
The Application of Reversible Intramolecular Sulfonamide Ligation to Modulate Reactivity in Organometallic Ruthenium(II) Diamine Complexes



Abstract
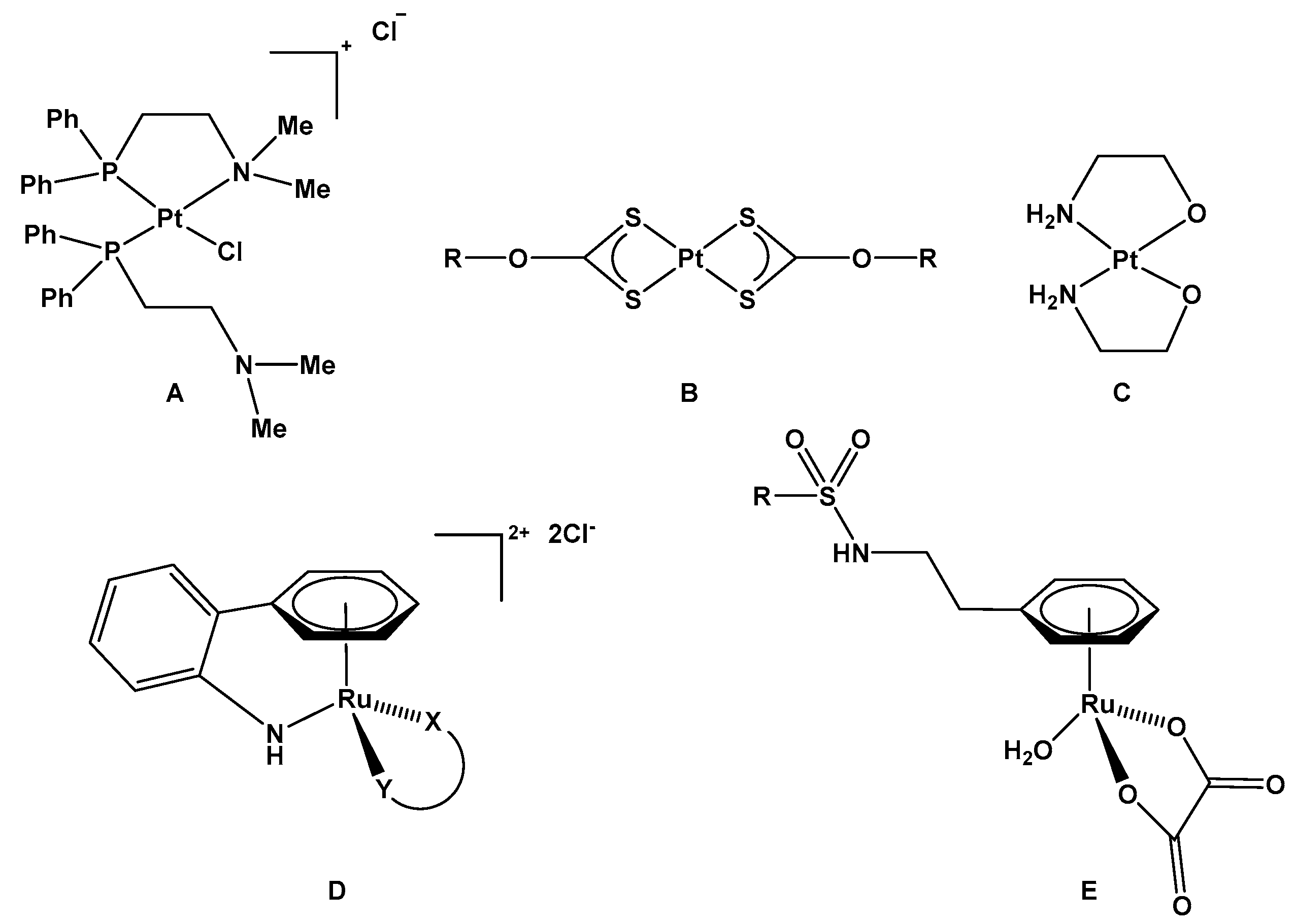
1. Introduction
2. Results and Discussion
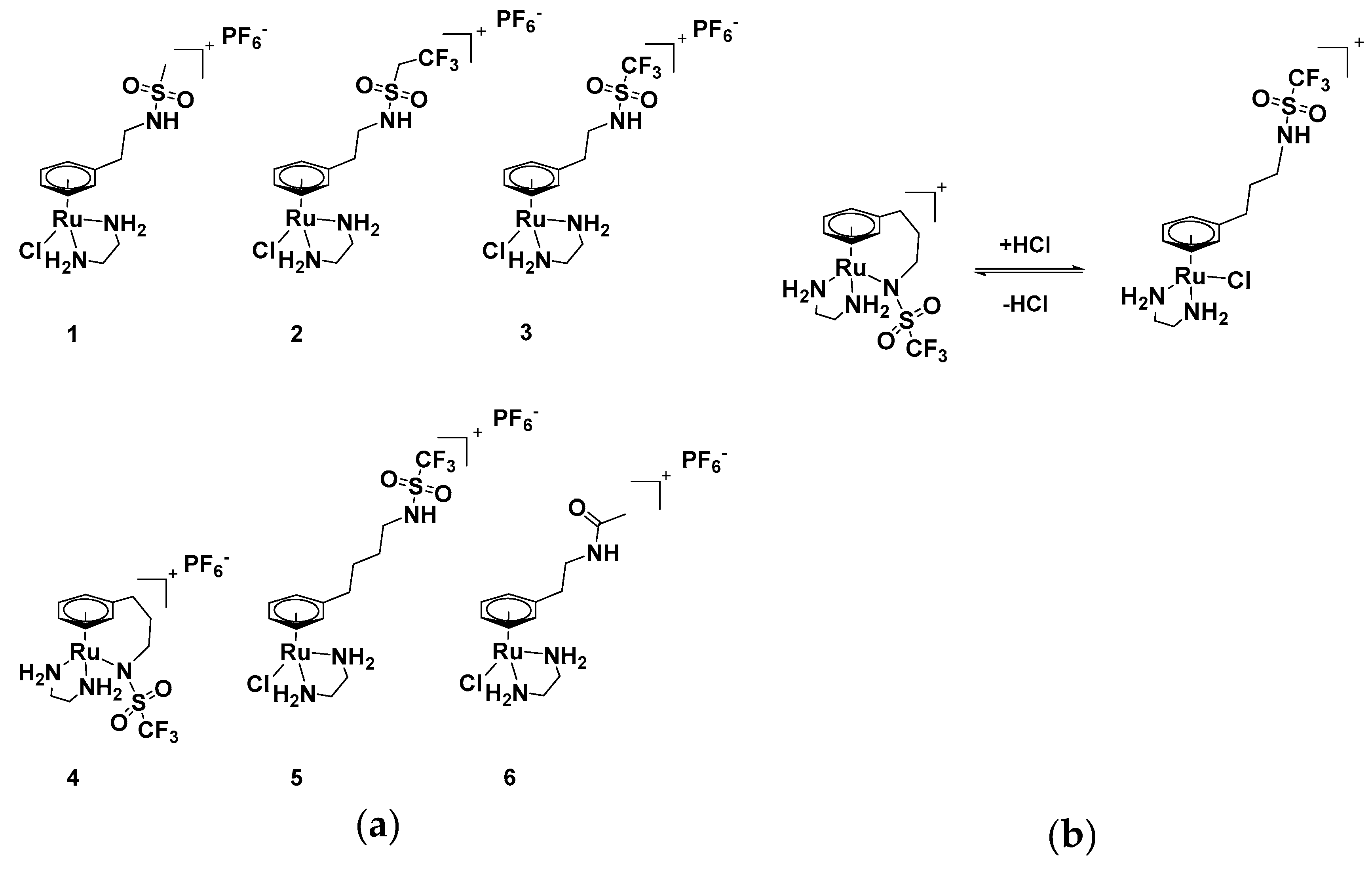
2.1. Synthesis and Characterisation
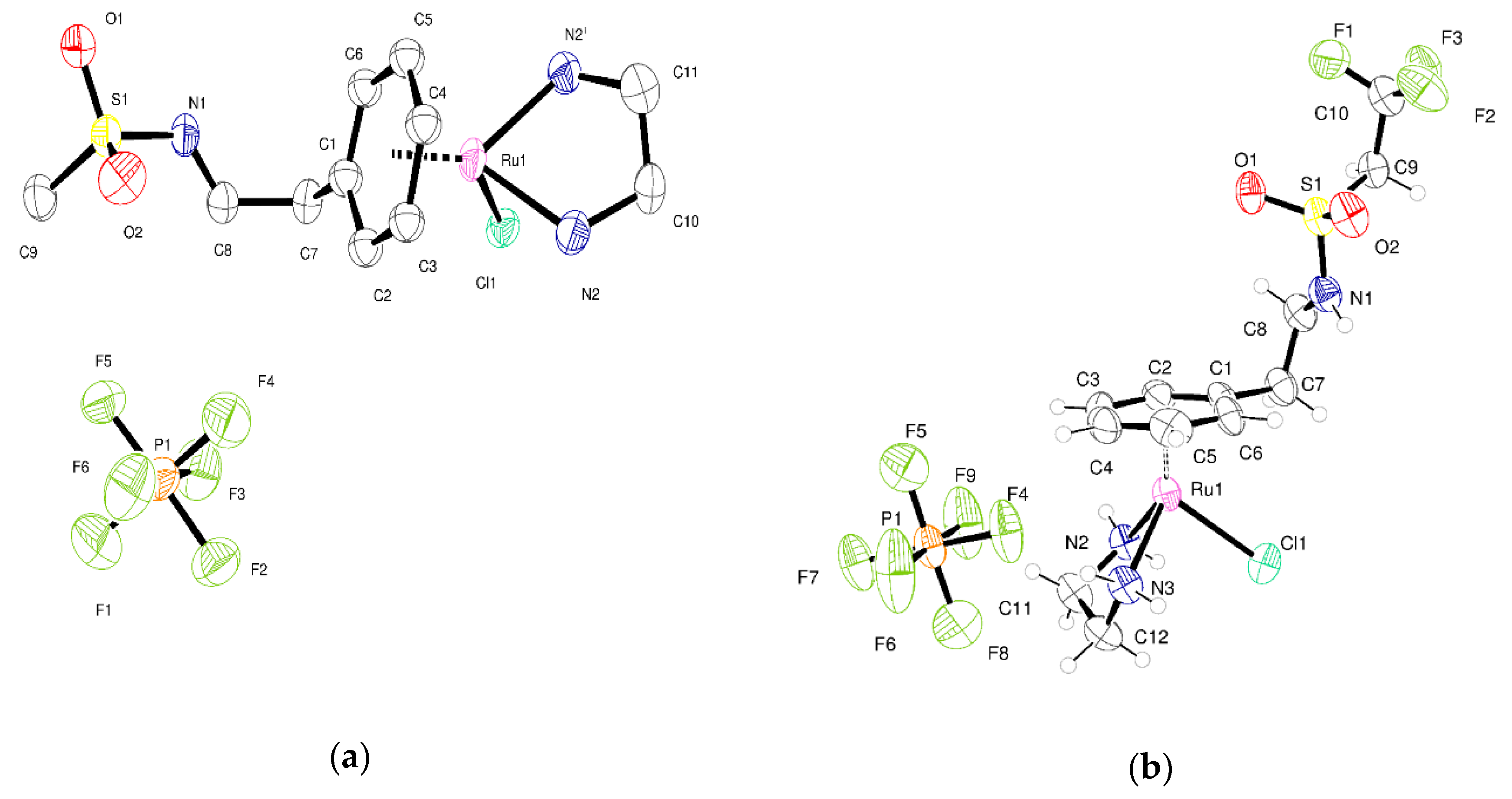
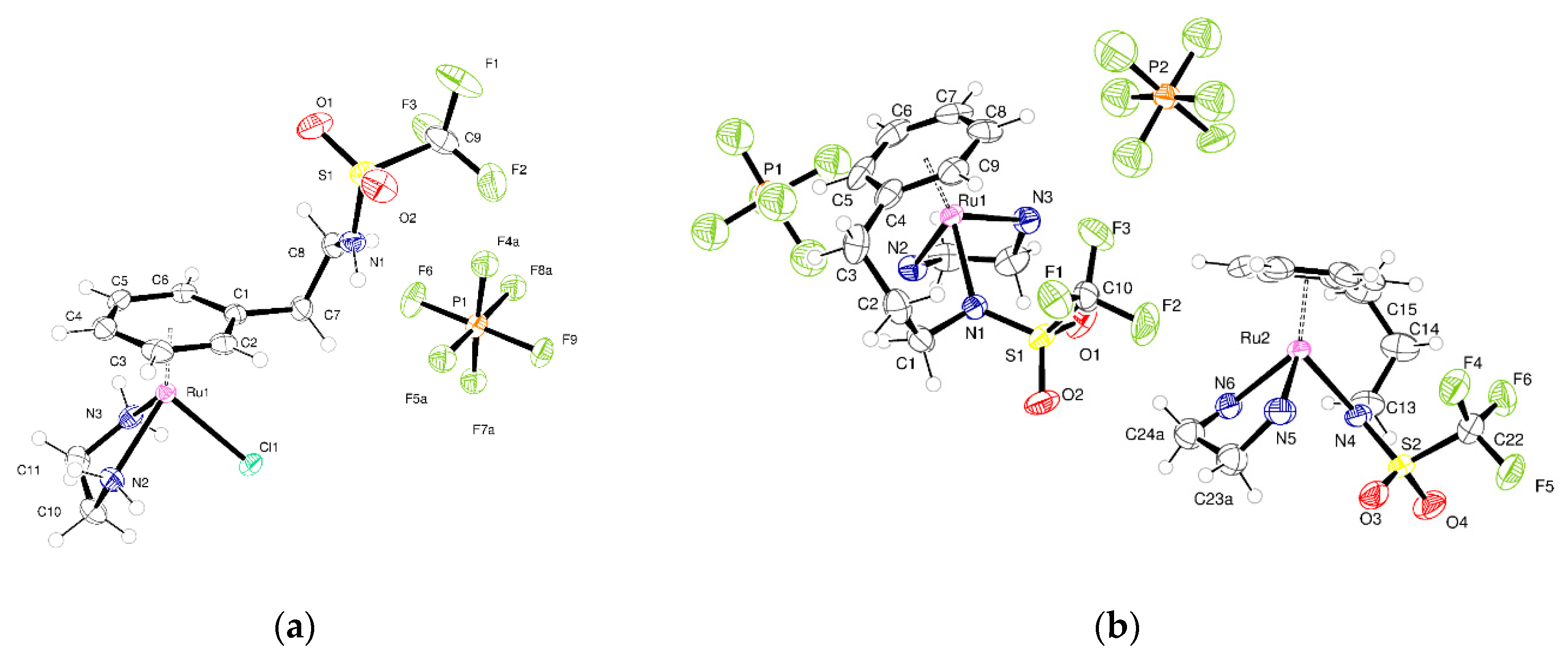
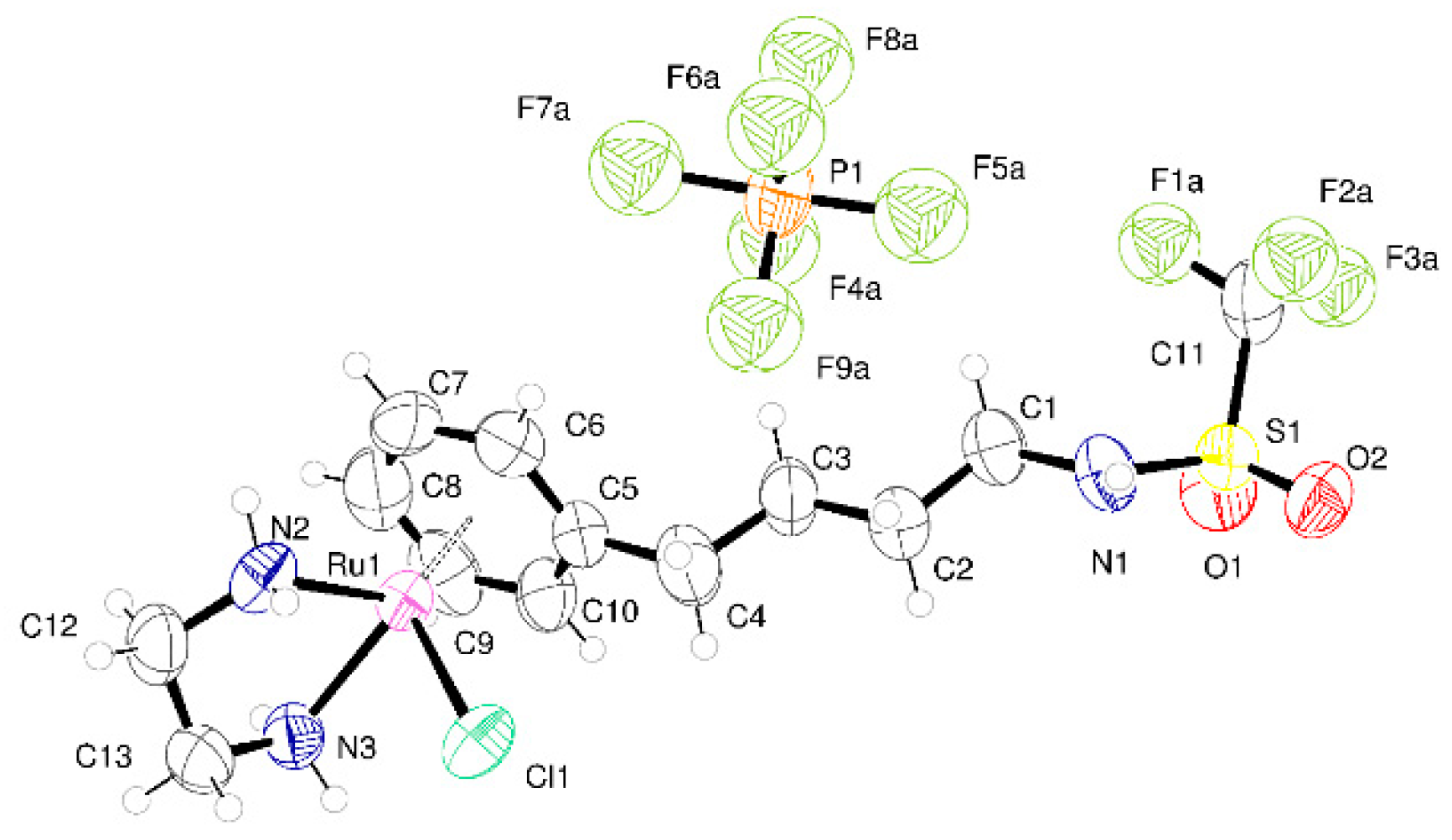
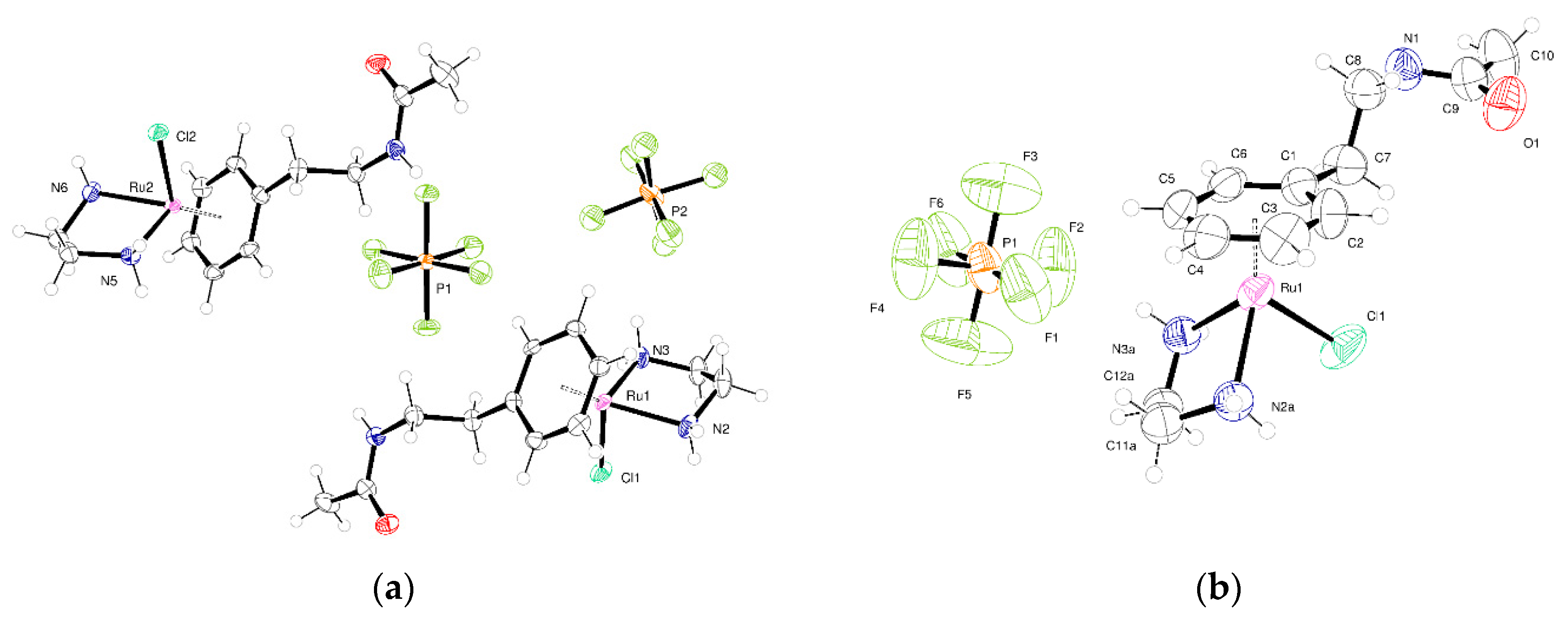
2.2. Crystallography
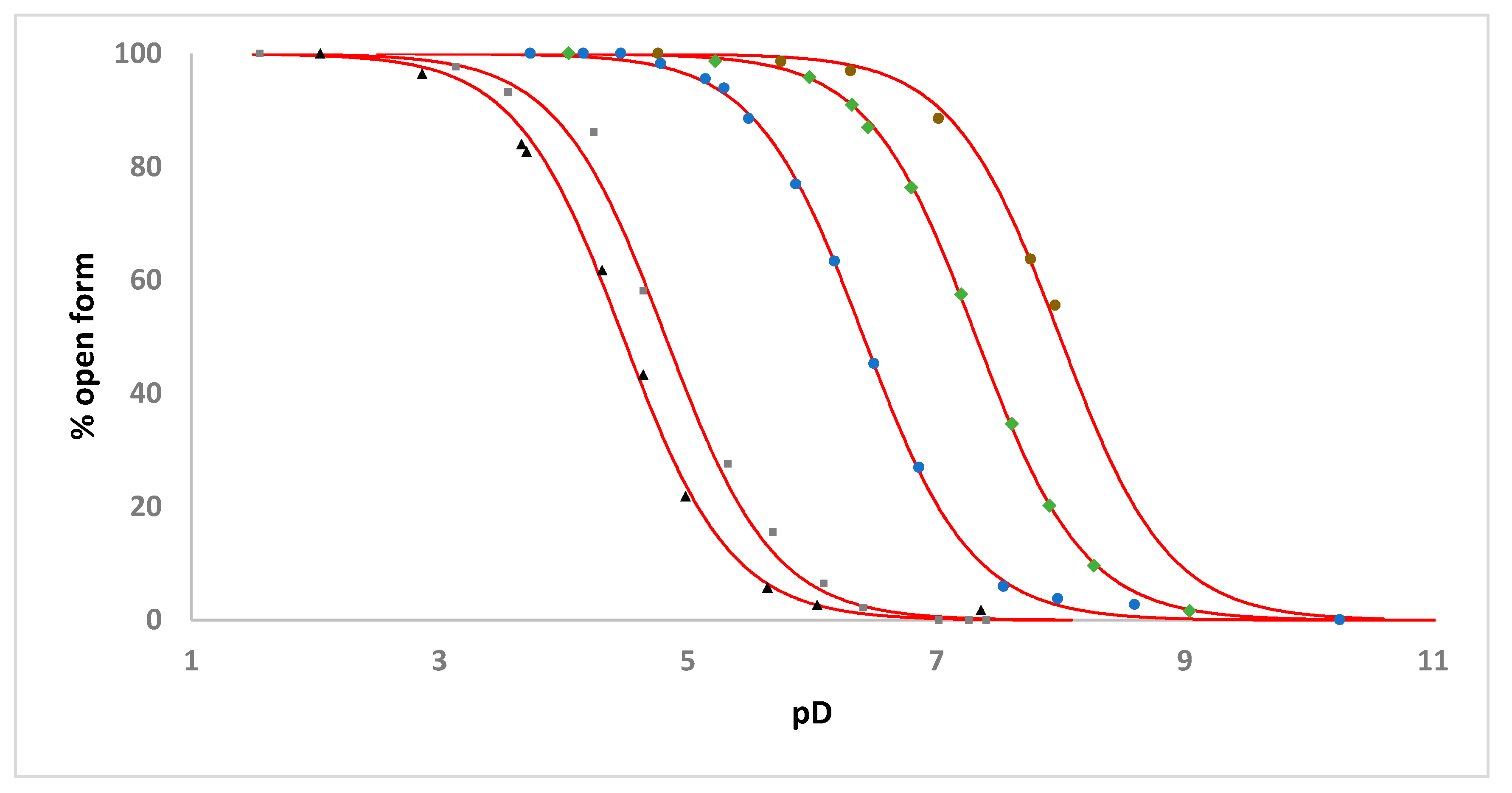
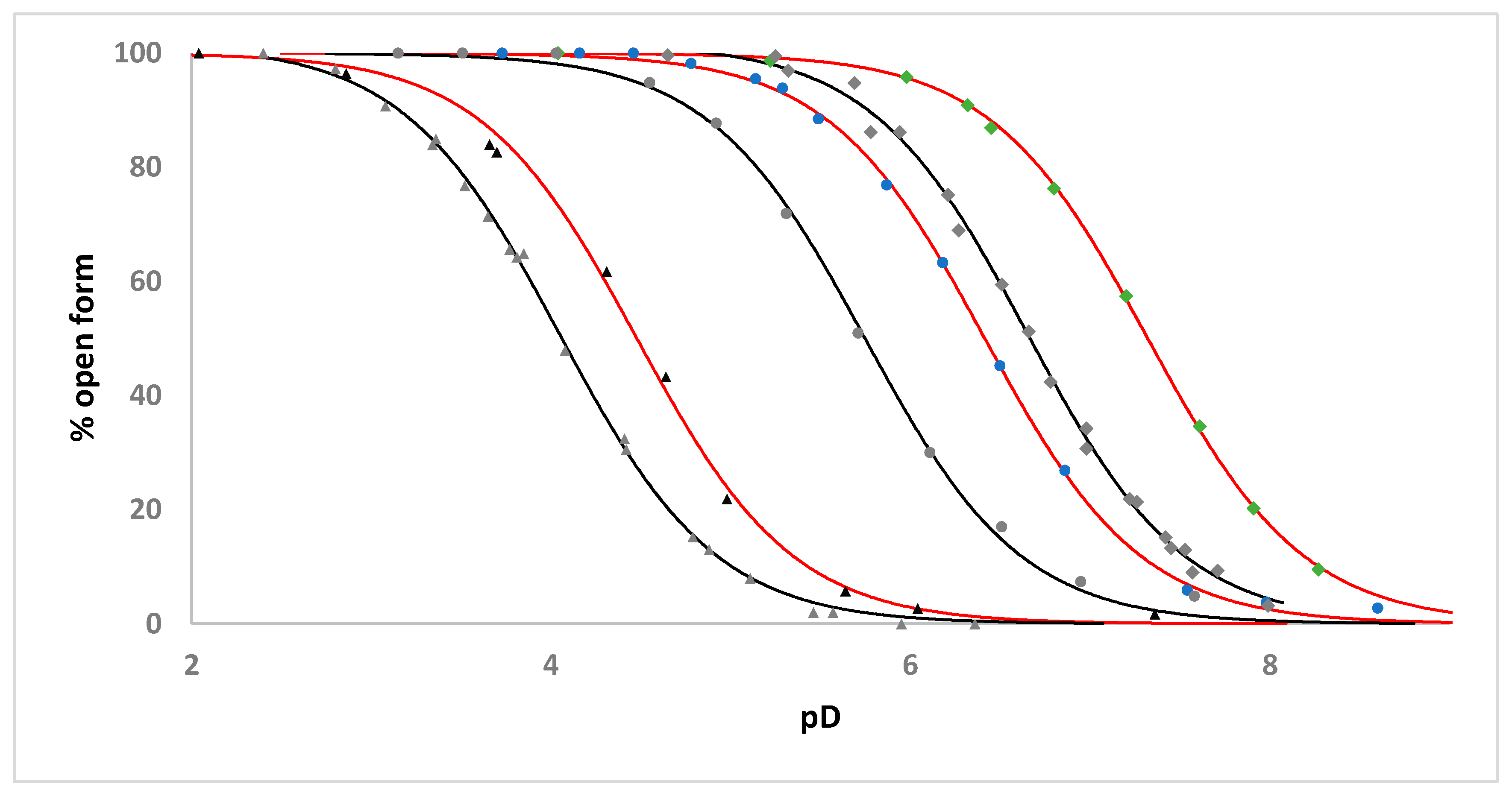
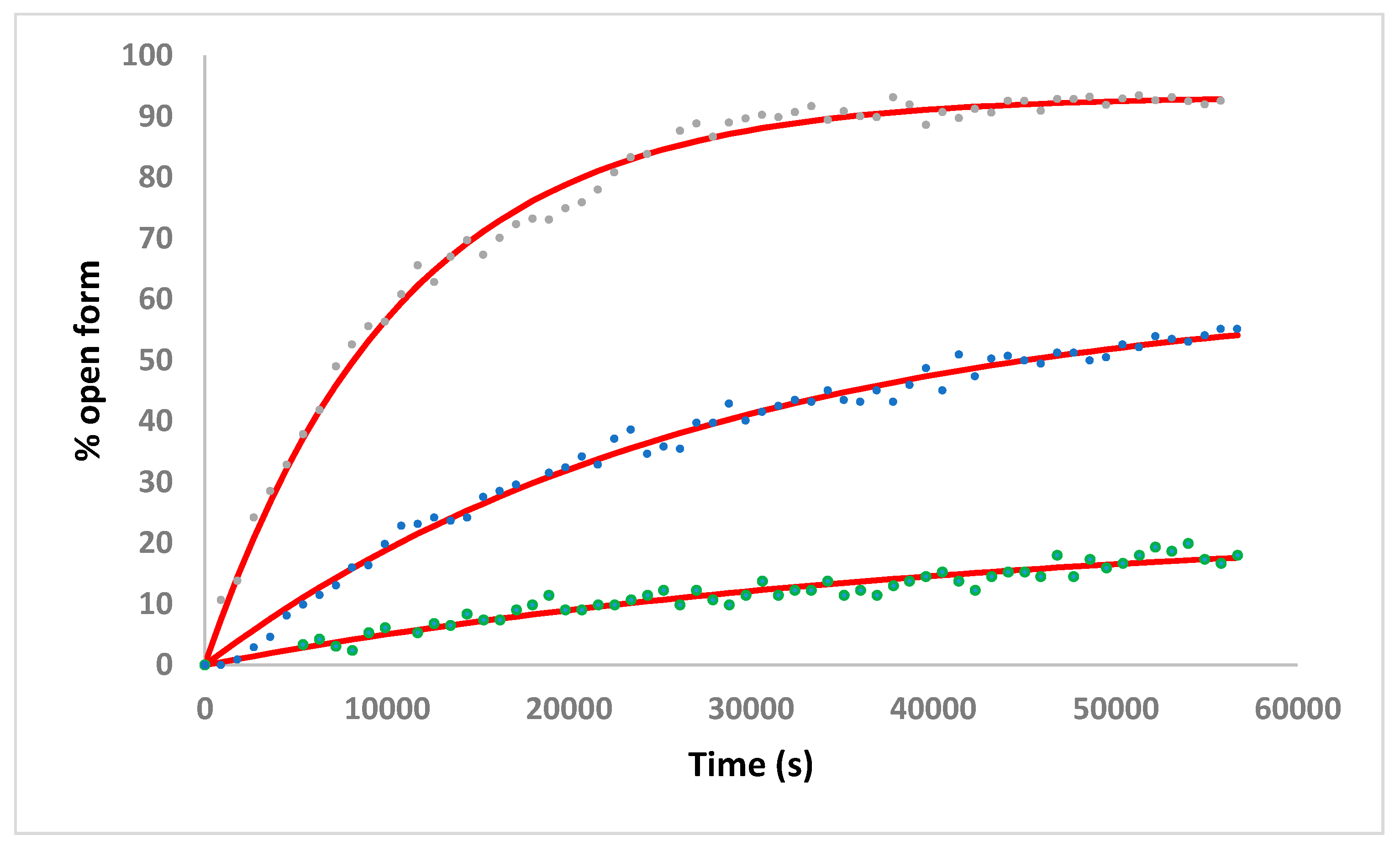
2.3. Solution Studies
2.4. Evaluation of In Vitro Anticancer Activity
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Instrumentation and Methods
4.2.1. NMR
4.2.2. Mass Spectrometry
4.2.3. X-ray Crystallography
4.2.4. Elemental Analysis
4.3. Synthesis
4.4. NMR Studies
4.5. Cell Culture
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Casini, A.; Vessières, A.; Meier-Menches, S.M. (Eds.) Metal-Based Anticancer Agents; The Royal Society of Chemistry: Croydon, UK, 2019. [Google Scholar]
- Zhang, P.; Sadler, P.J. Advances in the Design of Organometallic Anticancer Complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Murray, B.S.; Dyson, P.J. Recent Progress in the Development of Organometallics for the Treatment of Cancer. Curr. Opin. Chem. Biol. 2020, 56, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Habtemariam, A.; Watchman, B.; Potter, B.S.; Palmer, R.; Parsons, S.; Parkin, A.; Sadler, P.J. Control of Aminophosphine Chelate Ring-Opening in Pt(II) and Pd(II) Complexes: Potential Dual-Mode Anticancer Agents. Dalton Trans. 2001, 8, 1306–1318. [Google Scholar] [CrossRef]
- Habtemariam, A.; Sadler, P.J. Design of Chelate Ring-Opening Platinum Anticancer Complexes: Reversible Binding to Guanine. Chem. Commun. 1996, 15, 1785–1786. [Google Scholar] [CrossRef]
- Friebolin, W.; Schilling, G.; Zöller, M.; Amtmann, E. Synthesis and Structure−Activity Relationship of Novel Antitumoral Platinum Xanthate Complexes. J. Med. Chem. 2004, 47, 2256–2263. [Google Scholar] [CrossRef]
- Galanski, M.; Baumgartner, C.; Meelich, K.; Arion, V.B.; Fremuth, M.; Jakupec, M.A.; Schluga, P.; Hartinger, C.G.; Graf. v. Keyserlingk, N.; Keppler, B.K. Synthesis, Crystal Structure and pH Dependent Cytotoxicity of (SP-4-2)-bis(2-aminoethanolato-κ2 N,O)platinum(II)—A Representative of Novel pH Sensitive Anticancer Platinum Complexes. Inorg. Chim. Acta 2004, 357, 3237–3244. [Google Scholar] [CrossRef]
- Zorbas-Seifried, S.; Hartinger, C.G.; Meelich, K.; Galanski, M.; Keppler, B.K.; Zorbas, H. DNA Interactions of pH-Sensitive, Antitumor Bis(aminoalcohol)dichloroplatinum(II) Complexes. Biochemistry 2006, 45, 14817–14825. [Google Scholar] [CrossRef]
- Scaffidi-Domianello, Y.Y.; Legin, A.A.; Jakupec, M.A.; Arion, V.B.; Kukushkin, V.Y.; Galanski, M.; Keppler, B.K. Synthesis, Characterization, and Cytotoxic Activity of Novel Potentially pH-Sensitive Nonclassical Platinum(II) Complexes Featuring 1,3-Dihydroxyacetone Oxime Ligands. Inorg. Chem. 2011, 50, 10673–10681. [Google Scholar] [CrossRef]
- Scrase, T.G.; O’Neill, M.J.; Peel, A.J.; Senior, P.W.; Matthews, P.D.; Shi, H.; Boss, S.R.; Barker, P.D. Selective Lability of Ruthenium(II) Arene Amino Acid Complexes. Inorg. Chem. 2015, 54, 3118–3124. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Peña, F.; Pizarro, A.M. Control of Reversible Activation Dynamics of [Ru{η6:κ1--C6H5(C6H4)NH2}(XY)]n+ and the Effect of Chelating-Ligand Variation. Chem. Eur. J. 2017, 23, 16231–16241. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Peña, F.; Infante-Tadeo, S.; Habtemariam, A.; Pizarro, A.M. Reversible pH-Responsive Behavior of Ruthenium(II) Arene Complexes with Tethered Carboxylate. Inorg. Chem. 2018, 57, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.J.; Savoie, H.; Boyle, R.W.; Murray, B.S. pH-Dependent Modulation of Reactivity in Ruthenium(II) Organometallics. Organometallics 2018, 37, 294–297. [Google Scholar] [CrossRef]
- Morris, R.E.; Aird, R.E.; del Socorro Murdoch, P.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of Cancer Cell Growth by Ruthenium(II) Arene Complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef]
- Habtemariam, A.; Melchart, M.; Fernández, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-Activity Relationships for Cytotoxic Ruthenium(II) Arene Complexes Containing N,N-, N,O-, and O,O-Chelating Ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Skjærvø, S.L.; Høydalsvik, K.; Blichfeld, A.B.; Einarsrudand, M.-A.; Grande, T. Thermal Evolution of the Crystal Structure and Phase Transitions of KNbO3. R. Soc. Open Sci. 2018, 5, 180368. [Google Scholar] [CrossRef]
- Glasoe, P.K.; Long, F.A.J. Use of Glass Electrodes to Measure Acidities in Deuteium Oxide. Phys. Chem. 1960, 64, 188–190. [Google Scholar] [CrossRef]
- Bradbury, J.H.; Chapman, B.E.; Pellegrino, F.A. Hydrogen-Deuterium Exchange Kinetics of the C-2 Protons of Imidazole and Histidine Compounds. J. Am. Chem. Soc. 1973, 95, 6139–6140. [Google Scholar] [CrossRef]
- Canel, E.; Gültepe, A.; Doğan, A.; Kiliç, E. The Determination of Protonation Constants of Some Amino Acids and Their Esters by Potentiometry in Different Media. J. Soln. Chem. 2006, 35, 5–19. [Google Scholar] [CrossRef]
- Chen, H.; Parkinson, J.A.; Morris, R.E.; Sadler, P.J. Highly Selective Binding of Organometallic Ruthenium Ethylenediamine Complexes to Nucleic Acids: Novel Recognition Mechanisms. J. Am. Chem. Soc. 2003, 125, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Blessing, R. An Empirical Correction for Absorption Anisotropy. Acta Cryst. 1995, A51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement With SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Ito, M.; Endo, Y.; Ikariya, T. Well-Defined Triflylamide-Tethered Arene−Ru(Tsdpen) Complexes for Catalytic Asymmetric Hydrogenation of Ketones. Organometallics 2008, 27, 6053–6055. [Google Scholar] [CrossRef]
- Reiner, T.; Jantke, D.; Miao, X.-H.; Marziale, A.N.; Kiefer, F.J.; Eppinger, J. Phenylalanine—A Biogenic Ligand with Flexible η6- and η6:κ1-Coordination at Ruthenium(II) Centres. Dalton Trans. 2013, 42, 8692–8703. [Google Scholar] [CrossRef]
- Crabb, T.A.; Wilkinson, J.R. Synthesis of Hexahydroisoquinolines. J. Chem. Soc. Perkin Trans. 1975, 1, 1465–1470. [Google Scholar] [CrossRef]
Sample Availability: Limited samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Apparent pKa | Apparent pKa for the Analogous [Ru(η6-C6H5CH2CH2NHR)(C2O4)(H2O)] Complex |
|---|---|---|
| 1 | 7.32 | 6.65 ‡ |
| 2 | 6.41 | 5.75 |
| 3 | 4.50 | 4.05 ‡ |
| 4 | 4.82 | - |
| 5 | 8.00 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemp, S.A.; Prior, T.J.; Savoie, H.; Boyle, R.W.; Murray, B.S. The Application of Reversible Intramolecular Sulfonamide Ligation to Modulate Reactivity in Organometallic Ruthenium(II) Diamine Complexes. Molecules 2020, 25, 244. https://doi.org/10.3390/molecules25020244
Kemp SA, Prior TJ, Savoie H, Boyle RW, Murray BS. The Application of Reversible Intramolecular Sulfonamide Ligation to Modulate Reactivity in Organometallic Ruthenium(II) Diamine Complexes. Molecules. 2020; 25(2):244. https://doi.org/10.3390/molecules25020244
Chicago/Turabian StyleKemp, Samuel A., Timothy J. Prior, Huguette Savoie, Ross W. Boyle, and Benjamin S. Murray. 2020. "The Application of Reversible Intramolecular Sulfonamide Ligation to Modulate Reactivity in Organometallic Ruthenium(II) Diamine Complexes" Molecules 25, no. 2: 244. https://doi.org/10.3390/molecules25020244
APA StyleKemp, S. A., Prior, T. J., Savoie, H., Boyle, R. W., & Murray, B. S. (2020). The Application of Reversible Intramolecular Sulfonamide Ligation to Modulate Reactivity in Organometallic Ruthenium(II) Diamine Complexes. Molecules, 25(2), 244. https://doi.org/10.3390/molecules25020244