Moriwaki, H.; Abe, H.; Miwa, T.; Han, J.; Soloshonok, V.A. Large-Scale Asymmetric Synthesis of Fmoc-(S)-2-Amino-6,6,6-Trifluorohexanoic Acid. ChemistryOpen 2019, 8, 701–704. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.L.; Takeda, R.; Sakamoto, T.; Miwa, T.; Minamitsuji, Y.; Moriwaki, H.; Abe, H.; Soloshonok, V.A. Practical Method for Preparation of (S)‑2-Amino-5,5,5-trifluoropentanoic Acid via Dynamic Kinetic Resolution. ACS Omega 2019, 4, 11844–11851. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, M.J. Dynamic kinetic resolution of amines and amino acids by enzyme–metal cocatalysis. ChemCatChem 2011, 3, 271–277. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Jiang, H.; Chen, K.; Liu, H. Application of nickel (II) complexes to the efficient synthesis of α- or β-amino acids. Chimia 2011, 65, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Popkov, A.; De Spiegeleer, B. Chiral nickel (II) complexes in the preparation of 11C-and 18F-labelled enantiomerically pure α-amino acids. Dalton Trans. 2012, 41, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- So, S.M.; Kim, H.; Mui, L.; Chin, J. Mimicking nature to make unnatural amino acids and chiral diamines. Eur. J. Org. Chem. 2012, 2, 229–241. [Google Scholar] [CrossRef]
- D’Arrigo, P.; Cerioli, L.; Servi, S.; Viani, F.; Tessaroa, D. Synergy between catalysts: Enzymes and bases. DKR of non-natural amino acids derivatives. Cat. Sci. Technol. 2012, 2, 1606–1616. [Google Scholar] [CrossRef]
- D’Arrigo, P.; Cerioli, L.; Fiorati, A.; Servi, S.; Viani, F.; Tessaro, D. Naphthyl-l-α-amino acids via chemo-enzymatic dynamic kinetic resolution. Tetrahedron 2012, 23, 938–944. [Google Scholar] [CrossRef]
- Periasamy, M.; Gurubrahamam, R.; Sanjeevakumar, N.; Dalai, M.; Alakonda, L.; Reddy, P.O.; Suresh, S.; Satishkumar, S.; Padmaja, M.; Reddy, M.N.; et al. Convenient methods for the synthesis of chiral amino alcohols and amines. Chimia 2013, 67, 23–29. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I. Asymmetric synthesis of quaternary α-amino acids and their phosphonate analogues. Asian J. Org. Chem. 2014, 3, 1234–1260. [Google Scholar] [CrossRef]
- Metz, A.E.; Kozlowski, M.C. Recent advances in asymmetric catalytic methods for the formation of acyclic α, α-disubstituted α-amino acids. J. Org. Chem. 2015, 80, 1–7. [Google Scholar] [CrossRef]
- He, G.; Wang, B.; Nack, W.A.; Chen, G. Syntheses and transformations of α-amino acids via palladium-catalyzed auxiliary-directed sp3 C–H functionalization. Acc. Chem. Res. 2016, 49, 635–645. [Google Scholar] [CrossRef]
- Bott, F.; Field, L.D.; Sternhell, S. Steric effects. A study of a rationally designed system. J. Am. Chem. Soc. 1980, 102, 5618–5626. [Google Scholar] [CrossRef]
- Lunazzi, L.; Mancinelli, M.; Mazzanti, A.; Lepri, S.; Ruzziconi, R.; Schlosser, M. Rotational barriers of biphenyls having heavy heteroatoms as ortho substituents: Experimental and theoretical determination of steric effects. Org. Biomol. Chem. 2012, 10, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- de Riggi, I.; Virgili, A.; de Moragas, M.; Jaime, C. Restricted rotation and NOE transfer: A conformational study of some substituted (9-anthry1)carbinol derivatives. J. Org. Chem. 1995, 60, 27–31. [Google Scholar] [CrossRef]
- Belot, V.; Farran, D.; Jean, M.; Albalat, M.; Vanthuyne, N.; Roussel, C. Steric scale of common substituents from rotational barriers of N-(o-substituted aryl)thiazoline-2-thione atropisomers. J. Org. Chem. 2017, 82, 10188–10200. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Kukhar, V.P.; Galushko, S.V.; Svistunova, N.Y.; Avilov, D.V.; Kuzmina, N.A.; Raevski, N.I.; Struchkov, Y.T.; Pisarevsky, A.P.; Belokon, Y.N. General Method for the Synthesis of Enantiomerically Pure β-Hydroxy-α-Amino Acids, Containing Fluorine Atoms in the Side Chains. Case of Stereochemical Distinction Between Methyl and Trifluoromethyl Groups. X-Ray Crystal and Molecular Structure of the Nickel(II) Complex of (2S,3S)-2-(Trifluoromethyl)threonine. J. Chem. Soc. Perkin Trans. 1993, 1, 3143–3155. [Google Scholar]
- Jagodzinska, M.; Huguenot, F.; Candiani, G.; Zanda, M. Assessing the bioisosterism of the trifluoromethyl group with a protease probe. ChemMedChem 2009, 4, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Walborsky, H.M.; Baum, M.E. Chemical Effects of the Trifluoromethyl Group: III. Synthesis of 2-Amino-4,4,4-trifluorobutyric Acid. J. Org. Chem. 1956, 21, 538–539. [Google Scholar] [CrossRef]
- Steglich, W.; Heininger, H.U.; Dworschak, H.; Weygand, F. A General Method for the Preparation of β-Perfluoroalkylalanines. Angew. Chem. Int. Ed. Engl. 1967, 6, 807–808. [Google Scholar] [CrossRef]
- Tsushima, T.; Kawada, K.; Ishihara, S.; Shiratori, O.; Higaki, J.; Hirata, M. Fluorine containing amino acids and their derivatives. 7. Synthesis and antitumor activity of α- and γ-substituted methotrexate analogs. Tetrahedron 1988, 44, 5375–5387. [Google Scholar] [CrossRef]
- Seebach, D.; Buerger, H.M.; Schickli, C.P. Stereoselektive Umsetzungen von rac-, (R), oder (S)-5-Alkyliden-2-t-butyl-3-methyl-4-oxo-1-imidazolidincarbonsäure-t-butylestern (chirale 2,3-Dehydroaminosäure-Derivate) und Herstellung einiger nichtproteinogener Aminosäuren. Ann. Chem. 1991, 7, 669–684. [Google Scholar]
- Schedel, H.; Dmowski, W.; Burger, K. New Stereoconservative Syntheses of β,β,β- and γ,γ,γ-Trifluoro-α-amino, α-Hydroxy, and α-Mercapto Acids and Their Incorporation into a Peptide and Depsipeptide Fragment. Synthesis 2000, 12, 1681–1688. [Google Scholar] [CrossRef]
- Kondratov, I.S.; Logvinenko, I.G.; Tolmachova, N.A.; Morev, R.N.; Kliachyna, M.A.; Clausen, F.; Daniliuc, C.G.; Haufe, G. Synthesis and physical chemical properties of 2-amino-4-(trifluoromethoxy) butanoic acid–α CF3O-containing analogue of natural lipophilic amino acids. Org. Biomol. Chem. 2017, 15, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.T.; Huber, E.M.; de Bruin, G.; Heinemeyer, W.; Maurits, E.; Espinal, C.; Du, Y.; Janssens, M.; Weyburne, E.S.; Kisselev, A.F.; et al. Structure-Based Design of Inhibitors Selective for Human Proteasome β2c or β2i Subunits. J. Med. Chem. 2019, 62, 1626–1642. [Google Scholar] [CrossRef] [PubMed]
- Franko, N.; Grammatoglou, K.; Campanini, B.; Costantino, G.; Jirgensons, A.; Mozzarelli, A. Inhibition of O-acetylserine sulfhydrylase by fluoroalanine derivatives. J. Enzyme Inhib. Med. Chem. 2018, 33, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Gambini, L.; Salem, A.F.; Udompholkul, P.; Tan, X.F.; Baggio, C.; Shah, N.; Aronson, A.; Song, J.; Pellecchia, M. Structure-Based Design of Novel EphA2 Agonistic Agents with Nanomolar Affinity in Vitro and in Cell. ACS Chem. Biol. 2018, 13, 2633–2644. [Google Scholar]
- Papaioannou, N.; Fink, S.J.; Miller, T.A.; Shipps, G.W., Jr.; Travins, J.M.; Ehmann, D.E.; Rae, A.; Ellard, J.M. Preparation of Substituted Imidazopyridines as Inhibitors of Plasma Kallikrein. Patent W.O. 2019,178,129, 19 September 2019. [Google Scholar]
- Bilcer, G.M.; Kelly, T.A. Preparation of Hexahydro(di)azepino[3,2,1-hi]indole-, Tetrahydro-7H-6-oxa-9a-Azabenzo[cd]azulene- and Tetrahydro-7H-6-thia-9a-azabenzo[cd]azulene-Containing Peptides as Granzyme B Directed Imaging and Therapy. Patent W.O. 2019,160,916, 22 August 2019. [Google Scholar]
- Boss, C.; Cren, S.; Kimmerlin, T.; Lotz-Jenne, C.; Pothier, J.; Tidten-Luksch, N. Preparation of Imidazo[5,1-b]thiazole Derivatives as Inhibitors of Indoleamine 2,3-Dioxygenase and/or Tryptophan 2,3-Dioxygenase. Patent W.O. 2019,138,107, 18 July 2019. [Google Scholar]
- Nicolaou, K.C.; Erande, R.D.; Vourloumis, D.; Pulukuri, K.K.; Rigol, S. Preparation of Tubulysin Analogues as Anticancer Agents and Payloads for Antibody-Drug Conjugates Useful for Treatment of Cancer. Patent W.O. 2019,108,685, 6 June 2019. [Google Scholar]
- Saiah, E.; Vlasuk, G. Preparation of Amino Acid as Modulators of Sestrin-Gator Interaction. Patent W.O. 2018,200,625, 1 November 2018. [Google Scholar]
- Wang, S.; Zhou, H.; Lu, J.; Liu, L.; Sun, Y.; Bernard, D. Preparation of Small Molecule DCN1 Inhibitors and Therapeutic Methods Using the Same. Patent W.O. 2018,183,411, 4 October 2018. [Google Scholar]
- Bacon, E.M.; Chin, E.; Cottell, J.J.; Katana, A.A.; Kato, D.; Link, J.O.; Shapiro, N.; Trejo, M.; Teresa, A.; Yang, Z.Y. Preparation of Azapeptide Atazanavir Analogs Useful for Treating HIV Infections. Patent W.O. 2018,145,021, 9 August 2018. [Google Scholar]
- Röschenthaler, G.V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Poliashko, K.O.; Kukhar, V.P.; Rozhenko, A.B.; Soloshonok, V.A.; Sorochinsky, A.E. Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun. 2012, 48, 11519–11521. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Belokon, Y.N.; Kuzmina, N.A.; Maleev, V.I.; Svistunova, N.Y.; Solodenko, V.A.; Kukhar, V.P. Asymmetric Synthesis of Phosphorus Analogs of Dicarboxylic α-Amino Acids. J. Chem. Soc. Perkin Trans. 1992, 1, 1525–1529. [Google Scholar] [CrossRef]
- Qiu, W.; Gu, X.; Soloshonok, V.A.; Carducci, M.D.; Hruby, V.J. Stereoselective Synthesis of Conformationally Constrained Reverse Turn Dipeptide Mimetics. Tetrahedron Lett. 2001, 42, 145–148. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α-Disubstituted-α-Amino Acids. Synthesis 2010, 14, 2319–2344. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally Convenient Asymmetric Synthesis of (S)- and (R)-3-Amino-4,4,4-trifluorobutanoic Acid. Part II: Enantioselective Biomimetic Transamination of 4,4,4-Trifluoro-3-oxo-N-[(R)-1-phenylethyl)butanamide. J. Fluor. Chem. 2006, 127, 930–935. [Google Scholar] [CrossRef]
- Bravo, P.; Farina, A.; Kukhar, V.P.; Markovsky, A.L.; Meille, S.V.; Soloshonok, V.A.; Sorochinsky, A.E.; Viani, F.; Zanda, M.; Zappala, C. Stereoselective Additions of α-Lithiated Alkyl p-Tolylsulfoxides to N-PMP Fluoroalkyl Aldimines. An Efficient Approach to Enantiomerically Pure Fluoro-Amino Compounds. J. Org. Chem. 1997, 62, 3424–3425. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Kukhar, V.P. Biomimetic Base-Catalyzed [1,3]-Proton Shift Reaction. A Practical Synthesis of β-Fluoroalkyl-Amino Acids. Tetrahedron 1996, 52, 6953–6964. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical purifications via Self-Disproportionation of Enantiomers by achiral chromatography; Case study of a series of α-CF3-containing secondary alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019, 51, 865–889. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Han, J.; Kitagawa, O.; Aceña, J.L.; Klika, K.D.; Soloshonok, V.A. A comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography. RSC Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Recent approaches for asymmetric synthesis of a-amino acids via homologation of Ni(II) complexes. Amino Acids 2017, 49, 1487–1520. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V. Asymmetric synthesis of -amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of -amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 2: Aldol, Mannich addition reactions, deracemization and (S) to (R) interconversion of -amino acids. Amino Acids 2013, 45, 1017–1033. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases; Part 1: Alkyl halide alkylations. Amino Acids 2013, 45, 691–718. [Google Scholar] [CrossRef]
- Ellis, T.K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V.A. The Design, Synthesis and Evaluation of a New Generation of Modular Nucleophilic Glycine Equivalents for the Efficient Synthesis of Sterically Constrained-Amino Acids. J. Org. Chem. 2006, 71, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Ueki, H.; Ellis, T.K.; Yamada, T.; Ohfune, Y. Application of Modular Nucleophilic Glycine Equivalents for Truly Practical Asymmetric Synthesis of -Substituted Pyroglutamic Acids. Tetrahedron Lett. 2005, 46, 1107–1110. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J.L.; et al. Chemical Dynamic Kinetic Resolution and (S)/(R)-Interconversion of Unprotected α-Amino Acids. Angew. Chem. Int. Ed. 2014, 53, 12214–12217. [Google Scholar] [CrossRef] [PubMed]
- Jörres, M.; Aceña, J.L.; Soloshonok, V.A.; Bolm, C. Asymmetric Carbon-Carbon Bond Formations under Solvent-Less Conditions in Ball Mills. ChemCatChem 2015, 7, 1265–1269. [Google Scholar] [CrossRef]
- Jörres, M.; Chen, X.; Aceña, J.L.; Merkens, C.; Bolm, C.; Liu, H.; Soloshonok, V.A. Asymmetric Synthesis of a-Amino Acids under Operationally Convenient Conditions. Adv. Synth. Catal. 2014, 356, 2203–2208. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Abe, H.; Soloshonok, V.A. Second-order asymmetric transformation and its application for the practical synthesis of α-amino acids. Org. Biomol. Chem. 2018, 16, 4968–4972. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Yamamoto, J.; Sato, T.; Moriwaki, H.; Izawa, K.; Abe, H.; Soloshonok, V.A. Tandem alkylation—Second-order asymmetric transformation protocol for preparation of phenylalanine-type tailor-made α-amino acids. ACS Omega 2018, 3, 9729–9737. [Google Scholar] [CrossRef]
- Han, J.; Takeda, R.; Sato, T.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. Optical Resolution of Rimantadine. Molecules 2019, 24, 1828. [Google Scholar] [CrossRef]
- Nian, Y.; Wang, J.; Zhou, S.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V.A.; Liu, H. Recyclable Ligands for the Non-Enzymatic Dynamic Kinetic Resolution of Challenging α-Amino Acids. Angew. Chem. Int. Ed. 2015, 54, 12918–12922. [Google Scholar] [CrossRef]
- Romoff, T.T.; Palmer, A.B.; Mansour, N.; Creighton, C.J.; Miwa, T.; Ejima, Y.; Moriwaki, H.; Soloshonok, V.A. Scale-up Synthesis of (R)- and (S)-N-(2-benzoyl-4-chlorophenyl)-1-(3,4-dichlorobenzyl)pyrrolidine-2-carboxamide hydrochloride, a Versatile Reagent for Preparation of Tailor-made α- and β-Amino Acids in Enantiomerically Pure Form. Org. Process Res. Dev. 2017, 21, 732–739. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ellis, T.K.; Ueki, H.; Ono, T. Resolution/Deracemization of Chiral-Amino Acids Using Resolving Reagents with Flexible Stereogenic Centers. J. Am. Chem. Soc. 2009, 131, 7208–7209. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Ueki, H.; Aceña, J.L.; Ellis, T.K.; Moriwaki, H.; Soloshonok, V.A. Chemical approach for interconversion of (S)- and (R)-α-amino acids. Org. Biomol. Chem. 2013, 11, 4503–4507. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Ueki, H.; Aceña, J.L.; Ellis, T.K.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Chemical deracemization and (S) to (R) interconversion of some fluorine-containing α-amino acids. J. Fluor. Chem. 2013, 152, 114–118. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, J.; Chen, X.; Aceña, J.L.; Soloshonok, V.A.; Liu, H. Chemical Kinetic Resolution of Unprotected-Substituted-Amino Acids Using Recyclable Chiral Ligands. Angew. Chem. Int. Ed. 2014, 53, 7883–7886. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.K.; Hochla, V.M.; Soloshonok, V.A. Efficient Synthesis of 2-Aminoindane-2-Carboxylic Acid via Dialkylation of Nucleophilic Glycine Equivalent. J. Org. Chem. 2003, 68, 4973–4976. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.K.; Martin, C.H.; Tsai, G.M.; Ueki, H.; Soloshonok, V.A. Efficient Synthesis of Sterically Constrained Symmetrically, -Disubstituted-Amino Acids under Operationally Convenient Conditions. J. Org. Chem. 2003, 68, 6208–6214. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Avilov, D.V.; Kukhar, V.P. Highly Diastereoselective Asymmetric Aldol Reactions of Chiral Ni(II)-Complex of Glycine with Trifluoromethyl Ketones. Tetrahedron 1996, 7, 1547–1550. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Avilov, D.V.; Kukhar, V.P. Asymmetric Aldol Reactions of Trifluoromethyl Ketones with a Chiral Ni(II) Complex of Glycine: Stereocontrolling Effect of the Trifluoromethyl Group. Tetrahedron 1996, 52, 12433–12442. [Google Scholar] [CrossRef]
- Kawamura, A.; Moriwaki, H.; Röschenthaler, G.V.; Kawada, K.; Aceña, J.L.; Soloshonok, V.A. Synthesis of (2S,3S)-β-(trifluoromethyl)-α,β-diamino acid by Mannich addition of glycine Schiff base Ni(II) complexes to N-tert-butylsulfinyl-3,3,3-trifluoroacetaldimine. J. Fluor. Chem. 2015, 171, 67–72. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Avilov, D.V.; Kukhar, V.P.; Meervelt, L.V.; Mischenko, N. Highly Diastereoselective aza-Aldol Reactions of a Chiral Ni(II) Complex of Glycine with Imines. An Efficient Asymmetric Approach to 3-Perfluoroalkyl-2,3-Diamino Acids. Tetrahedron Lett. 1997, 38, 4671–4674. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J. (S)- or (R)-N-(E-enoyl)-4-phenyl-1,3-oxazolidin-2-ones: Ideal Michael Acceptors to Afford a Virtually Complete Control of Simple and Face Diastereoselectivity in Addition Reactions with Glycine Derivatives. Org. Lett. 2000, 2, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V.; Yamazaki, T. Rational Design of Highly Diastereoselective, Organic Base-Catalyzed, Room Temperature Michael Addition Reactions. J. Org. Chem. 2000, 65, 6688–6696. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V.A. Efficient Asymmetric Synthesis of Novel 4-Substituted and Configurationally Stable Analogs of Thalidomide. Org. Lett. 2006, 8, 5625–5628. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Xie, C.; Mei, H.; Takeda, R.; Kawamura, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Han, J.; Aceña, J.L.; et al. Asymmetric synthesis of (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid by sequential SN2–SN2′ dialkylation of (R)-N-(benzyl)proline-derived glycine Schiff base Ni(II) complex. RSC Adv. 2015, 5, 1051–1058. [Google Scholar] [CrossRef]
- Kawashima, A.; Shu, S.; Takeda, R.; Kawamura, A.; Sato, T.; Moriwaki, H.; Wang, J.; Izawa, K.; Aceña, J.L.; Soloshonok, V.A.; et al. Advanced asymmetric synthesis of (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid by alkylation/cyclization of newly designed axially chiral Ni(II) complex of glycine Schiff base. Amino Acids 2016, 48, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, D.; Zhou, S.; Soloshonok, V.A.; Liu, H. Asymmetric Synthesis of Sterically Constrained Linear Trifluoromethyl Containing Amino Acids via Alkylation of Chiral Equivalents of Nucleophilic Glycine and Alanine. J. Org. Chem. 2011, 76, 684–687. [Google Scholar] [CrossRef]
- Tang, X.; Soloshonok, V.A.; Hruby, V.J. Convenient Asymmetric Synthesis of Enantiomerically Pure 2’,6’-Dimethyltyrosine (DMT) via Alkylation of Chiral Nucleophilic Glycine Equivalent. Tetrahedron 2000, 11, 2917–2925. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ono, T.; Ueki, H.; Vanthuyne, N.; Balaban, T.S.; Bürck, J.; Fliegl, H.; Klopper, W.; Naubron, J.V.; Tam, T.T.; et al. Ridge-tile-like chiral topology: Synthesis, resolution and complete chiroptical characterization of enantiomers of edge-sharing binuclear square planar complexes of Ni(II) bearing achiral ligands. J. Am. Chem. Soc. 2010, 132, 10477–10483. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H. Design, Synthesis and Characterization of Binuclear Ni(II) Complexes with Inherent Helical Chirality. J. Am. Chem. Soc. 2007, 129, 2426–2427. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Scale | Conditions | Results |
|---|---|---|---|
| 1 | (S)-6 80 g | ICH2CF3 (1.05 eq. 29.32 g), KOH (96.8% assay, 1.05 eq. 8.14 g), MeOH (0.9 v, 72 mL), DMF (10 v, 800 mL), rt (room temperature), 1 h | (S)(2S)-7 73.7 g, 81.1% yield, >99% de (diasteromeric excess) |
| 2 | (S)-6 250 g | ICH2CF3 (1.05 eq. 91.61 g), KOH (96.8% assay, 1.05 eq. 25.42 g), MeOH (0.9 v, 225 mL), DMF (10 v, 2500 mL), rt, 1 h | (S)(2S)-7 229.1 g, 80.7% yield, 99.9% de |
| 3 | (S)-6 250 g | ICH2CF3 (1.05 eq. 91.61 g), KOH (96.8% assay, 1.05 eq. 25.42 g), MeOH (0.9 v, 225 mL), DMF (10 v, 2500 mL), rt, 1 h | (S)(2S)-7 228.5 g, 80.4% yield, >99% de |
| Entry | (S)(2S)-7 | Conditions | (S)-12 |
|---|---|---|---|
| 1 | 10 g | 6N HCl (5 eq. 12.2 mL), DME (2 v, 20 mL), 50 °C, 1 h added H2O (2 v, 20 mL), 30 °C, 2 h then EDTA•2Na (1.02 eq. 5.55 g), 48% NaOH (4.2 eq. 5.1 g) Na2CO3 (1.3 eq. 2.02 g), Fmoc-OSu (1 eq. 4.94 g) MeCN (4 v, 9.2 mL), rt, 3 h | 4.26 g, 76.7% yield 99.0% ee (enantiomeric excess) |
| 2 | 220 g | 6N HCl (5 eq. 268 mL), DME (2 v, 440 mL), 50 °C, 1 h added H2O (2 v, 440 mL), 30 °C, 2 h then EDTA•2Na (1.02 eq. 122.2 g), 48% NaOH (5.4 eq. 143.6 g) Na2CO3 (1.3 eq. 44.35 g), Fmoc-OSu (1 eq. 108.57 g) MeCN (4 v, 200 mL), rt, 4 h | 92.7 g, 75.9% yield 98.8% ee |
| 3 | 220 g | 6N HCl (5 eq. 268 mL), DME (2 v, 440 mL), 50 °C, 1 h added H2O (2 v, 440 mL), 30 °C, 1.5 h then EDTA•2Na (1.02 eq. 122.2 g), 48% NaOH (5.2 eq. 138 g) Na2CO3 (1.3 eq. 44.35 g), Fmoc-OSu (1 eq. 108.57 g) MeCN (4 v, 200 mL), rt, 4 h | 99.5 g, 81.5% yield 98.4% ee |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Takeda, R.; Liu, X.; Konno, H.; Abe, H.; Hiramatsu, T.; Moriwaki, H.; Soloshonok, V.A. Preparative Method for Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid. Molecules 2019, 24, 4521. https://doi.org/10.3390/molecules24244521
Han J, Takeda R, Liu X, Konno H, Abe H, Hiramatsu T, Moriwaki H, Soloshonok VA. Preparative Method for Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid. Molecules. 2019; 24(24):4521. https://doi.org/10.3390/molecules24244521
Chicago/Turabian StyleHan, Jianlin, Ryosuke Takeda, Xinyi Liu, Hiroyuki Konno, Hidenori Abe, Takahiro Hiramatsu, Hiroki Moriwaki, and Vadim A. Soloshonok. 2019. "Preparative Method for Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid" Molecules 24, no. 24: 4521. https://doi.org/10.3390/molecules24244521
APA StyleHan, J., Takeda, R., Liu, X., Konno, H., Abe, H., Hiramatsu, T., Moriwaki, H., & Soloshonok, V. A. (2019). Preparative Method for Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid. Molecules, 24(24), 4521. https://doi.org/10.3390/molecules24244521