Solubility and Bioavailability Enhancement of Oridonin: A Review


Abstract
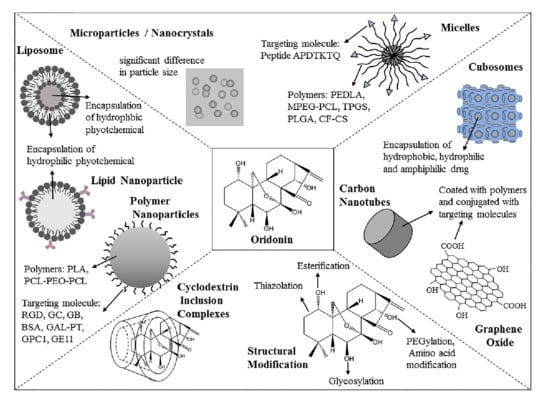
1. Introduction
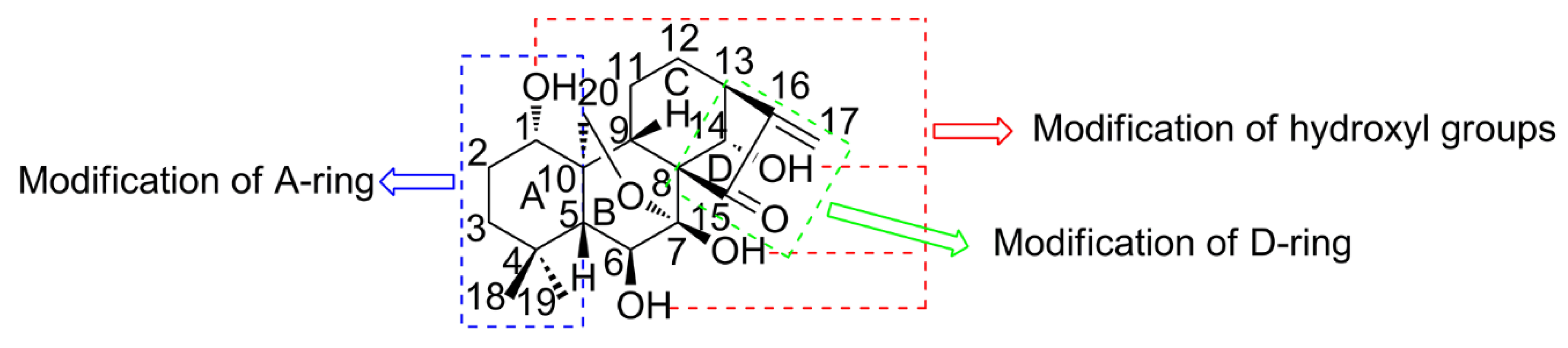
2. Strategies for Structural Modification
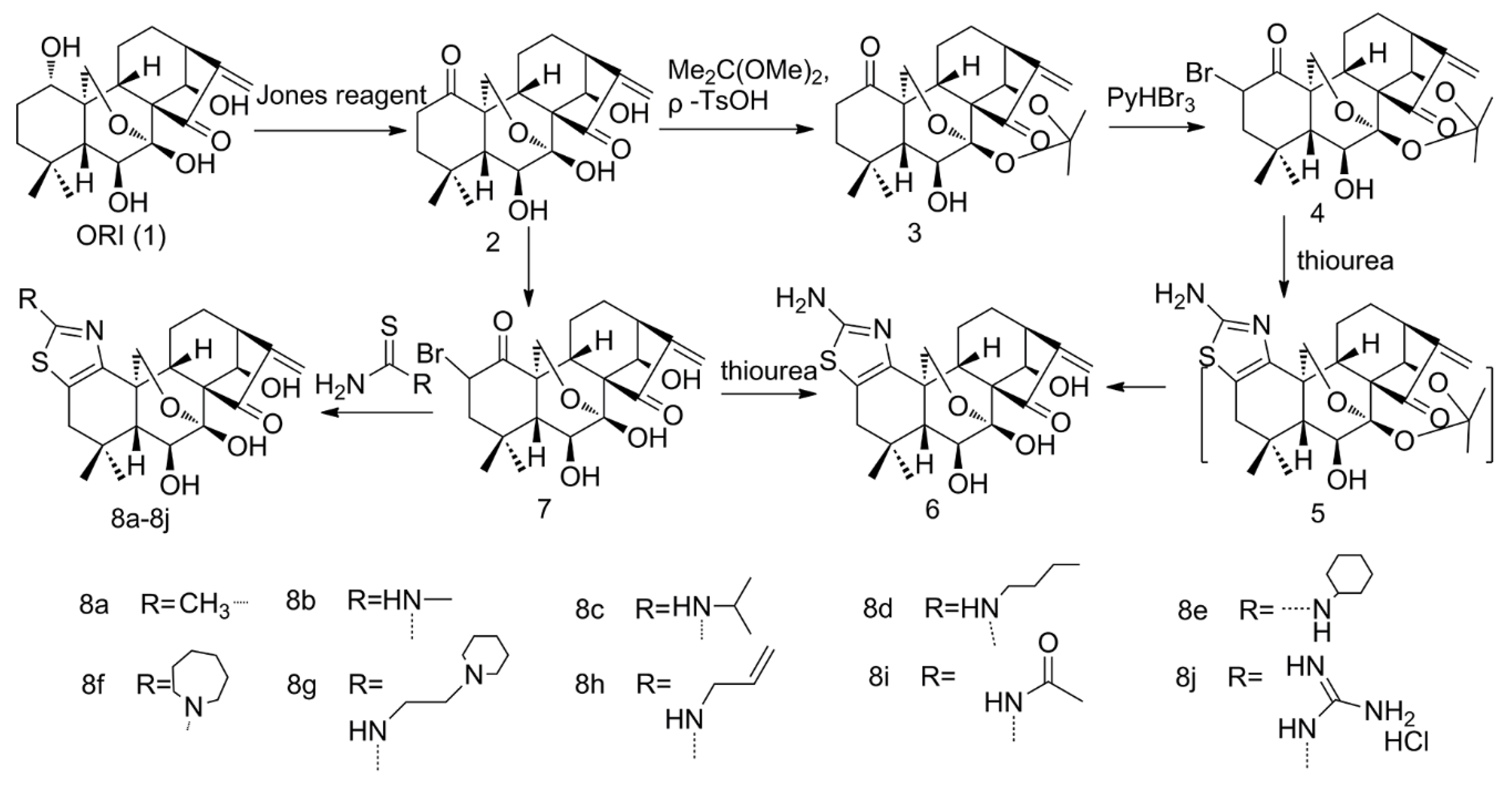
2.1. Thiazolation of ORI
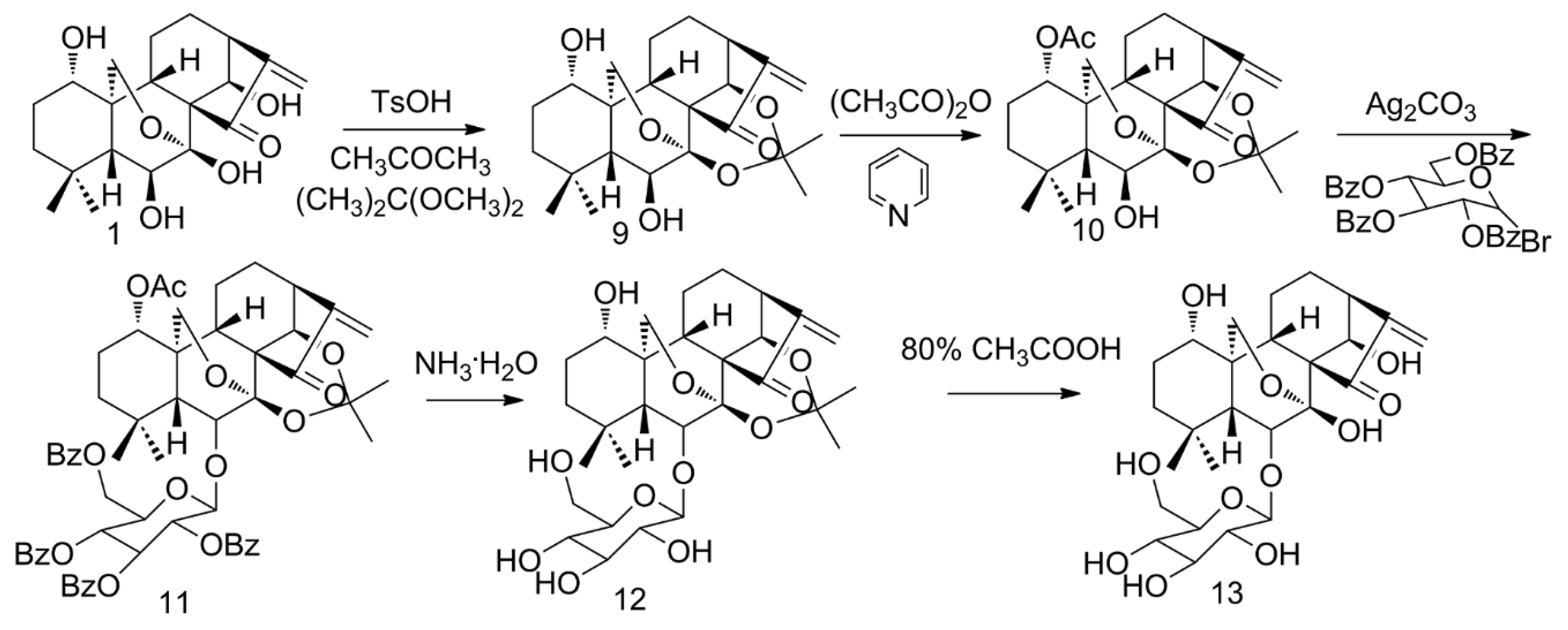
2.2. Glycosylation of ORI
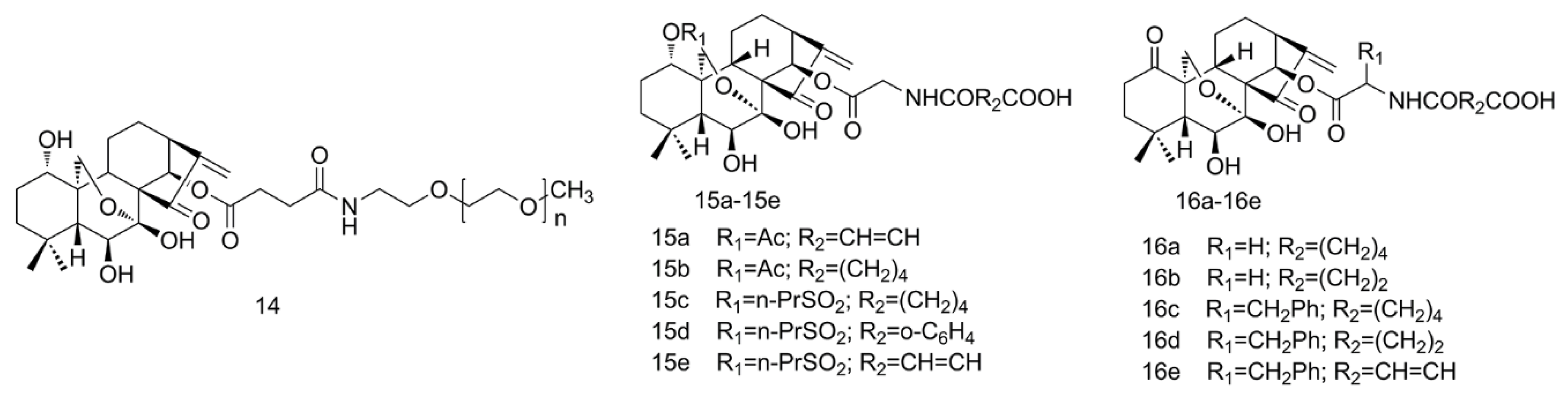
2.3. PEGylation and Esterification of ORI
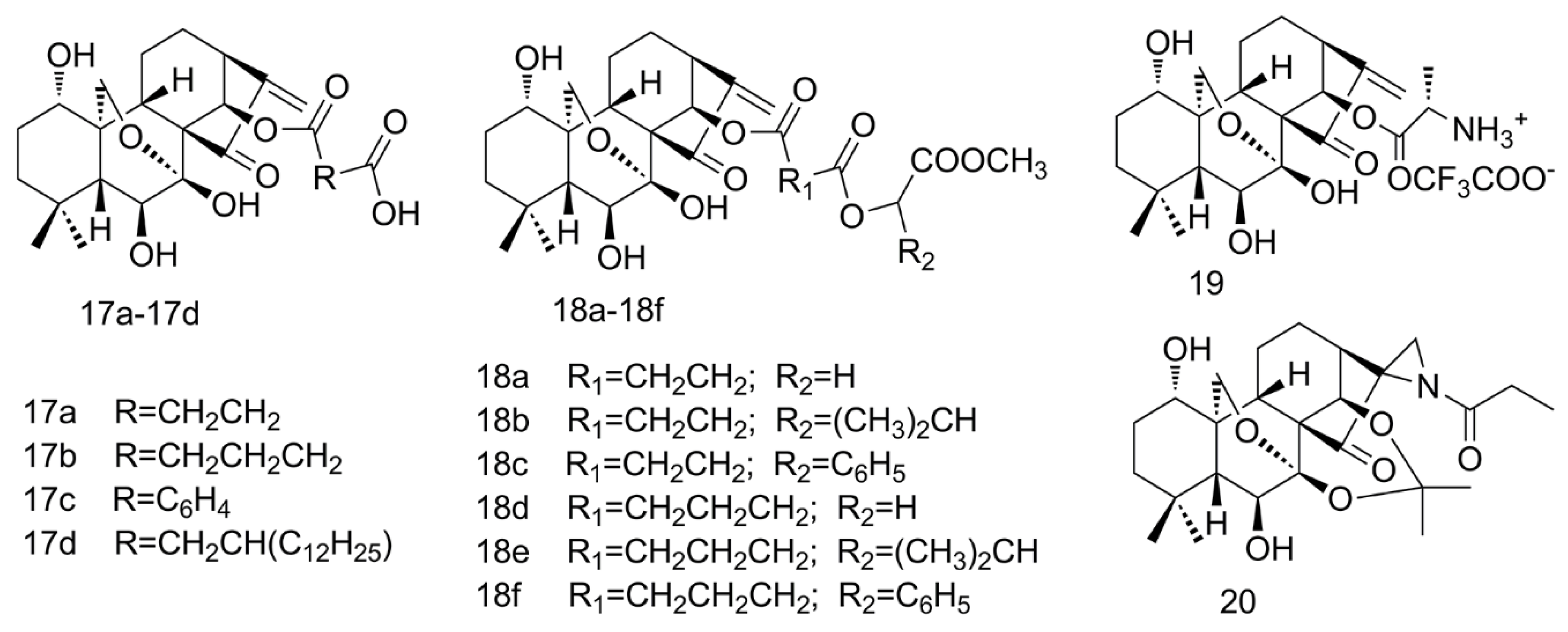
2.4. Amino Acid Modification of ORI
2.5. Aziridination of ORI
3. Strategizes for Pharmaceutical Formulations
3.1. Cyclodextrin Inclusion Complexes
3.2. Microparticles
3.3. Nanocrystals
3.4. Nanoparticles
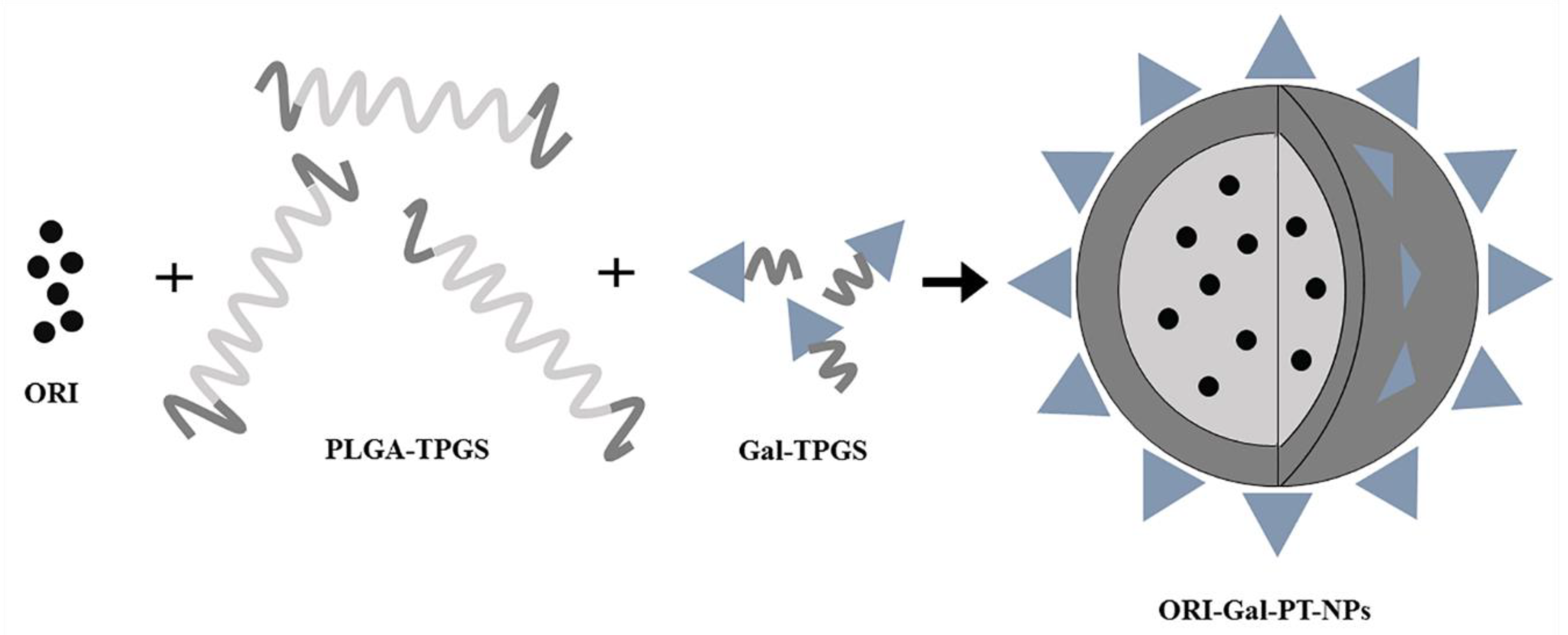
3.4.1. Polymer Nanoparticles
3.4.2. Lipid Nanoparticles
Solid Lipid Nanoparticles
Nanostructured Lipid Carriers
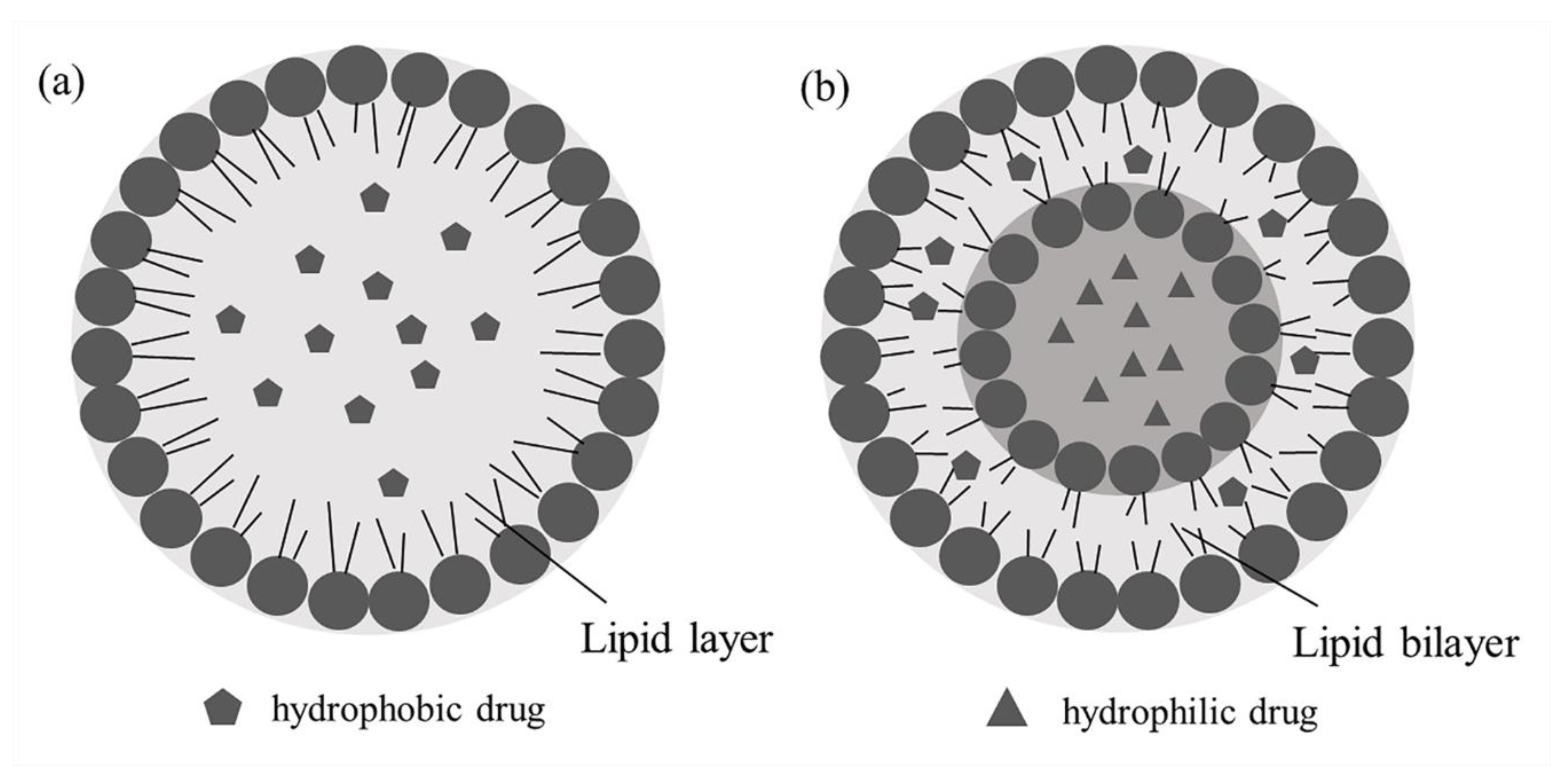
3.5. Liposomes
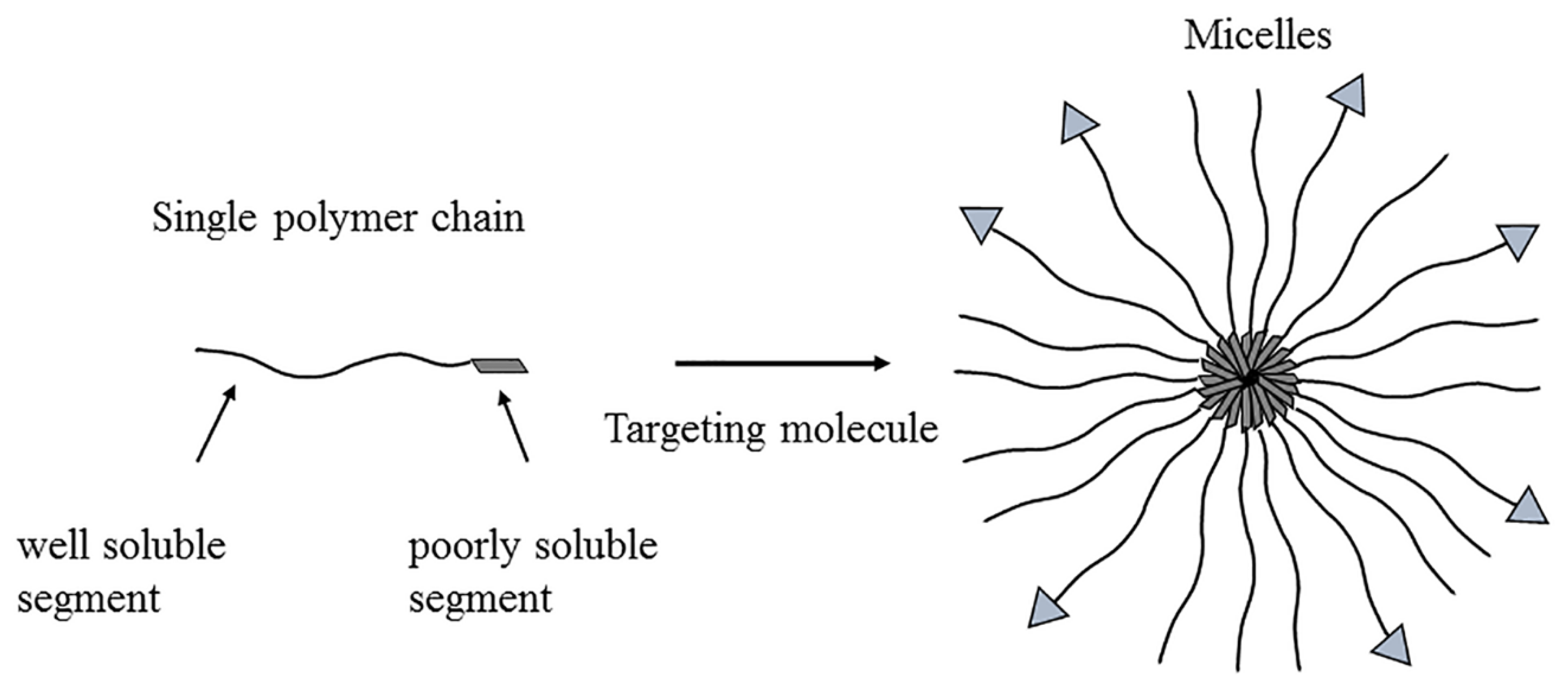
3.6. Micelles
3.7. Combination of Formulations
3.8. Others
4. Conclusions
Funding
Conflicts of Interest
References
- Osawa, K.; Yasuda, H.; Maruyama, T.; Morita, H.; Takeya, K.; Itokawa, H. Antibacterial trichorabdal diterpenes from Rabdosia trichocarpa. Phytochemistry 1994, 36, 1287–1291. [Google Scholar] [CrossRef]
- Zhang, J.X.; Han, Q.B.; Zhao, A.H.; Sun, H.D. Diterpenoids from Isodon japonica. Fitoterapia 2003, 74, 435–438. [Google Scholar] [CrossRef]
- Li, C.Y.; Wang, E.Q.; Cheng, Y.; Bao, J.K. Oridonin: An active diterpenoid targeting cell cycle arrest, apoptotic and autophagic pathways for cancer therapeutics. Int. J. Biochem. Cell Biol. 2011, 43, 701–704. [Google Scholar] [CrossRef]
- Tian, W.; Chen, S.Y. Recent advances in the molecular basis of anti-neoplastic mechanisms of oridonin. Chin. J. Integr. Med. 2013, 19, 315–320. [Google Scholar] [CrossRef]
- Xu, S.; Pei, L.; Li, D.; Yao, H.; Cai, H.; Yao, H.; Wu, X.; Xu, J. Synthesis and antimycobacterial evaluation of natural oridonin and its enmein-type derivatives. Fitoterapia 2014, 99, 300–306. [Google Scholar] [CrossRef]
- Xue, Y.; Wang, Y.; Feng, D.; Lin, S.; Xu, L. Multiple -modulation effects of Oridonin on the production of proinflammatory cytokines and neurotrophic factors in LPS-activated microglia. Int. Immunopharmacol. 2009, 9, 360–365. [Google Scholar] [CrossRef]
- Liu, G.A.; Ding, L.; Yang, Y.; Yang, H.; Yang, Q.M.; Wang, H.Q. Anti-oxidative action of ent-kaurene diterpenoids. Res. Chem. Intermediat. 2006, 32, 787–794. [Google Scholar] [CrossRef]
- Wang, L.X.; Sun, Y.; Chen, C.; Huang, X.Y.; Lin, Q.; Qian, G.Q.; Dong, W.; Chen, Y.F. Effects and mechanism of oridonin on pulmonary hypertension induced by chronic hypoxia-hypercapnia in rats. Chin. Med. J. 2009, 122, 1380–1387. [Google Scholar]
- Chen, R.Y.; Xu, B.; Chen, S.F.; Chen, S.S.; Zhang, T.; Ren, J.; Xu, J. Effect of oridonin-mediated hallmark changes on inflammatory pathways in human pancreatic cancer (BxPC−3) cells. World. J. Gastroenterol. 2014, 20, 14895–14903. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550–2561. [Google Scholar] [CrossRef]
- Xu, J.; Wold, E.A.; Ding, Y.; Shen, Q.; Zhou, J. Therapeutic potential of oridonin and its analogs: From anticancer and antiinflammation to neuroprotection. Molecules 2018, 23, 474. [Google Scholar] [CrossRef]
- Hu, A.P.; Du, J.M.; Li, J.Y.; Liu, J.W. Oridonin promotes CD4+ /CD25+ Treg differentiation, modulates Th1/Th2 balance and induces HO−1 in rat splenic lymphocytes. Inflamm. Res. 2008, 57, 163–170. [Google Scholar] [CrossRef]
- Zang, K.H.; Shao, Y.Y.; Zuo, X.; Rao, Z.; Qin, H.Y. Oridonin alleviates visceral hyperalgesia in a rat model of postinflammatory irritable bowel syndrome: Role of colonic enterochromaffin cell and serotonin availability. J. Med. Food 2016, 19, 586–592. [Google Scholar] [CrossRef]
- Song, M.; Liu, X.; Liu, K.; Zhao, R.; Huang, H.; Shi, Y.; Zhang, M.; Zhou, S.; Xie, H.; Chen, H.; et al. Targeting AKT with oridonin inhibits growth of esophageal squamous cell carcinoma in vitro and patient-derived xenografts in vivo. Mol. Cancer Ther. 2018, 17, 1540–1553. [Google Scholar] [CrossRef]
- Li, S.; Shi, D.; Zhang, L.; Yang, F.; Cheng, G. Oridonin enhances the radiosensitivity of lung cancer cells by upregulating Bax and downregulating Bcl-2. Exp. Ther. Med. 2018, 16, 4859–4864. [Google Scholar] [CrossRef]
- Zhang, J.F.; Liu, J.J.; Liu, P.Q.; Lin, D.J.; Li, X.D.; Chen, G.H. Oridonin inhibits cell growth by induction of apoptosis on human hepatocelluar carcinoma BEL-7402 cells. Hepatol. Res. 2006, 35, 104–110. [Google Scholar] [CrossRef]
- Ming, M.; Sun, F.Y.; Zhang, W.T.; Liu, J.K. Therapeutic effect of oridonin on mice with prostate cancer. Asian Pac. J. Trop. Med. 2016, 9, 182–185. [Google Scholar] [CrossRef]
- Li, C.; Wang, Q.; Shen, S.; Wei, X.; Li, G. Oridonin inhibits VEGF-A-associated angiogenesis and epithelial-mesenchymal transition of breast cancer in vitro and in vivo. Oncol. Lett. 2018, 16, 2289–2298. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, Q.; Huang, D.; He, L.; Zhang, H.; Hu, B.; Peng, H.; Ren, D. ROS/JNK/c-Jun axis is involved in oridonin-induced caspase-dependent apoptosis in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2019, 513, 594–601. [Google Scholar] [CrossRef]
- Ding, C.; Zhang, Y.; Chen, H.; Yang, Z.; Wild, C.; Ye, N.; Ester, C.D.; Xiong, A.; White, M.A.; Shen, Q.; et al. Oridonin ring A-based diverse constructions of enone functionality: Identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J. Med. Chem. 2013, 56, 8814–8825. [Google Scholar] [CrossRef]
- Node, M.; Sai, M.; Fuji, K.; Fujita, E.; Takeda, S.; Unemi, N. Antitumor activity of diterpenoids, trichorabdals A, B, and C, and the related compounds: Synergism of two active sites. Chem. Pharm. Bull. 1983, 31, 1433–1436. [Google Scholar] [CrossRef][Green Version]
- Sun, H.D.; Lin, Z.W.; Niu, F.D.; Shen, P.Q.; Pan, L.T.; Lin, L.Z.; Cordell, G.A. Diterpenoids from Isodon eriocalyx var. laxiflora. Phytochemistry 1995, 38, 1451–1455. [Google Scholar] [CrossRef]
- Yang, Y.C.; Wei, M.C. Kinetic and characterization studies for three bioactive compounds extracted from Rabdosia rubescens using ultrasound. Food Bioprod. Process 2015, 94, 101–113. [Google Scholar] [CrossRef]
- Xu, W.; Sun, J.; Zhang, T.T.; Ma, B.; Cui, S.M.; Chen, D.W.; He, Z.G. Pharmacokinetic behaviors and oral bioavailability of oridonin in rat plasma. Acta. Pharmacol. Sin. 2006, 27, 1642–1646. [Google Scholar] [CrossRef]
- Chen, S.; Liu, J.; Zhang, H. Efficacy of rabdosia rubescens in the treatment of gingivitis. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2009, 29, 659–663. [Google Scholar] [CrossRef]
- Cao, S.; Huang, Y.; Zhang, Q.; Lu, F.; Donkor, P.O.; Zhu, Y.; Qiu, F.; Kang, N. Molecular mechanisms of apoptosis and autophagy elicited by combined treatment with oridonin and cetuximab in laryngeal squamous cell carcinoma. Apoptosis 2019, 24, 33–45. [Google Scholar] [CrossRef]
- Zhu, L.; Li, M.; Liu, X.; Jin, Y. Drug-loaded PLGA electrospraying porous microspheres for the local therapy of primary lung cancer via pulmonary delivery. ACS Omega 2017, 2, 2273–2279. [Google Scholar] [CrossRef]
- Ding, Y.; Ding, C.Y.; Ye, N.; Liu, Z.Q.; Wold, E.A.; Chen, H.Y.; Wild, C.; Shen, Q.; Zhou, J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016, 122, 102–117. [Google Scholar] [CrossRef]
- Cheng, W.Y.; Huang, C.H.; Ma, W.F.; Tian, X.J.; Zhang, X.J. Recent Development of oridonin derivatives with diverse pharmacological activities. Mini Rev. Med. Chem. 2019, 19, 114–124. [Google Scholar] [CrossRef]
- Hutchinson, I.; Jennings, S.A.; Vishnuvajjala, B.R.; Westwell, A.D.; Stevens, M.F.G. Antitumor benzothiazoles. 16 Synthesis and pharmaceutical properties of antitumor 2-(4-aminophenyl)-benzothiazole amino acid prodrugs. J. Med. Chem. 2002, 45, 744–747. [Google Scholar] [CrossRef]
- Ding, C.; Zhang, Y.; Chen, H.; Yang, Z.; Wild, C.; Chu, L.; Liu, H.; Shen, Q.; Zhou, J. Novel nitrogen-enriched oridonin analogues with thiazole-fused a-ring: Protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J. Med. Chem. 2013, 56, 5048–5058. [Google Scholar] [CrossRef]
- Cummins, C.B.; Wang, X.F.; Xu, J.M.; Hughes, B.D.; Ding, Y.; Chen, H.Y.; Zhou, J.; Radhakrishnan, R.S. Antifibrosis Effect of Novel Oridonin Analog CYD0618 Via Suppression of the NF-κB Pathway. J. Surg. Res. 2018, 232, 283–292. [Google Scholar] [CrossRef]
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014, 343, 37–45. [Google Scholar] [CrossRef]
- YAN, X.B.; Lei, M.; Zhang, Y.J.; Liu, H.M. Synthesis of oridonin glucopyranoside. J. Org. Chem. 2005, 25, 222–224. [Google Scholar]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta. 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Greenwald, R.B. PEG drugs: An overview. J. Control. Release 2001, 74, 159–171. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. Bio. Drugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, D.; Zhao, Z.; Jia, L.; Zheng, D.; Liu, G.; Hao, L.; Zhang, Q.; Tian, X.; Li, C.; et al. Synthesis characterization, in vitro and in vivo evaluation of PEGylated oridonin conjugates. Int. J. Pharm. 2013, 456, 80–86. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Ran, Q.; Wang, L.; Liu, J.; Wang, Z.; Wu, X.; Hua, W.; Yuan, S.; Zhang, L.; et al. Synthesis and biological evaluation of novel 1-O- and 14-O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 4741–4744. [Google Scholar] [CrossRef]
- Wu, G.Y. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef]
- Vig, B.S.; Huttunen, K.M.; Laine, K.; Rautio, J. Amino acids as promoieties in prodrug design and development. Adv. Drug Deliv. Rev. 2013, 65, 1370–1385. [Google Scholar] [CrossRef]
- Wang, L.; Ran, Q.; Li, D.H.; Yao, H.Q.; Zhang, Y.H.; Yuan, S.T.; Zhang, L.Y.; Shen, M.Q.; Xu, J.Y. Synthesis and anti-tumor activity of 14-O-derivatives of natural oridonin. Chin. J. Nat. Med. 2011, 9, 194–198. [Google Scholar]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef]
- Sun, P.Y.; Wu, G.L.; Qiu, Z.J.; Chen, Y.J. L-alanine-(14-oridonin) Ester Trifluoroacetate as Well as Preparation Method and Application. Chinese Patent CN 104,017,000 A, 3 September 2014. [Google Scholar]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; Gonzalez-Paez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.J.; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [Google Scholar] [CrossRef]
- Ding, Y.; Li, D.F.; Ding, C.Y.; Wang, P.Y.; Liu, Z.Q.; Wold, E.A.; Ye, N.; Chen, H.Y.; White, M.A.; Shen, Q.; et al. Regio- and Stereospecific Synthesis of Oridonin D-Ring Aziridinated Analogues for the Treatment of Triple-Negative Breast Cancer via Mediated Irreversible Covalent Warheads. J. Med. Chem. 2018, 61, 2737–2752. [Google Scholar] [CrossRef]
- Brewster, M.E.; Vandecruys, R.; Peeters, J.; Neeskens, P.; Verreck, G.; Loftsson, T. Comparative interaction of 2-hydroxypropyl-β-cyclodextrin and sulfobutylether-β-cyclodextrin with itraconazole: Phase-solubility behavior and stabilization of supersaturated drug solutions. Eur. J. Pharm. Sci. 2008, 34, 94–103. [Google Scholar] [CrossRef]
- Grau, M.J.; Kayser, O.; Müller, R.H. Nanosuspensions of poorly soluble drugs—Reproducibility of small scale production. Int. J. Pharm. 2000, 196, 155–159. [Google Scholar] [CrossRef]
- Prof, D.I.; Wolfram, S. Cyclodextrin inclusion compounds in research and industry. Angew. Chem. Int. Ed. 1980, 19, 344–362. [Google Scholar]
- Liu, W.; Zhao, B.; Li, Y.C.; Liu, H.M. NMR spectra and structures of oridonin derivatives complexes with β-cyclodextrin. Magn. Reson. Chem. 2011, 49, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Pralhad, T.; Rajendrakumar, K. Study of freeze-dried QC cyclodextrin binary systems by DSC, FT-IR, X-ray diffraction and SEM analysis. J. Pharm. Biomed. Anal. 2004, 34, 333–339. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, W.; Sun, J.; Liu, X.; Zhao, Y.; Sun, Y.; Zhang, T.; He, Z. Characterization and in vivo evaluation of an inclusion complex of oridonin and 2-hydroxypropyl-beta-cyclodextrin. Drug Dev. Ind. Pharm. 2008, 34, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Halasz, K.; Kelly, S.J.; Iqbal, M.T.; Pathak, Y.; Sutariya, V. Micro/nanoparticle delivery systems for ocular diseases. Assay Drug Dev. Technol. 2019, 17, 152–166. [Google Scholar] [CrossRef]
- He, J.; Cao, Y.; Yuan, Q. Preparation and Photocytotoxicity in vitro of oridonin-porphyrin-chitosan microspheres. J. Org. Chem. 2017, 37, 759–766. [Google Scholar]
- Zhu, L.; Li, M.; Liu, X.; Du, L.; Jin, Y. Inhalable oridonin-loaded poly(lactic–glycolic) acid large porous microparticles for treatment of primary non-small cell lung cancer. Acta Pharm. Sin. B 2017, 7, 80–90. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Hu, B. The anti-tumor effect of folate-targeted liposome microbubbles loaded with oridonin as ultrasound-triggered tumor-targeted therapeutic carrier system. J. Drug Target. 2017, 25, 83–91. [Google Scholar] [CrossRef]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenization. Eur. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Panmai, S.; Wu, Y.H. Nanosizing-oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef]
- Roa, W.H.; Azarmi, S.; Al-Hallak, M.H.D.K.; Finlay, W.H.; Magliocco, A.M.; Löbenberg, R. Inhalable nanoparticles, a noninvasive approach to treat lung cancer in a mouse model. J. Control. Release 2011, 150, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Miconazole nanosuspensions: Influence of formulation variables on particle size reduction and physical stability. Int. J. Pharm. 2010, 396, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Patravale, V.B.; Date, A.A.; Kulkarni, R.M. Nanosuspension: A promsing drug delivery strategy. J. Pharm. Pharmacol. 2004, 56, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.P.; Müller, R.H. Production and characterisation of highly concentrated nanosuspensions by high pressure homogenisation. Int. J. Pharm. 2001, 214, 21–24. [Google Scholar] [CrossRef]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, D.; Chen, M.; Zheng, T.; Wang, S. Preparation and characterization of an oridonin nanosuspension for solubility and dissolution velocity enhancement. Drug Dev. Ind. Pharm. 2007, 33, 1332–1339. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, D.; Chen, M.; Duan, C.; Dai, W.; Jia, L.; Zhao, W. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int. J. Pharm. 2008, 355, 321–327. [Google Scholar] [CrossRef]
- Lou, H.; Zhang, X.; Gao, L.; Feng, F.; Wang, J.; Wei, X.; Yu, Z.; Zhang, D.; Zhang, Q. In vitro and in vivo antitumor activity of oridonin nanosuspension. Int. J. Pharm. 2009, 379, 181–186. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Xue, W.; Yang, Y.; Xu, D.; Zhao, Y.; Lou, H. Effects of oridonin nanosuspension on cell proliferation and apoptosis of human prostatic carcinoma PC-3 cell line. Int. J. Nanomed. 2010, 5, 735–742. [Google Scholar] [CrossRef]
- Lou, H.; Gao, L.; Wei, X.; Zhang, Z.; Zheng, D.; Zhang, D.; Zhang, X.; Li, Y.; Zhang, Q. Oridonin nanosuspension enhances anti-tumor efficacy in SMMC−7721 cells and H22 tumor bearing mice. Colloids Surf. B Biointerfaces 2011, 87, 319–325. [Google Scholar] [CrossRef]
- Feng, F.F.; Zhang, D.R.; Tian, K.L.; Lou, H.Y.; Qi, X.L.; Wang, Y.C.; Duan, C.X.; Jia, L.J.; Wang, F.H.; Liu, Y.; et al. Growth inhibition and induction of apoptosis in MCF−7 breast cancer cells by oridonin nanosuspension. Drug Deliv. 2011, 18, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, D.; Xu, X.; Feng, F.; Ren, G.; Chu, Q.; Zhang, Q.; Tian, K. Oridonin nanosuspension was more effective than free oridonin on G2/M cell cycle arrest and apoptosis in the human pancreatic cancer PANC-1 cell line. Int. J. Nanomed. 2012, 7, 1793–1804. [Google Scholar]
- Liu, P.; Rong, X.; Laru, J.; Van, V.B.; Kiesvaara, J.; Hirvonen, J.; Laaksonen, T.; Peltonen, L. Nanosuspensions of poorly soluble drugs: Preparation and development by wet milling. Int. J. Pharm. 2011, 411, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, A.; Liu, P.; Puranen, J.; Rönkkö, S.; Laaksonen, T.; Kalesnykas, G.; Oksala, O.; Ilkka, J.; Laru, J.; Järvinen, K.; et al. Brinzolamide nanocrystal formulations for ophthalmic delivery: Reduction of elevated intraocular pressure in vivo. Int. J. Pharm. 2014, 467, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, T.; Lan, Y.; Wu, B.; Shi, Z. Nanosuspensions containing oridonin/HP-β-cyclodextrin inclusion complexes for oral bioavailability enhancement via improved dissolution and permeability. AAPS PharmSciTech 2016, 17, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, D.; Tan, T. Studies on the oridonin-loaded poly (d,l-lactic acid) nanoparticles in vitro and in vivo. Int. J. Biol. Macromol. 2007, 40, 153–158. [Google Scholar] [CrossRef]
- Liu, Y.T.; Feng, S.S. Surfactant chain length effects on nanoparticles of biodegradable polymers for targeted drug delivery. AIChE J. 2012, 58, 3289–3297. [Google Scholar] [CrossRef]
- Feng, N.; Wu, P.; Li, Q.; Mei, Y.; Shi, S.; Yu, J.; Xu, J.; Liu, Y.; Wang, Y. Oridonin-loaded poly(ε-caprolactone)-poly (ethylene oxide)-poly(ε-caprolactone) copolymer nanoparticles: Preparation, characterization, and antitumor activity on mice with transplanted hepatoma. J. Drug Target. 2008, 16, 479–485. [Google Scholar] [CrossRef]
- Mei, Y.; Xu, J.; Zhao, J.; Feng, N.; Liu, Y.; Wei, L. An HPLC method for determination of oridonin in rabbits using isopsoralen as an internal standard and its application to pharmacokinetic studies for oridonin-loaded nanoparticles. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 869, 138–141. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, J.H.; Liu, Y.; Feng, N.P.; Zhang, Y.T. RGD-modified poly (D, L-lactic acid) nanoparticles enhance tumor targeting of oridonin. Int. J. Nanomed. 2012, 7, 211–219. [Google Scholar]
- Tian, Q.; Zhang, C.N.; Wang, X.H.; Wang, W.; Huang, W.; Cha, R.T.; Wang, C.H.; Yuan, Z.; Liu, M.; Wan, H.Y.; et al. Glycyrrhetinic acid-modified chitosan/poly (ethylene glycol) nanoparticles for liver-targeted delivery. Biomaterials 2010, 31, 4748–4756. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Duan, C.; Zhang, D.; Jia, L.; Liu, G.; Liu, Y.; Wang, F.; Li, C.; Guo, H.; Zhang, Q. Galactosylated chitosan nanoparticles for epatocyte-targeted delivery of oridonin. Int. J. Pharm. 2012, 436, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, D.; Guo, H.; Hao, L.; Zheng, D.; Liu, G.; Shen, J.; Tian, X.; Zhang, Q. Preparation and characterization of galactosylated bovine serum albumin nanoparticles for liver-targeted delivery of oridonin. Int. J. Pharm. 2013, 448, 79–86. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, D.R.; Guo, Y.Y.; Guo, H.J.; Li, T.T.; Hao, L.L.; Zheng, D.D.; Liu, G.P.; Zhang, Q. Galactosylated bovine serum albumin nanoparticles for parenteral delivery of oridonin: Tissue distribution and pharmacokinetic studies. J. Microencapsul. 2014, 31, 573–578. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Liu, G.; Guo, H.; Li, C.; Zhang, Y.; Zhang, F.; Zhao, Z.; Cheng, H. Novel galactosylated biodegradable nanoparticles for hepatocyte-delivery of oridonin. Int. J. Pharm. 2016, 502, 47–60. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, Z.; Shi, J.; Wang, F.; Luan, Y. Co-delivery of docetaxel and chloroquine via PEO-PPO-PCL/TPGS micelles for overcoming multidrug resistance. Int. J. Pharm. 2015, 495, 932–939. [Google Scholar] [CrossRef]
- Shi, C.H.; Zhang, Z.Q.; Wang, F.; Ji, X.Q.; Zhao, Z.X.; Luan, Y.X. Docetaxel-loaded PEO–PPO–PCL/TPGS mixed micelles for overcoming multidrug resistance and enhancing antitumor efficacy. J. Mater. Chem. B 2015, 3, 4259–4271. [Google Scholar] [CrossRef]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Luo, Y.F.; Chen, D.W.; Ren, L.X.; Zhao, X.L.; Qin, J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J. Control. Release 2006, 114, 53–59. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Zhang, D.; Tan, T.; Gao, L. Preparation of oridonin-loaded solid lipid nanoparticles and studies on them in vitro and in vivo. Nanotechnology 2006, 17, 5821–5828. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.P.; Chen, R.E.; Wang, Y.P.; Li, H.; Wang, Y.T.; Chen, M.W. Oridonin loaded solid lipid nanoparticles enhanced antitumor activity in MCF-7 cells. J. Nanomater. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- Joshi, M.; Pathak, S.; Sharma, S.; Patravale, V. Design and in vivo pharmacodynamics evaluation of nanostructured lipid carriers for parenteral delivery of artemether: Nanoject. Int. J. Pharm. 2008, 346, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Zheng, L.; Yu, L.; Zhang, Q.; Liu, W. Preparation and characterization of monocaprate nanostructured lipid carriers. Colloids Surf. A Physicochem. Eng. Asp. 2007, 311, 106–111. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, L.L.; Du, Y.Z.; You, J.; Hu, F.Q.; Zeng, S. Preparation and characteristics of nanostructured lipid carriers for control-releasing progesterone by melt-emulsification. Colloids Surf. B Biointerfaces 2007, 60, 174–179. [Google Scholar] [CrossRef]
- Müller, R.H.; Petersen, R.D.; Hommoss, A.; Pardeike, J. Nanostructured lipid carriers (NLC) in cosmetic dermal products. Adv. Drug Deliv. Rev. 2007, 59, 522–530. [Google Scholar] [CrossRef]
- Han, F.; Li, S.; Yin, R.; Liu, H.; Xu, L. Effect of surfactants on the formation and characterization of a new type of colloidal drug delivery system: Nanostructured lipid carriers. Colloids Surf. A Physicochem. Eng. Asp. 2008, 315, 210–216. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Selvamuthukumar, S.; Velmurugan, R. Nanostructured lipid carriers: A potential drug carrier for cancer chemotherapy. Lipids Health Dis. 2012, 11, 159–173. [Google Scholar] [CrossRef]
- Patel, D.; Dasgupta, S.; Dey, S.; Ramani, Y.R.; Ray, S.; Mazumder, B. Nanostructured lipid carriers (NLC)-based gel for the topical delivery of aceclofenac preparation, characterization, and in vivo evaluation. Sci. Pharm. 2012, 80, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, D.; Duan, C.; Jia, L.; Wang, Y.; Feng, F.; Zhang, Q. Preparation and characteristics of oridonin-loaded nanostructured lipid carriers as a controlled-release delivery system. J. Microencapsul. 2010, 27, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Shen, J.; Zhang, D.; Duan, C.; Liu, G.; Zheng, D.; Tian, X.; Liu, Y.; Zhang, Q. In vitro and in vivo evaluation of oridonin-loaded long circulating nanostructured lipid carriers. Int. J. Biol. Macromol. 2012, 50, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Dai, W.; Zhang, D.; Duan, C.; Jia, L.; Liu, Y.; Zhang, Q. In vivo studies on the oridonin-loaded nanostructured lipid carriers. Drug Deliv. 2012, 19, 286–291. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, X.; Ye, Y.; Zhang, T.; Wang, H.; Ma, Z.; Wu, B. Nanostructured lipid carriers used for oral delivery of oridonin:an effect of ligand modification on absorption. Int. J. Pharm. 2015, 479, 391–398. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.Q.; Liu, J.G.; Zhang, M.Y.; Yu, M.L.; Feng, N.P. Wheat germ agglutinin modification of lipid-polymer hybrid nanoparticles: Enhanced cellular uptake and bioadhesion. RSC Adv. 2016, 6, 36125–36135. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Liang, J.; Zhang, M.; Li, Z.; Wang, Z.; Dang, B.; Feng, N. Mucosal transfer of wheat germ agglutinin modified lipid-polymer hybrid nanoparticles for oral delivery of oridonin. Nanomedicine 2017, 13, 2219–2229. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalentions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Zhuang, Q.; Liu, X.; Sun, Z.; Wang, H.; Jiang, J. A validated UPLC-MS/MS method to determine free and total irinotecan and its two metabolites in human plasma after intravenous administration of irinotecan hydrochloride liposome injection. J. Pharm. Biomed. Anal. 2019, 170, 112–123. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, Y.; Hou, X.; Li, Z.; Zhang, K.; Shao, Q.; Feng, N. Improved oral bioavailability of notoginsenoside R1 with sodium glycocholate-mediated liposomes: Preparation by supercritical fluid technology and evaluation in vitro and in vivo. Int. J. Pharm. 2018, 552, 360–370. [Google Scholar] [CrossRef]
- Ferreira, K.S.A.; Santos, B.M.A.D.; Lucena, N.P.; Ferraz, M.S.; Carvalho, R.S.F.; Duarte, A.P., Jr.; Magalhães, N.S.S.; Lira, R.P.C. Ocular delivery of moxifloxacin-loaded liposomes. Arq. Bras. Oftalmol. 2018, 81, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, L.; Li, R.; Zhang, L.; Wang, J.; Yu, Y. Metabolic profile of lung-targeted docetaxel liposomes in rabbits, rats and mice. Xenobiotica 2019, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xia, Q.; Li, Y.; He, Z.; Li, Z.; Guo, T.; Wu, Z.; Feng, N. CD44 assists the topical anti-psoriatic efficacy of curcumin-loaded hyaluronan-modified ethosomes: A new strategy for clustering drug in inflammatory skin. Theranostics 2019, 9, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef]
- Wang, C.; Wei, Y.; Yu, L.; Zhang, L. The effect of stealth liposomes on pharmacokinetics, tissue distribution and anti-tumor activity of oridonin. PDA J. Pharm. Sci. Technol. 2009, 63, 409–416. [Google Scholar]
- Sun, X.Y.; Qu, C.X.; Lin, H.; Dong, J.Y. In vitro and in vivo evaluation of freeze-dried oridonin-loaded PEGylated liposomes. Lat. Am. J. Pharm. 2014, 33, 1144–1150. [Google Scholar]
- Wang, C.J.; Zhu, G.J.; Yu, L.; Shi, B.H. Preparation, in vitro, and in vivo antitumor activity of folate receptor-targeted nanoliposomes containing oridonin. Drug Dev. Res. 2013, 74, 43–49. [Google Scholar] [CrossRef]
- Guo, B.H.; Cheng, Y.; Lin, L.P.; Lin, D.H.; Wu, W. Preparation and characterization of galactose-modified liposomes by a nonaqueous enzymatic reaction. J. Liposome Res. 2011, 21, 255–260. [Google Scholar] [CrossRef]
- Guo, B.; Cheng, Y.; Li, N.; Li, X.; Jin, M.; Li, T.; Li, J. In vitro and in vivo studies of galactose-modified liver-targeting liposomes. J. Drug Target. 2013, 21, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, M.; Zhang, L.; Khougaz, K.; Eisenberg, A. Micellization of ionic block copolymers in three 28 dimensions. Acc. Chem. Res. 1996, 29, 53–72. [Google Scholar] [CrossRef]
- Li, W.; Peng, H.; Ning, F.; Yao, L.; Luo, M.; Zhao, Q.; Xiong, H. Amphiphilic chitosan derivative-based core-shell micelles: Synthesis, characterisation and properties for sustained release of vitamin D3. Food. Chem. 2014, 152, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Rösler, A.; Vandermeulen, G.W.M.; Klok, H.A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2001, 53, 95–108. [Google Scholar] [CrossRef]
- Shuai, X.; Ai, H.; Nasongkla, N.; Kim, S.; Gao, J. Micellar carriers based on block copolymers of poly(ε-caprolactone) and poly (ethylene glycol) for doxorubicin delivery. J. Control. Release 2004, 98, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef]
- Zhang, W.T.; Wang, D.K.; He, X.X.; Pan, Y.; Huang, Q.B.; Han, L.; Lin, L.F.; Liu, X.T. Preparation and characterization of oridonin-loaded block copolymer micelles. Chin. J. New Drugs 2009, 18, 1560–1565. [Google Scholar]
- Xue, B.; Wang, Y.; Tang, X.; Xie, P.; Wang, Y.; Luo, F.; Wu, C.; Qian, Z. Biodegradable self-assembled MPEG-PCL micelles for hydrophobic oridonin delivery in vitro. J. Biomed. Nanotechnol. 2012, 8, 80–89. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Hua, H.Y.; Zhao, Y.; Zhang, Z.Z. Cytotoxicity and cellular kinetics of oridonin-loaded cholesterol formate-graft chitosan copolymer nano-micelles. J. Control. Release 2013, 172, E90–E91. [Google Scholar] [CrossRef]
- Dian, L.; Yu, E.; Chen, X.; Wen, X.; Zhang, Z.; Qin, L.; Wang, Q.; Li, G.; Wu, C. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale. Res. Lett. 2014, 9, 684–695. [Google Scholar] [CrossRef]
- Hou, J.; Wang, J.; Sun, E.; Yang, L.; Yan, H.M.; Jia, X.B.; Zhang, Z.H. Preparation and evaluation of icariside II-loaded binary mixed micelles using Solutol HS15 and Pluronic F127 as carriers. Drug Deliv. 2016, 23, 3248–3256. [Google Scholar] [CrossRef]
- Chang, T.; Gosain, P.; Stenzel, M.H.; Lord, M.S. Drug-loading of poly (ethylene glycol methyl ether methacrylate) (PEGMEMA)—Based micelles and mechanisms of uptake in colon carcinoma cells. Colloids. Surf. B. Biointerfaces 2016, 144, 257–264. [Google Scholar] [CrossRef]
- Ke, Z.; Zhang, Z.; Wu, H.; Jia, X.; Wang, Y. Optimization and evaluation of Oridonin-loaded Soluplus-Pluronic P105 mixed micelles for oral administration. Int. J. Pharm. 2017, 518, 193–202. [Google Scholar] [CrossRef]
- Fang, X.B.; Xu, Y.Q.; Chan, H.F.; Wang, C.M.; Zheng, Q.; Xiao, F.; Chen, M.W. A Redox-sensitive and RAGE-targeting nanocarrier for hepatocellular carcinoma therapy. Mol. Pharm. 2016, 13, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Letzner, H. New materials and methods for clinically and esthetically acceptable restorations. Quintessenz Zahntech. 1988, 14, 1093–1106. [Google Scholar] [PubMed]
- Bland, S. New materials and methods for next generation technologies. Mater. Today 2016, 19, 243. [Google Scholar]
- Shi, X.; Peng, T.; Huang, Y.; Mei, L.; Gu, Y.; Huang, J.; Han, K.; Li, G.; Hu, C.; Pan, X.; et al. Comparative studies on glycerol monooleate- and phytantriol-based cubosomes containing oridonin in vitro and in vivo. Pharm. Dev. Technol. 2017, 22, 322–329. [Google Scholar] [CrossRef]
- Wang, C.J.; Li, A. Preparation, Characterization, and in vitro and vivo antitumor activity of oridonin-conjugated multiwalled carbon nanotubes functionalized with carboxylic group. J. Nanomater. 2016, 10, 1687–1693. [Google Scholar]
- Chen, X.Y.; Shi, Y.L.; Yang, D.; Hu, J.H.; Yang, P.Y. Preparation of polystyrene functionalized graphene by atom transfer nitroxide radical coupling reaction. Acta Chim. Sin. 2012, 70, 817–821. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Li, Y.J.; Shi, P.; Wang, B.C.; Huang, X.Y. Functionalized graphene oxide as a nanocarrier for loading and delivering of oridonin. J. Org. Chem. 2013, 33, 573–580. [Google Scholar] [CrossRef][Green Version]
- Chai, D.D.; Hao, B.J.; Hu, R.; Zhang, F.; Yan, J.; Sun, Y.; Huang, X.Y.; Zhang, Q.X.; Jiang, H. Delivery of Oridonin and Methotrexate via PEGylated Graphene Oxide. ACS Appl. Mater. Interfaces 2019, 11, 22915–22924. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Sabouni, R.; Husseini, G.A. Anti-cancer drug delivery using metal organic frameworks (MOFs). Curr. Med. Chem. 2017, 24, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Dong, X.; Wang, W.; Sai, N.; Yang, C.; You, L.; Huang, H.; Yin, X.; Ni, J. Biocompatible Fe-based micropore metal-organic frameworks as sustained-release anticancer drug carriers. Molecules 2018, 23, 2490. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.S.; Luo, J.Y.; Cai, M.R.; Qin, L.Y.; Wang, Y.B.; Gao, L.L.; Huang, P.Q.; Yu, Y.C.; Ding, Y.M.; Dong, X.X.; et al. Investigation of Metal-Organic Framework−5 (MOF−5) as an Antitumor Drug Oridonin Sustained Release Carrier. Molecules 2019, 24, 3369. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Chen, R.; Chen, X.; Zhang, H.; Song, L.; Cui, W.; Zhang, J.; Ye, D.; Zhang, Y.; Wang, Z. Oridonin-loaded and GPC1-targeted gold nanoparticles for multimodal imaging and therapy in pancreatic cancer. Int. J. Nanomed. 2018, 13, 6809–6827. [Google Scholar] [CrossRef]
- Pi, J.; Jiang, J.; Cai, H.; Yang, F.; Jin, H.; Yang, P.; Cai, J.; Chen, Z.W. GE11 peptide conjugated selenium nanoparticles for EGFR targeted oridonin delivery to achieve enhanced anticancer efficacy by inhibiting EGFR-mediated PI3K/AKT and Ras/Raf/MEK/ERK pathways. Drug Deliv. 2017, 24, 1549–1564. [Google Scholar] [CrossRef]
- Jiang, J.H.; Pi, J.; Jin, H.; Cai, J.Y. Functional graphene oxide as cancer-targeted drug delivery system to selectively induce oesophageal cancer cell apoptosis. Artif. Cells Nanomed. Biotechnol. 2018, 46, S297–S307. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Liu, T.; Zhao, L.; Zhao, J.; Feng, N. Development and in-vivo assessment of the bioavailability of oridonin solid dispersions by the gas anti-solvent technique. Int. J. Pharm. 2011, 411, 172–177. [Google Scholar] [CrossRef]
- Duan, C.X.; Zhang, D.R.; Wang, F.H.; Zheng, D.D.; Jia, L.J.; Feng, F.F.; Liu, Y.; Wang, Y.C.; Tian, K.L.; Wang, F.S.; et al. Chitosan-g-poly(N-isopropylacrylamide) based nanogels for tumor extracellular targeting. Int. J. Pharm. 2011, 409, 252–259. [Google Scholar] [CrossRef]
- Duan, C.; Gao, J.; Zhang, D.; Jia, L.; Liu, Y.; Zheng, D.; Liu, G.; Tian, X.; Wang, F.; Zhang, Q. Galactose-decorated pH-responsive nanogels for hepatoma-targeted delivery of oridonin. Biomacromolecules 2011, 12, 4335–4343. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, Y.; Feng, N.; Xu, J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int. J. Pharm. 2008, 355, 269–276. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activities | Mechanism of Actions | Ref. |
|---|---|---|
| Anti-cancer | Repressing cell cycle, down-regulating telomerase activity, inhibiting cell membrane sodium pump activity, inducing tumor cell apoptosis | [3,4] |
| Anti-bacterial | - | [5] |
| Neuroregulation | Upregulating the production of the neurotrophic factor and nerve growth factor | [6] |
| Anti-oxidation | Scavenging active oxygen free radicals | [7] |
| Depressurization | Inducing pulmonary artery smooth muscle cell apoptosis | [8] |
| Anti-inflammation | Down-regulating the inflammatory factors IL-figure1β, IL-6, and IL−33; Blocking the interaction between NLRP3 and NEK7, inhibiting NLRP3 inflammasome activation | [9,10,11] |
| Immune-modulating | Promoting CD4+ /CD25+ Treg differentiation; modulating Th1/Th2 balance | [12] |
| Analgesia | Reducing colonic EC cell hyperplasia and 5-HT availability | [13] |
| Research Objects | Biological Activity or Mechanism | Ref. |
|---|---|---|
| New Zealand white rabbits, Kunming strain mice | Particle size affects pharmacokinetics and tissue distribution | [68] |
| K562 cell, S-180 tumor-burdened mice | Significantly more toxic to K562 cells than free ORI (p < 0.01) | [69] |
| PC-3 cell | Inducing early apoptosis and enhancing growth suppression | [70] |
| SMMC-7721 cell, H22 tumor-bear mice | Arresting cells in the G2/M phase, tumor volume response ORI-N has a higher anti-tumor effect | [71] |
| MCF-7 cell | Reducing the expression of Bcl-2 and increasing Bax | [72] |
| PANC-1 cell | Suppressing the expression of B1 and p-cdc2 (T161) on G2/M cell phase | [73] |
| SD-rats | Improving dissolution and permeability by interaction with absorptive epithelia and anti-drug efflux. | [74] |
| Delivery Systems | Administration | Results | Ref. |
|---|---|---|---|
| ORI-PLA-NPs | Intravenous administration in mice at a dose of 25 mg/kg | High concentration of ORI in liver, lung and spleen | [78] |
| ORI-PCL-PEO-PCL-NPs | Intravenous administration in H22 tumor-bear mice at a dose of 8 mg/kg | Tumor volume and weight decreased, and the survival rate increased to 169.6% | [80,81] |
| ORI-PLA-RGD-NPs | Intravenous administration in the H22 tumor-bear mice at a dose equivalent to 20 mg/kg of ORI | Enhanced targeting effect; tumor volume and weight were significantly reduced (p < 0.01), with average survival length extended from 27 days to 41 days compared with ORI-PLA-NP | [82] |
| ORI-GC-NPs | Intravenous administration in mice at a dose of 1 mg/kg | Prolonging MRT0–t from 3.415 h to 14.042 h, liver AUC0–t increasing by 6.4-fold compared with ORI solution | [84] |
| ORI-GB-NPs | Intravenous administration on Wistar rats at a dose of 14 mg/kg and Kunming strain mice at 20 mg/kg | The retarded in vitro release with increasing of crosslinking agent glutaraldehyde; liver active targeting; enhancing the drug plasma concentration and prolonging the circulation time | [85,86] |
| ORI-Gal-PT-NPs | Treating HepG2 cells at concentrations of 5.0, 7.5, 10 and 12.5 mg/mL | The apoptosis increasing by 4−5.6% compared with ORI solution | [87] |
| Materials | Preparation Methods | Administration | Results | Ref. |
|---|---|---|---|---|
| Cholesterol and soy lecithin, CPA, PEG2000-DSPE | Thin-film ultrasonic dispersion method | Mice injected via the tail vein at 33.5 mg/kg | Enhancing T1/2 from 0.65 h to 24.89 h, 85.4% tumor inhibition rate | [118] |
| PEG | Ethanol injection method | H22 tumor-bearing mice | Drug accumulation in tumor cells, prolonged plasma residence | [119] |
| CH, soy bean lecithin, folic acid, N,N’-dicyclohexyl carbodiimide, PEG2000-DSPE | Thin-film ultrasonic dispersion method | Intravenous administration in tumor-bearing nude BALB/C mice at a dose of 1.5 × 10−2 g/kg/day | Body weight gain, 85.6% tumor inhibition | [120] |
| EPC, CH, NOH, | Ethanol injection method | Intravenous administration in mice at a dose of 30 mg/kg | Enhancing MRT0–t from 1.69 h to 9.40 h, Enhancing AUC0–t by 4.58-fold, 61.18% Te | [121] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, S.; Dai, M.; Nai, J.; Zhu, L.; Sheng, H. Solubility and Bioavailability Enhancement of Oridonin: A Review. Molecules 2020, 25, 332. https://doi.org/10.3390/molecules25020332
Zhang Y, Wang S, Dai M, Nai J, Zhu L, Sheng H. Solubility and Bioavailability Enhancement of Oridonin: A Review. Molecules. 2020; 25(2):332. https://doi.org/10.3390/molecules25020332
Chicago/Turabian StyleZhang, Yuanyuan, Shaohua Wang, Mengmeng Dai, Jijuan Nai, Liqiao Zhu, and Huagang Sheng. 2020. "Solubility and Bioavailability Enhancement of Oridonin: A Review" Molecules 25, no. 2: 332. https://doi.org/10.3390/molecules25020332
APA StyleZhang, Y., Wang, S., Dai, M., Nai, J., Zhu, L., & Sheng, H. (2020). Solubility and Bioavailability Enhancement of Oridonin: A Review. Molecules, 25(2), 332. https://doi.org/10.3390/molecules25020332