
25 Years of Collaboration with A Genius: Deciphering Adenine Nucleotide Ca2+ Mobilizing Second Messengers Together with Professor Barry Potter

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
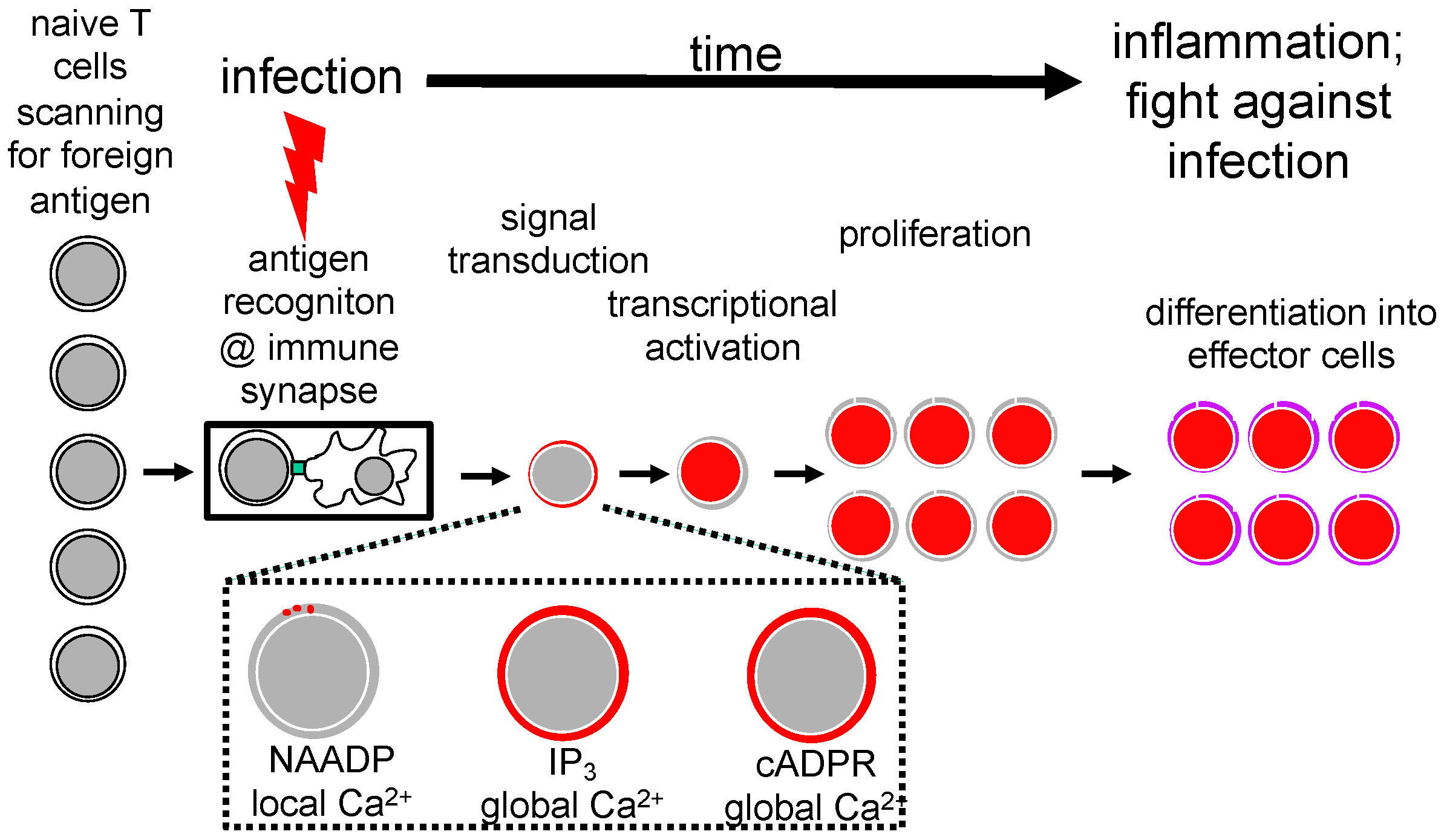
2. Stages of T Cell Activation and T Cell Ca2+ Signaling
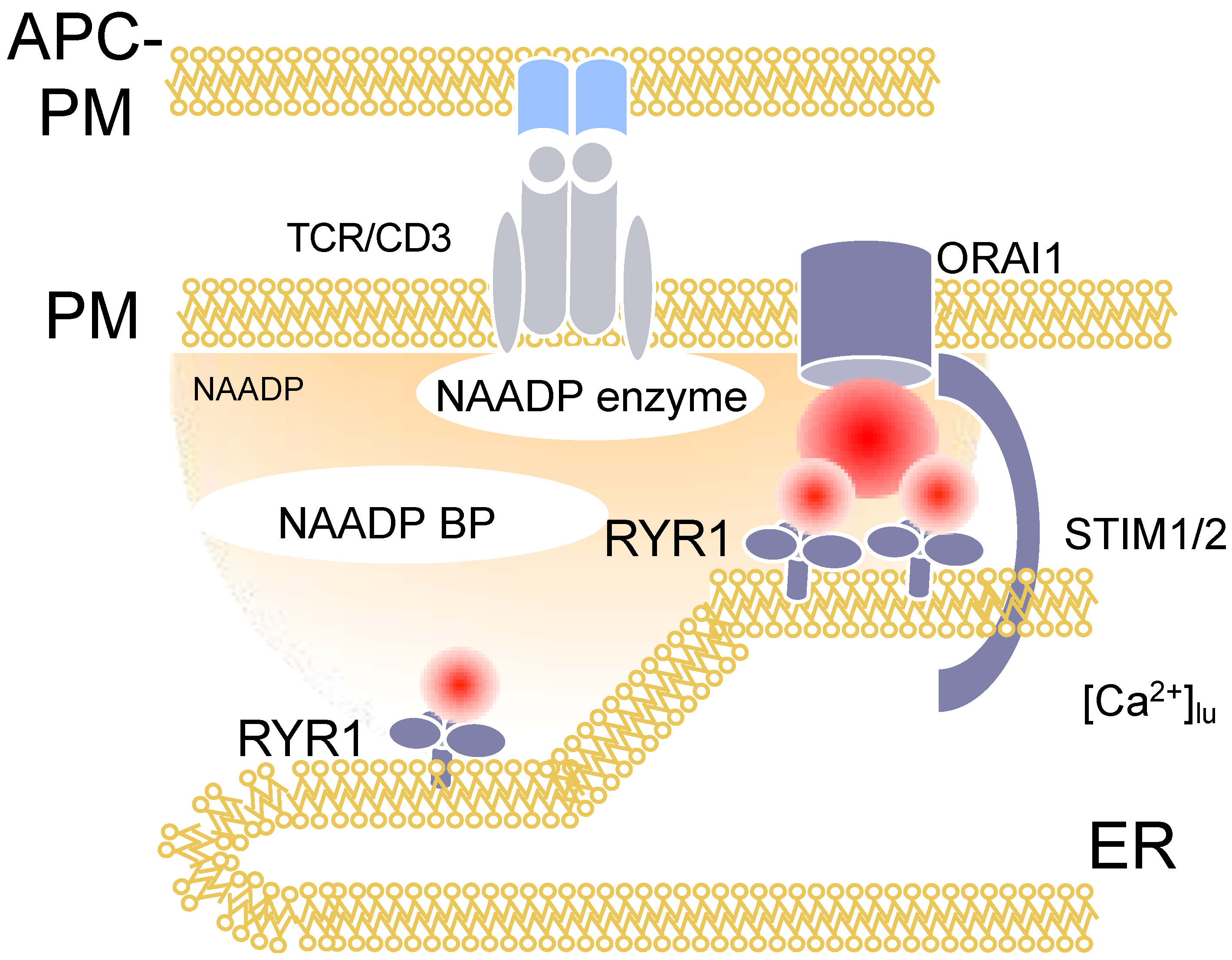
3. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP)
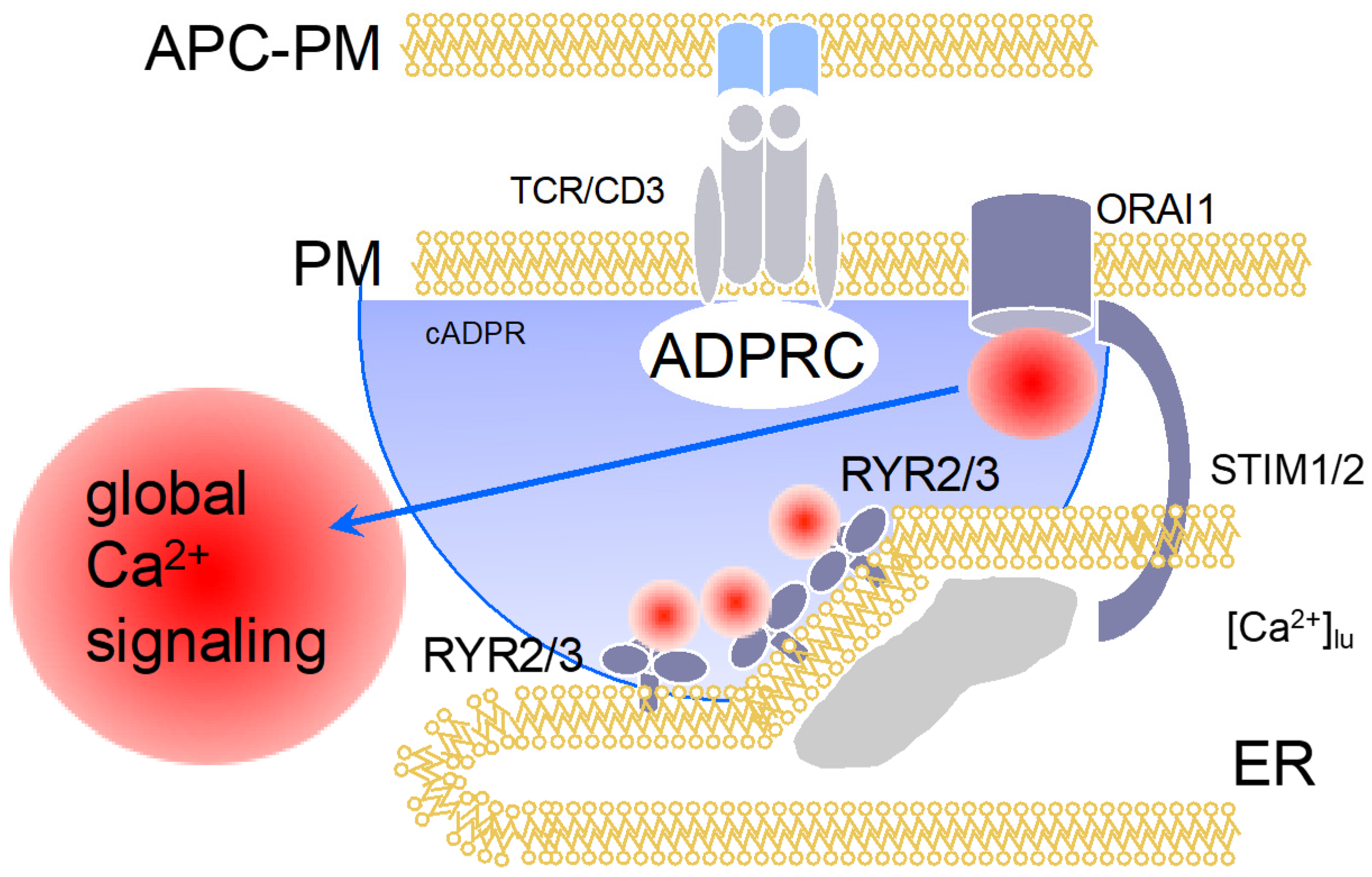
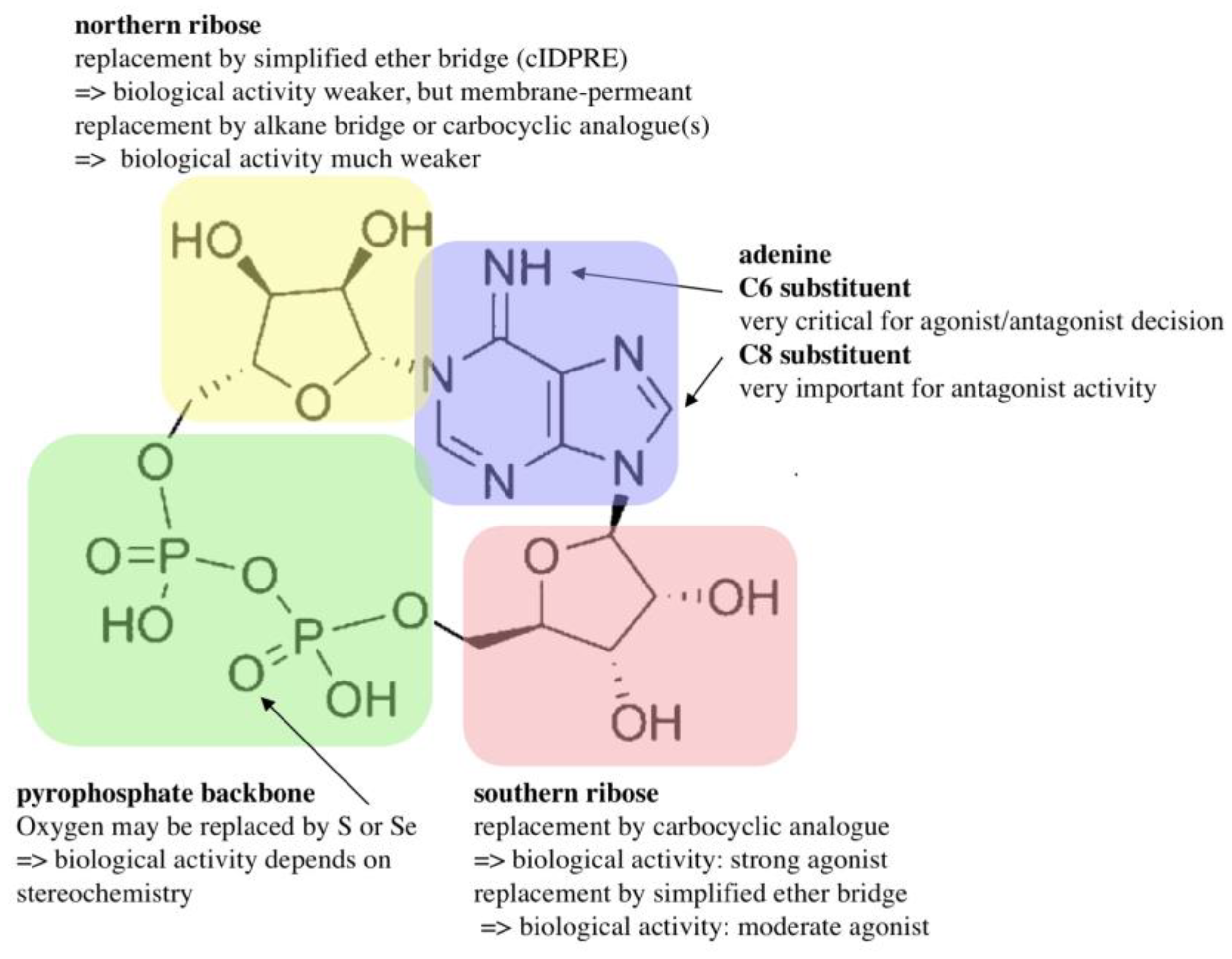
4. Cyclic Adenosine Diphosphoribose (cADPR)
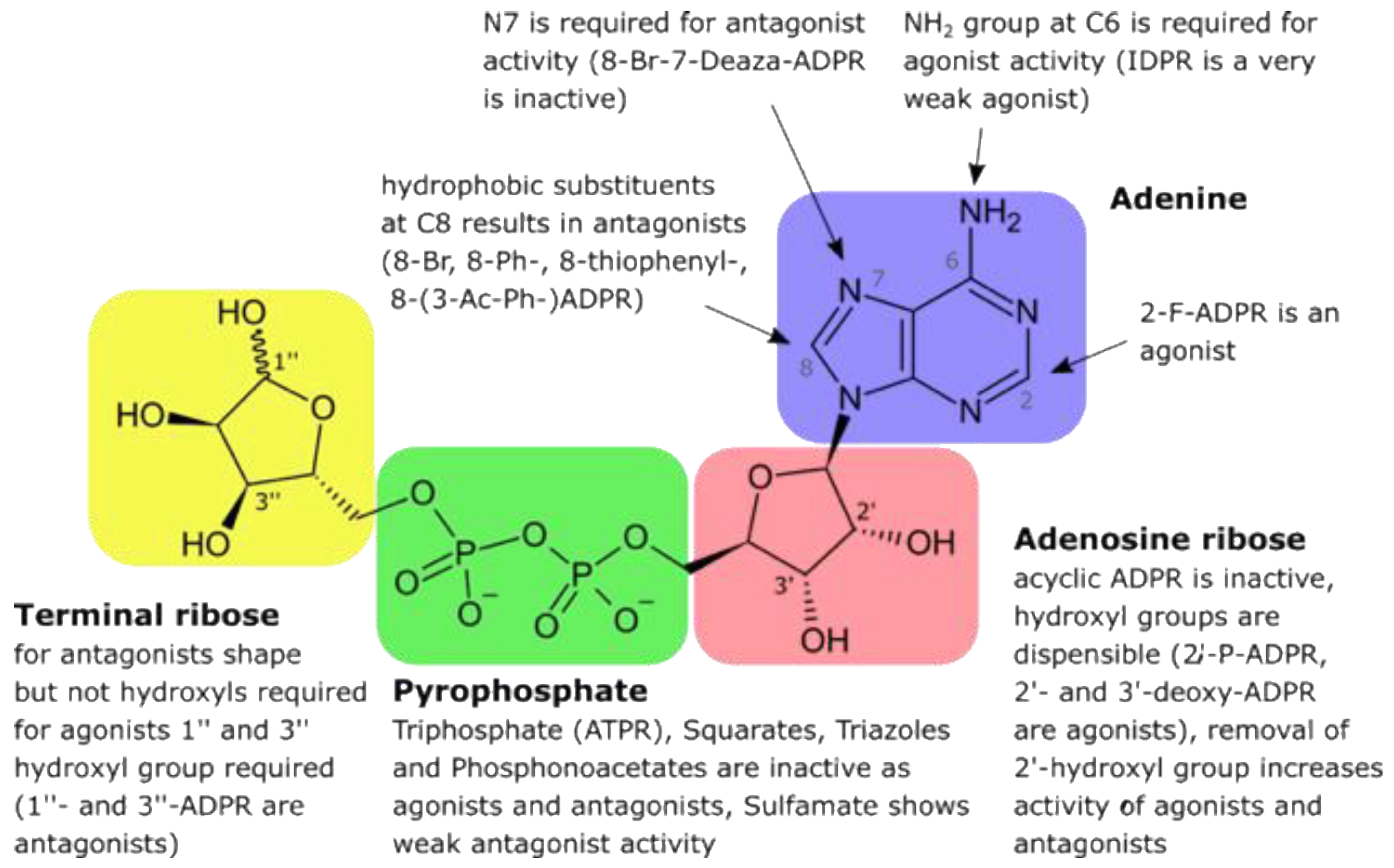
5. Adenosine Diphospho-Ribose (ADPR) and 2′-Deoxy-ADPR (2dADPR)
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cambridge Dictionary. Available online: https://dictionary.cambridge.org/de/worterbuch/englisch/genius (accessed on 11 September 2020).
- Guse, A.H.; da Silva, C.P.; Emmrich, F.; Ashamu, G.A.; Potter, B.V.; Mayr, G.W. Characterization of cyclic adenosine diphosphate-ribose-induced Ca2+ release in T lymphocyte cell lines. J. Immunol. 1995, 155, 3353–3359. [Google Scholar]
- Lee, H.C.; Aarhus, R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Streb, H.; Irvine, R.F.; Berridge, M.J.; Schulz, I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 1983, 306, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar]
- Wolf, I.M.; Diercks, B.P.; Gattkowski, E.; Czarniak, F.; Kempski, J.; Werner, R.; Schetelig, D.; Mittrücker, H.W.; Schumacher, V.; von Osten, M.; et al. Frontrunners of T cell activation: Initial, localized Ca2+-signals mediated by NAADP and the type 1 ryanodine receptor. Sci. Signal. 2015, 8, ra102. [Google Scholar] [CrossRef]
- Diercks, B.P.; Werner, R.; Weidemüller, P.; Czarniak, F.; Hernandez, L.; Lehmann, C.; Rosche, A.; Krüger, A.; Kaufmann, U.; Vaeth, M.; et al. ORAI1, STIM1/2, and RYR1 shape subsecond Ca2+ microdomains upon T cell activation. Sci. Signal. 2018, 11, eaat0358. [Google Scholar] [CrossRef]
- Dammermann, W.; Zhang, B.; Nebel, M.; Cordiglieri, C.; Odoardi, F.; Kirchberger, T.; Kawakami, N.; Dowden, J.; Schmid, F.; Dornmair, K.; et al. NAADP-mediated Ca2+-signaling via type 1 ryanodine receptor in T cells revealed by a synthetic NAADP antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10678–10683. [Google Scholar] [CrossRef]
- Walseth, T.F.; Lin-Moshier, Y.; Weber, K.; Marchant, J.S.; Slama, J.T.; Guse, A.H. Nicotinic Acid Adenine Dinucleotide 2′-Phosphate (NAADP) Binding Proteins in T-Lymphocytes. Messenger 2012, 1, 86–94. [Google Scholar] [CrossRef]
- Cordiglieri, C.; Odoardi, F.; Zhang, B.; Nebel, M.; Kawakami, N.; Klinkert, W.E.; Lodygin, D.; Lühder, F.; Breunig, E.; Schild, D.; et al. Nicotinic acid adenine dinucleotide phosphate-mediated calcium signalling in effector T cells regulates autoimmunity of the central nervous system. Brain 2010, 133, 1930–1943. [Google Scholar] [CrossRef]
- Nebel, M.; Schwoerer, A.P.; Warszta, D.; Siebrands, C.C.; Limbrock, A.C.; Swarbrick, J.M.; Fliegert, R.; Weber, K.; Bruhn, S.; Hohenegger, M.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated calcium signaling and arrhythmias in the heart evoked by β-adrenergic stimulation. J. Biol. Chem. 2013, 288, 16017–16030. [Google Scholar] [CrossRef]
- Hermann, J.; Bender, M.; Schumacher, D.; Woo, M.S.; Shaposhnykov, A.; Rosenkranz, S.C.; Kuryshev, V.; Meier, C.; Guse, A.H.; Friese, M.A.; et al. Contribution of NAADP to Glutamate-Evoked Changes in Ca2+ Homeostasis in Mouse Hippocampal Neurons. Front. Cell Dev. Biol. 2020, 8, 496. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; Roth, E.; Emmrich, F. Intracellular Ca2+-pools in Jurkat T-lymphocytes. Biochem. J. 1993, 291, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; Emmrich, F. T-cell receptor-mediated metabolism of inositol polyphosphates in Jurkat T-lymphocytes. Identification of a d-myo-inositol 1,2,3,4,6-pentakisphosphate-2-phosphomonoesterase activity, a D-myo-inositol 1,3,4,5,6-pentakisphosphate-1/3-phosphatase activity and a d/l-myo-inositol 1,2,4,5,6-pentakisphosphate-1/3-kinase activity. J. Biol. Chem. 1991, 266, 24498–24502. [Google Scholar] [PubMed]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Guse, A.H.; Silva, C.P.; Weber, K.; Ashamu, G.A.; Potter, B.V.; Mayr, G.W. Regulation of cADP-ribose-induced Ca2+ release by Mg2+ and inorganic phosphate. J. Biol. Chem. 1996, 271, 23946–23953. [Google Scholar] [CrossRef]
- Guse, A.H.; Berg, I.; da Silva, C.P.; Potter, B.V.; Mayr, G.W. Ca2+-entry induced by cyclic ADP-ribose in intact T-lymphocytes. J. Biol. Chem. 1997, 272, 8546–8550. [Google Scholar] [CrossRef]
- da Silva, C.P.; Potter, B.V.; Mayr, G.W.; Guse, A.H. Quantification of intracellular levels of cyclic ADP-ribose by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1998, 707, 43–50. [Google Scholar] [CrossRef]
- Guse, A.H.; da Silva, C.P.; Berg, I.; Skapenko, A.L.; Weber, K.; Heyer, P.; Hohenegger, M.; Ashamu, G.A.; Schulze-Koops, H.; Potter, B.V.; et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 1999, 398, 70–73. [Google Scholar] [CrossRef]
- Sethi, J.K.; Empson, R.M.; Bailey, V.C.; Potter, B.V.; Galione, A. 7-Deaza-8-bromo-cyclic ADP-ribose, the first membrane-permeant, hydrolysis-resistant cyclic ADP-ribose antagonist. J. Biol. Chem. 1997, 272, 16358–16363. [Google Scholar] [CrossRef]
- Bennett, D.L.; Cheek, T.R.; Berridge, M.J.; De Smedt, H.; Parys, J.B.; Missiaen, L.; Bootman, M.D. Expression and function of ryanodine receptors in nonexcitable cells. J. Biol. Chem. 1996, 271, 6356–6362. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; Cakir-Kiefer, C.; Fukuoka, M.; Shuto, S.; Weber, K.; Bailey, V.C.; Matsuda, A.; Mayr, G.W.; Oppenheimer, N.; Schuber, F.; et al. Novel hydrolysis-resistant analogues of cyclic ADP-ribose: Modification of the “northern” ribose and calcium release activity. Biochemistry 2002, 41, 6744–6751. [Google Scholar] [CrossRef] [PubMed]
- Bailey, V.C.; Fortt, S.M.; Summerhill, R.J.; Galione, A.; Potter, B.V. Cyclic aristeromycin diphosphate ribose: A potent and poorly hydrolysable Ca2+-mobilising mimic of cyclic adenosine diphosphate ribose. FEBS Lett. 1996, 379, 227–230. [Google Scholar] [CrossRef]
- Wagner, G.K.; Black, S.; Guse, A.H.; Potter, B.V. First enzymatic synthesis of an N1-cyclised cADPR (cyclic-ADP ribose) analogue with a hypoxanthine partial structure: Discovery of a membrane permeant cADPR agonist. Chem. Commun. (Camb.) 2003, 15, 1944–1945. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.K.; Riley, A.M.; Rosenberg, H.J.; Taylor, C.W.; Guse, A.H.; Potter, B.V. Analogues of cyclic adenosine 5′-diphosphate ribose and adenophostin A, nucleotides in cellular signal transduction. Nucleic Acids Res. Suppl. 2003, 3, 1–2. [Google Scholar] [CrossRef]
- Wagner, G.K.; Guse, A.H.; Potter, B.V. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5′-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [Google Scholar] [CrossRef]
- Kirchberger, T.; Wagner, G.; Xu, J.; Cordiglieri, C.; Wang, P.; Gasser, A.; Fliegert, R.; Bruhn, S.; Flügel, A.; Lund, F.E.; et al. Cellular effects and metabolic stability of N1-cyclic inosine diphosphoribose and its derivatives. Br. J. Pharmacol. 2006, 149, 337–344. [Google Scholar] [CrossRef]
- Kirchberger, T.; Moreau, C.; Wagner, G.K.; Fliegert, R.; Siebrands, C.C.; Nebel, M.; Schmid, F.; Harneit, A.; Odoardi, F.; Flügel, A.; et al. 8-Bromo-cyclic inosine diphosphoribose: Towards a selective cyclic ADP-ribose agonist. Biochem. J. 2009, 422, 139–149. [Google Scholar] [CrossRef]
- Kudoh, T.; Fukuoka, M.; Ichikawa, S.; Murayama, T.; Ogawa, Y.; Hashii, M.; Higashida, H.; Kunerth, S.; Weber, K.; Guse, A.H.; et al. Synthesis of stable and cell-type selective analogues of cyclic ADP-ribose, a Ca2+-mobilizing second messenger. Structure--activity relationship of the N1-ribose moiety. J. Am. Chem. Soc. 2005, 127, 8846–8855. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Sakaguchi, N.; Kudoh, T.; Takano, S.; Uehara, M.; Murayama, T.; Sakurai, T.; Hashii, M.; Higashida, H.; Weber, K.; et al. Design and synthesis of cyclic ADP-4-thioribose as a stable equivalent of cyclic ADP-ribose, a calcium ion-mobilizing second messenger. Angew. Chem. Int. Ed. Engl. 2013, 52, 6633–6637. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Takano, S.; Sakaguchi, N.; Kudoh, T.; Murayama, T.; Sakurai, T.; Hashii, M.; Higashida, H.; Weber, K.; Guse, A.H.; et al. Design, Synthesis, and Chemical and Biological Properties of Cyclic ADP-4-Thioribose as a Stable Equivalent of Cyclic ADP-Ribose. Messenger 2014, 3, 35–51. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guse, A.H. Structure activity relationship of cyclic ADPribose, an update. J. Chin. Pharm. Sci. 2013, 22, 127–136. [Google Scholar] [CrossRef]
- Moreau, C.; Kirchberger, T.; Swarbrick, J.M.; Bartlett, S.J.; Fliegert, R.; Yorgan, T.; Bauche, A.; Harneit, A.; Guse, A.H.; Potter, B.V. Structure-activity relationship of adenosine 5′-diphosphoribose at the transient receptor potential melastatin 2 (TRPM2) channel: Rational design of antagonists. J. Med. Chem. 2013, 56, 10079–10102. [Google Scholar] [CrossRef]
- Fliegert, R.; Bauche, A.; Wolf Pérez, A.M.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. [Google Scholar] [CrossRef]
- Baszczyňski, O.; Watt, J.M.; Rozewitz, M.D.; Guse, A.H.; Fliegert, R.; Potter, B.V.L. Synthesis of Terminal Ribose Analogues of Adenosine 5′-Diphosphate Ribose as Probes for the Transient Receptor Potential Cation Channel TRPM2. J. Org. Chem. 2019, 84, 6143–6157. [Google Scholar] [CrossRef] [PubMed]
- Baszczyňski, O.; Watt, J.M.; Rozewitz, M.D.; Fliegert, R.; Guse, A.H.; Potter, B.V.L. Synthesis of phosphonoacetate analogues of the second messenger adenosine 5′-diphosphate ribose (ADPR). RSC Adv. 2020, 10, 1776–1785. [Google Scholar] [CrossRef]





© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guse, A.H. 25 Years of Collaboration with A Genius: Deciphering Adenine Nucleotide Ca2+ Mobilizing Second Messengers Together with Professor Barry Potter. Molecules 2020, 25, 4220. https://doi.org/10.3390/molecules25184220
Guse AH. 25 Years of Collaboration with A Genius: Deciphering Adenine Nucleotide Ca2+ Mobilizing Second Messengers Together with Professor Barry Potter. Molecules. 2020; 25(18):4220. https://doi.org/10.3390/molecules25184220
Chicago/Turabian StyleGuse, Andreas H. 2020. "25 Years of Collaboration with A Genius: Deciphering Adenine Nucleotide Ca2+ Mobilizing Second Messengers Together with Professor Barry Potter" Molecules 25, no. 18: 4220. https://doi.org/10.3390/molecules25184220
APA StyleGuse, A. H. (2020). 25 Years of Collaboration with A Genius: Deciphering Adenine Nucleotide Ca2+ Mobilizing Second Messengers Together with Professor Barry Potter. Molecules, 25(18), 4220. https://doi.org/10.3390/molecules25184220