Confinement Effects on Glass-Forming Aqueous Dimethyl Sulfoxide Solutions

 ,
,  ,
, {kind=link}
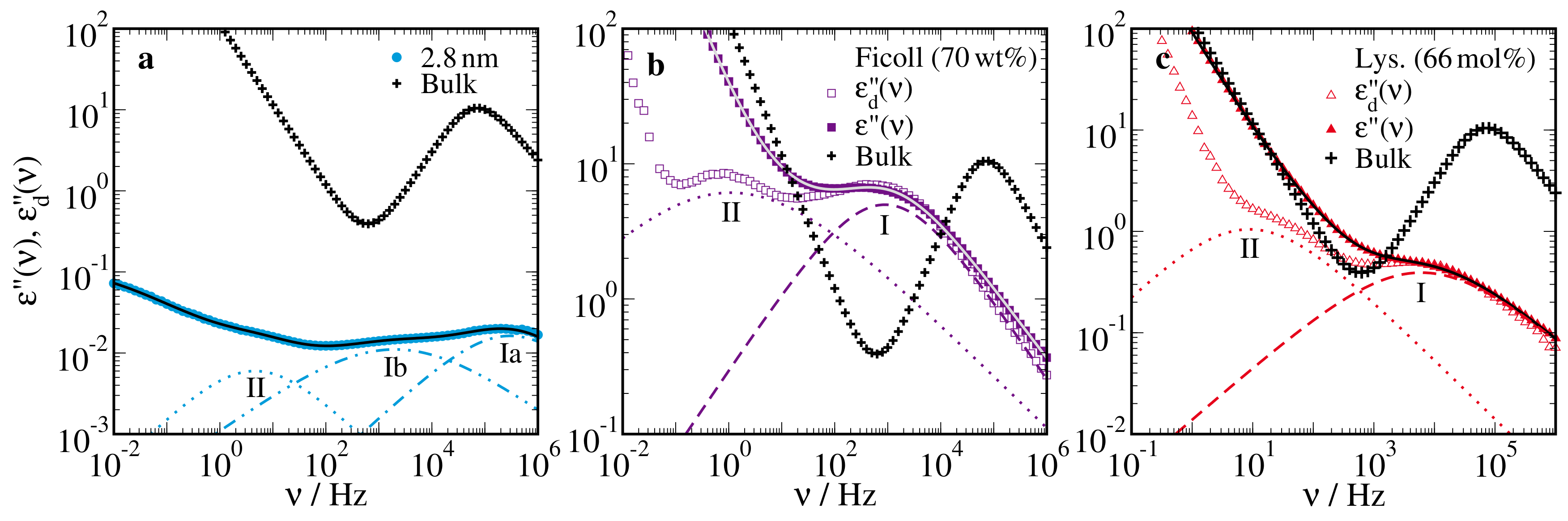{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Broadband Dielectric Spectroscopy
2.2. H NMR
2.2.1. H Spin-Lattice Relaxation
2.2.2. H Solid-Echo Intensities
2.2.3. H Stimulated-Echo Experiments
2.3. Rotational Correlation Times
2.4. Self-Diffusion Coefficients
3. Conclusions
4. Materials & Methods
4.1. Sample Preparation
4.2. Broadband Dielectric Spectroscopy and Nuclear Magnetic Resonance Experiments
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BDS | Broadband dielectric spectroscopy |
| CC | Cole–Cole |
| DMSO | Dimethyl sulfoxide |
| MWS | Maxwell–Wagner–Sillars |
| NMR | Nuclear magnetic resonance |
| SLR | Spin-lattice relaxation |
| SED | Stokes–Einstein–Debye |
| SEI | Solid-echo intensity |
| STE | Stimulated echo |
| SFG | Static field gradient |
| VFT | Vogel–Fulcher–Tammann |
References
- Bhushan, B.; Israelachvili, J.N.; Landman, U. Nanotribology: Friction, wear and lubrication at the atomic scale. Nature 1995, 374, 607–616. [Google Scholar] [CrossRef]
- Eijkel, J.C.T.; van den Berg, A. Nanofluidics: What is it and what can we expect from it? Microfluid. Nanofluid. 2005, 1, 249–267. [Google Scholar] [CrossRef]
- Bocquet, L.; Charlaix, E. Nanofluidics, from bulk to interfaces. Chem. Soc. Rev. 2010, 39, 1073–1095. [Google Scholar] [CrossRef] [PubMed]
- Haywood, D.G.; Saha-Shah, A.; Baker, L.A.; Jacobson, S.C. Fundamental Studies of Nanofluidics: Nanopores, Nanochannels, and Nanopipets. Anal. Chem. 2014, 87, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J. 1978, 22, 283–294. [Google Scholar] [CrossRef]
- Takata, K. Aquaporins: Water channel proteins of the cell membrane. Prog. Histochem. Cytochem. 2004, 39, 1–83. [Google Scholar] [CrossRef]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Pissis, P.; Laudat, J.; Daoukaki-Diamanti, D.; Kyritsis, A. Structure and Dynamic Properties of Water Confined in Small Volumes. In Hydrogen Bond Networks; Springer: Dordrecht, The Netherlands, 1994; pp. 425–432. [Google Scholar] [CrossRef]
- Yoshida, K.; Yamaguchi, T.; Kittaka, S.; Bellissent-Funel, M.C.; Fouquet, P. Thermodynamic, structural, and dynamic properties of supercooled water confined in mesoporous MCM-41 studied with calorimetric, neutron diffraction, and neutron spin echo measurements. J. Chem. Phys. 2008, 129, 054702. [Google Scholar] [CrossRef]
- Swenson, J.; Bergman, R.; Howells, W.S. Quasielastic neutron scattering of two-dimensional water in a vermiculite clay. J. Chem. Phys. 2000, 113, 2873–2879. [Google Scholar] [CrossRef]
- Forrest, J.A.; Dalnoki-Veress, K. The glass transition in thin polymer films. Adv. Colloid Interface Sci. 2001, 94, 167–195. [Google Scholar] [CrossRef]
- Cerveny, S.; Alegrìa, Á.; Colmenero, J. Universal features of water dynamics in solutions of hydrophilic polymers, biopolymers, and small glass-forming materials. Phys. Rev. E 2008, 77, 031803. [Google Scholar] [CrossRef] [PubMed]
- Blochowicz, T.; Gouirand, E.; Fricke, A.; Spehr, T.; Stühn, B.; Frick, B. Accelerated dynamics of supercooled glycerol in soft confinement. Chem. Phys. Lett. 2009, 475, 171–174. [Google Scholar] [CrossRef]
- Swenson, J.; Cerveny, S. Dynamics of deeply supercooled interfacial water. J. Phys. Condens. Matter 2014, 27, 033102. [Google Scholar] [CrossRef] [PubMed]
- Cerveny, S.; Mallamace, F.; Swenson, J.; Vogel, M.; Xu, L. Confined Water as Model of Supercooled Water. Chem. Rev. 2016, 116, 7608–7625. [Google Scholar] [CrossRef]
- Ball, P. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef]
- Ippolito, J.A.; Alexander, R.S.; Christianson, D.W. Hydrogen bond stereochemistry in protein structure and function. J. Mol. Biol. 1990, 215, 457–471. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N. Water Mediation in Protein Folding and Molecular Recognition. Annu. Rev. Biophys. Biomol. Struct 2006, 35, 389–415. [Google Scholar] [CrossRef]
- Fumino, K.; Peppel, T.; Geppert-Rybczyńska, M.; Zaitsau, D.H.; Lehmann, J.K.; Verevkin, S.P.; Köckerling, M.; Ludwig, R. The influence of hydrogen bonding on the physical properties of ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 14064. [Google Scholar] [CrossRef]
- Alcoutlabi, M.; McKenna, G.B. Effects of confinement on material behaviour at the nanometre size scale. J. Phys. Condens. Matter 2005, 17, R461–R524. [Google Scholar] [CrossRef]
- Zheng, W.; Simon, S.L. Confinement effects on the glass transition of hydrogen bonded liquids. J. Chem. Phys. 2007, 127, 194501. [Google Scholar] [CrossRef] [PubMed]
- Swenson, J.; Jansson, H.; Bergman, R. Relaxation Processes in Supercooled Confined Water and Implications for Protein Dynamics. Phys. Rev. Lett. 2006, 96, 247802. [Google Scholar] [CrossRef] [PubMed]
- Alba-Simionesco, C.; Coasne, B.; Dosseh, G.; Dudziak, G.; Gubbins, K.E.; Radhakrishnan, R.; Sliwinska-Bartkowiak, M. Effects of confinement on freezing and melting. J. Phys. Condens. Matter 2006, 18, R15–R68. [Google Scholar] [CrossRef] [PubMed]
- Richert, R. Dielectric spectroscopy and dynamics in confinement. Eur. Phys. J. Spec. Top. 2010, 189, 37–46. [Google Scholar] [CrossRef]
- Vogel, M. NMR Studies on Simple Liquids in Confinement. Eur. Phys. J. Spec. Top. 2010, 189, 47–64. [Google Scholar] [CrossRef]
- Buntkowsky, G.; Vogel, M.; Winter, R. Properties of Hydrogen-Bonded Liquids at Interfaces. Z. Phys. Chem. 2018, 232, 937–972. [Google Scholar] [CrossRef]
- Demuth, D.; Sattig, M.; Steinrücken, E.; Weigler, M.; Vogel, M. 2H NMR Studies on the Dynamics of Pure and Mixed Hydrogen-Bonded Liquids in Confinement. Z. Phys. Chem. 2018, 232, 1059–1087. [Google Scholar] [CrossRef]
- Buntkowsky, G.; Vogel, M. Small Molecules, Non-Covalent Interactions, and Confinement. Molecules 2020, 25, 3311. [Google Scholar] [CrossRef]
- Matsubara, H.; Pichierri, F.; Kurihara, K. Mechanism of Diffusion Slowdown in Confined Liquids. Phys. Rev. Lett. 2012, 109, 197801. [Google Scholar] [CrossRef]
- Klameth, F.; Henritzi, P.; Vogel, M. Static and dynamic length scales in supercooled liquids: Insights from molecular dynamics simulations of water and tri-propylene oxide. J. Chem. Phys. 2014, 140, 144501. [Google Scholar] [CrossRef]
- Geske, J.; Harrach, M.; Heckmann, L.; Horstmann, R.; Klameth, F.; Müller, N.; Pafong, E.; Wohlfromm, T.; Drossel, B.; Vogel, M. Molecular Dynamics Simulations of Water, Silica, and Aqueous Mixtures in Bulk and Confinement. Z. Phys. Chem. 2018, 232, 1187–1225. [Google Scholar] [CrossRef]
- Arndt, M.; Stannarius, R.; Gorbatschow, W.; Kremer, F. Dielectric investigations of the dynamic glass transition in nanopores. Phys. Rev. E 1996, 54, 5377–5390. [Google Scholar] [CrossRef] [PubMed]
- Gradmann, S.; Medick, P.; Rössler, E.A. Glassy Dynamics in Nanoconfinement as Revealed by 31P NMR. J. Phys. Chem. B 2009, 113, 8443–8445. [Google Scholar] [CrossRef] [PubMed]
- Mallamace, F.; Branca, C.; Corsaro, C.; Leone, N.; Spooren, J.; Stanley, H.E.; Chen, S.H. Dynamical Crossover and Breakdown of the Stokes-Einstein Relation in Confined Water and in Methanol-Diluted Bulk Water. J. Phys. Chem. B 2010, 114, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Weigler, M.; Winter, E.; Kresse, B.; Brodrecht, M.; Buntkowsky, G.; Vogel, M. Static field gradient NMR studies of water diffusion in mesoporous silica. Phys. Chem. Chem. Phys. 2020, 22, 13989–13998. [Google Scholar] [CrossRef] [PubMed]
- Schiro, G.; Cupane, A.; Pagnotta, S.E.; Bruni, F. Dynamic properties of solvent confined in silica gels studied by broadband dielectric spectroscopy. J. Non-Cryst. Solids 2007, 353, 4546–4551. [Google Scholar] [CrossRef]
- Elamin, K.; Jansson, H.; Swenson, J. Dynamics of aqueous binary glass-formers confined in MCM-41. Phys. Chem. Chem. Phys. 2015, 17, 12978–12987. [Google Scholar] [CrossRef]
- Sattig, M.; Elamin, K.; Reuhl, M.; Swenson, J.; Vogel, M. Dynamics of DiPGME-Water Mixtures in Mesoporous Silica. J. Phys. Chem. C 2017, 121, 6796–6806. [Google Scholar] [CrossRef]
- Mhanna, R.; Catrou, P.; Dutta, S.; Lefort, R.; Essafri, I.; Ghoufi, A.; Muthmann, M.; Zamponi, M.; Frick, B.; Morineau, D. Dynamic Heterogeneities in Liquid Mixtures Confined in Nanopores. J. Phys. Chem. B 2020, 124, 3152–3162. [Google Scholar] [CrossRef]
- Gelb, L.D.; Gubbins, K.E.; Radhakrishnan, R.; Sliwinska-Bartkowiak, M. Phase separation in confined systems. Rep. Prog. Phys. 1999, 62, 1573–1659. [Google Scholar] [CrossRef]
- Guo, X.Y.; Watermann, T.; Sebastiani, D. Local Microphase Separation of a Binary Liquid under Nanoscale Confinement. J. Phys. Chem. B 2014, 118, 10207–10213. [Google Scholar] [CrossRef] [PubMed]
- Harrach, M.F.; Drossel, B.; Winschel, W.; Gutmann, T.; Buntkowsky, G. Mixtures of Isobutyric Acid and Water Confined in Cylindrical Silica Nanopores Revisited: A Combined Solid-State NMR and Molecular Dynamics Simulation Study. J. Phys. Chem. C 2015, 119, 28961–28969. [Google Scholar] [CrossRef]
- Schmitz, R.; Müller, N.; Ullmann, S.; Vogel, M. A molecular dynamics simulations study on ethylene glycol-water mixtures in mesoporous silica. J. Chem. Phys. 2016, 145, 104703. [Google Scholar] [CrossRef] [PubMed]
- Muthulakshmi, T.; Dutta, D.; Maheshwari, P.; Pujari, P.K. Evidence for confinement induced phase separation in ethanol–water mixture: A positron annihilation study. J. Phys. Condens. Matter 2017, 30, 025001. [Google Scholar] [CrossRef]
- Rasmussen, D.H.; MacKenzie, A.P. Phase Diagram for the System Water–Dimethylsulphoxide. Nature 1968, 220, 1315–1317. [Google Scholar] [CrossRef]
- Packer, K.J.; Tomlinson, D.J. Nuclear spin relaxation and self-diffusion in the binary system, dimethylsulphoxide (DMSO)+water. Trans. Faraday Soc. 1971, 67, 1302. [Google Scholar] [CrossRef]
- Luzar, A.; Stefan, J. Dielectric behaviour of DMSO-water mixtures. A hydrogen-bonding model. J. Mol. Liq. 1990, 46, 221–238. [Google Scholar] [CrossRef]
- Borin, I.A.; Skaf, M.S. Molecular association between water and dimethyl sulfoxide in solution: A molecular dynamics simulation study. J. Chem. Phys. 1999, 110, 6412–6420. [Google Scholar] [CrossRef]
- Catalán, J.; Díaz, C.; García-Blanco, F. Characterization of Binary Solvent Mixtures of DMSO with Water and Other Cosolvents. J. Org. Chem. 2001, 66, 5846–5852. [Google Scholar] [CrossRef]
- Bordallo, H.N.; Herwig, K.W.; Luther, B.M.; Levinger, N.E. Quasi-elastic neutron scattering study of dimethyl-sulfoxide–water mixtures: Probing molecular mobility in a nonideal solution. J. Chem. Phys. 2004, 121, 12457. [Google Scholar] [CrossRef]
- Soper, A.K.; Luzar, A. A neutron diffraction study of dimethyl sulphoxide–water mixtures. J. Chem. Phys. 1992, 97, 1320–1331. [Google Scholar] [CrossRef]
- Vishnyakov, A.; Lyubartsev, A.P.; Laaksonen, A. Molecular Dynamics Simulations of Dimethyl Sulfoxide and Dimethyl Sulfoxide-Water Mixture. J. Phys. Chem. A 2001, 105, 1702–1710. [Google Scholar] [CrossRef]
- Wulf, A.; Ludwig, R. Structure and Dynamics of Water Confined in Dimethyl Sulfoxide. ChemPhysChem 2006, 7, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.; Chen, Z.; Li, H.; Zhang, L.; Ye, J.; Zhang, Q.; Zhuang, W. Molecular Mechanism of Water Reorientation Dynamics in Dimethyl Sulfoxide Aqueous Mixtures. J. Phys. Chem. B 2020, 124, 1806–1816. [Google Scholar] [CrossRef]
- Strauss, G.; Ingenito, E.P. Stabilization of liposome bilayers to freezing and thawing: Effects of cryoprotective agents and membrane proteins. Cryobiology 1980, 17, 508–515. [Google Scholar] [CrossRef]
- Lusceac, S.A.; Gainaru, C.; Ratzke, D.A.; Graf, M.F.; Vogel, M. Secondary Water Relaxation in a Water/Dimethyl Sulfoxide Mixture Revealed by Deuteron Nuclear Magnetic Resonance and Dielectric Spectroscopy. J. Phys. Chem. B 2011, 115, 11588–11596. [Google Scholar] [CrossRef]
- Wong, D.B.; Sokolowsky, K.P.; El-Barghouthi, M.I.; Fenn, E.E.; Giammanco, C.H.; Sturlaugson, A.L.; Fayer, M.D. Water Dynamics in Water/DMSO Binary Mixtures. J. Phys. Chem. B 2012, 116, 5479–5490. [Google Scholar] [CrossRef]
- Gao, M.; Held, C.; Patra, S.; Arns, L.; Sadowski, G.; Winter, R. Crowders and Cosolvents-Major Contributors to the Cellular Milieu and Efficient Means to Counteract Environmental Stresses. ChemPhysChem 2017, 18, 2951–2972. [Google Scholar] [CrossRef]
- Weißheit, S.; Kahse, M.; Kämpf, K.; Tietze, A.; Vogel, M.; Winter, R.; Thiele, C.M. Elastin-like Peptide in Confinement: FT-IR and NMR T1 Relaxation Data. Z. Phys. Chem. 2018, 232, 1239–1261. [Google Scholar] [CrossRef]
- Venturoli, D.; Rippe, B. Ficoll and Dextran vs. Globular Proteins for Testing Glomerular Permselectivity: Effects of Molecular Size, Shape, Charge, and Deformability. Am. J. Physiol. Ren. Physiol. 2005, 288, F605–F613. [Google Scholar] [CrossRef]
- Fissell, W.H.; Hofmann, C.L.; Smith, R.; Chen, M.H. Size and Conformation of Ficoll as Determined by Size-Exclusion Chromatography followed by Multiangle Light Scattering. Am. J. Physiol. Ren. Physiol. 2010, 298, F205–F208. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, C.J.F.; Bordewijk, P. Theory of Electric Polarization; Elsevier: Amsterdam, The Netherlands, 1978; Volume 2. [Google Scholar]
- Broadband Dielectric Spectroscopy; Kremer, F., Schönhals, A., Eds.; Springer: Berlin, Germany, 2003. [Google Scholar]
- Sjöström, J.; Swenson, J.; Bergman, R.; Kittaka, S. Investigating hydration dependence of dynamics of confined water: Monolayer, hydration water and Maxwell-Wagner processes. J. Chem. Phys. 2008, 128, 154503. [Google Scholar] [CrossRef] [PubMed]
- Jansson, H.; Bergman, R.; Swenson, J. Role of Solvent for the Dynamics and the Glass Transition of Proteins. J. Phys. Chem. B 2011, 115, 4099–4109. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Richert, R. Dielectric spectroscopy study of myoglobin in glycerol-water mixtures. Biochim. Biophys. Acta-Proteins Proteom. 2014, 1844, 323–329. [Google Scholar] [CrossRef]
- Nakanishi, M.; Sokolov, A.P. Protein dynamics in a broad frequency range: Dielectric spectroscopy studies. J. Non-Cryst. Solids 2015, 407, 478–485. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Z.; Du, X. Evolution of Dielectric Behavior of Regenerated Cellulose Film during Isothermal Dehydration Monitored in Real Time via Dielectric Spectroscopy. Polymers 2019, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Wübbenhorst, M.; van Turnhout, J. Analysis of complex dielectric spectra. I. One-dimensional derivative techniques and three-dimensional modelling. J. Non-Cryst. Solids 2002, 305, 40–49. [Google Scholar] [CrossRef]
- Panagopoulou, A.; Kyritsis, A.; Sabateri-Serra, R.; Gomez-Ribelles, J.L.; Shinyashiki, N.; Pissis, P. Glass transition and dynamics in BSA-water mixtures over wide ranges of composition studied by thermal and dielectric techniques. Biochim. Biophys. Acta-Proteins Proteom. 2011, 1814, 1984–1996. [Google Scholar] [CrossRef]
- Panagopoulou, A.; Kyritsis, A.; Vodina, M.; Pissis, P. Dynamics of uncrystallized water and protein in hydrated elastin studied by thermal and dielectric techniques. Biochim. Biophys. Acta-Proteins Proteom. 2013, 1834, 977–988. [Google Scholar] [CrossRef]
- Khodadadi, S.; Sokolov, A.P. Protein Dynamics: From Rattling in a Cage to Structural Relaxation. Soft Matter 2015, 11, 4984–4998. [Google Scholar] [CrossRef]
- Ngai, K.L.; Capaccioli, S.; Paciaroni, A. Dynamics of hydrated proteins and bio-protectants: Caged dynamics, α-relaxation, and β-relaxation. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 3553–3563. [Google Scholar] [CrossRef] [PubMed]
- Kämpf, K.; Demuth, D.; Zamponi, M.; Wuttke, J.; Vogel, M. Quasielastic neutron scattering studies on couplings of protein and water dynamics in hydrated elastin. J. Chem. Phys. 2020, 152, 245101. [Google Scholar] [CrossRef] [PubMed]
- Rosenstihl, M.; Kämpf, K.; Klameth, F.; Sattig, M.; Vogel, M. Dynamics of interfacial water. J. Non-Cryst. Solids 2015, 407, 449–458. [Google Scholar] [CrossRef]
- Brodrecht, M.; Klotz, E.; Lederle, C.; Breitzke, H.; Stühn, B.; Vogel, M.; Buntkowsky, G. A Combined Solid-State NMR, Dielectric Spectroscopy and Calorimetric Study of Water in Lowly Hydrated MCM-41 Samples. Z. Phys. Chem. 2018, 232, 1003–1015. [Google Scholar] [CrossRef]
- Lederle, C.; Sattig, M.; Vogel, M. Effects of Partial Crystallization on the Dynamics of Water in Mesoporous Silica. J. Phys. Chem. C 2018, 122, 15427–15434. [Google Scholar] [CrossRef]
- Herbers, C.R.; Sauer, D.; Vogel, M. 2H NMR studies of glycerol dynamics in protein matrices. J. Chem. Phys. 2012, 136, 124511. [Google Scholar] [CrossRef]
- Richert, R.; Agapov, A.; Sokolov, A.P. Appearance of a Debye process at the conductivity relaxation frequency of a viscous liquid. J. Chem. Phys. 2011, 134, 104508. [Google Scholar] [CrossRef]
- Cheng, S.; Mirigian, S.; Carrillo, J.M.Y.; Bocharova, V.; Sumpter, B.G.; Schweizer, K.S.; Sokolov, A.P. Revealing spatially heterogeneous relaxation in a model nanocomposite. J. Chem. Phys. 2015, 143, 194704. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K.; Spiess, H.W. Multidimensional Solid-State NMR and Polymers, 1st ed.; Academic Press: London, UK, 1994. [Google Scholar]
- Böhmer, R.; Diezemann, G.; Hinze, G.; Rössler, E. Dynamics of supercooled liquids and glassy solids. Prog. Nucl. Magn. Reson. Spectrosc. 2001, 39, 191–267. [Google Scholar] [CrossRef]
- Sattig, M.; Vogel, M. Dynamic Crossovers and Stepwise Solidification of Confined Water: A 2H NMR Study. J. Phys. Chem. Lett. 2013, 5, 174–178. [Google Scholar] [CrossRef]
- Sattig, M.; Reutter, S.; Fujara, F.; Werner, M.; Buntkowsky, G.; Vogel, M. NMR studies on the temperature-dependent dynamics of confined water. Phys. Chem. Chem. Phys. 2014, 16, 19229–19240. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Fella, V.; Huang, W.; Zhang, K.A.I.; Landfester, K.; Butt, H.J.; Vogel, M.; Floudas, G. Crystallization and Dynamics of Water Confined in Model Mesoporous Silica Particles: Two Ice Nuclei and Two Fractions of Water. Langmuir 2019, 35, 5890–5901. [Google Scholar] [CrossRef] [PubMed]
- Weigler, M.; Brodrecht, M.; Buntkowsky, G.; Vogel, M. Reorientation of Deeply Cooled Water in Mesoporous Silica: NMR Studies of the Pore-Size Dependence. J. Phys. Chem. B 2019, 123, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Reuhl, M.; Weigler, M.; Brodrecht, M.; Buntkowsky, G.; Vogel, M. Nuclear Magnetic Resonance and Broadband Dielectric Spectroscopy Studies on the Dynamics of Ethylene Glycol in Mesoporous Silica. J. Phys. Chem. C 2020. [Google Scholar] [CrossRef]
- Vogel, M. Origins of Apparent Fragile-to-Strong Transitions of Protein Hydration Waters. Phys. Rev. Lett. 2008, 101, 225701. [Google Scholar] [CrossRef]
- Lusceac, S.A.; Vogel, M.R.; Herbers, C.R. 2H and 13C NMR Studies on the Temperature-Dependent Water and Protein Dynamics in Hydrated Elastin, Myoglobin and Collagen. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 41–48. [Google Scholar] [CrossRef][Green Version]
- Kämpf, K.; Kremmling, B.; Vogel, M. Vanishing amplitude of backbone dynamics causes a true protein dynamical transition: 2H NMR studies on perdeuterated C-phycocyanin. Phys. Rev. E 2014, 89, 032710. [Google Scholar] [CrossRef]
- Fabri, D.; Williams, M.A.K.; Halstead, T.K. Water T2 relaxation in sugar solutions. Carbohydr. Res. 2005, 340, 889–905. [Google Scholar] [CrossRef]
- Spiess, H.W. Deuteron NMR methods for studying molecular order and motion in solid polymers and liquid crystalline polymers. Pure Appl. Chem. 1985, 57, 1617–1626. [Google Scholar] [CrossRef]
- Schmidt, C.; Kuhn, K.J.; Spiess, H.W. Distribution of correlation times in glassy polymers from pulsed deuteron NMR. Prog. Colloid Polym. Sci. 1985, 71, 71–76. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Capaccioli, S.; Ngai, K.L.; Ancherbak, S.; Bertoldo, M.; Ciampalini, G.; Thayyil, M.S.; Wang, L.M. The JG β-Relaxation in Water and Impact on the Dynamics of Aqueous Mixtures and Hydrated Biomolecules. J. Chem. Phys. 2019, 151, 034504. [Google Scholar] [CrossRef]
- Arndt, M.; Stannarius, R.; Groothues, H.; Hempel, E.; Kremer, F. Length Scale of Cooperativity in the Dynamic Glass Transition. Phys. Rev. Lett. 1997, 79, 2077–2080. [Google Scholar] [CrossRef]
- Xia, Y.; Cho, H.; Risbud, S.H.; Bartl, M.H.; Sen, S. Coexistence of Structural and Dynamical Heterogeneity in Liquids Under Nanoconfinement. Front. Phys. 2020, 8, 130. [Google Scholar] [CrossRef]
- Elamin, K.; Jansson, H.; Kittaka, S.; Swenson, J. Different behavior of water in confined solutions of high and low solute concentrations. Phys. Chem. Chem. Phys. 2013, 15, 18437. [Google Scholar] [CrossRef] [PubMed]
- Essafri, I.; Morineau, D.; Ghoufi, A. Microphase separation of a miscible binary liquid mixture under confinement at the nanoscale. Npj Comput. Mater. 2019, 5, 42. [Google Scholar] [CrossRef]
- Tanner, J.E. Use of the Stimulated Echo in NMR Diffusion Studies. J. Chem. Phys. 1970, 52, 2523–2526. [Google Scholar] [CrossRef]
- Callaghan, P.T. Principles of Nuclear Magnetic Resonance Microscopy; Clarendon Press: Oxford, UK, 1991. [Google Scholar]
- Fleischer, G.; Fujara, F. NMR as a Generalized Incoherent Scattering Experiment. In Solid-State NMR I Methods; Springer: Berlin, Germany, 1994; pp. 159–207. [Google Scholar] [CrossRef]
- Diffusion NMR of Confined Systems; Valiullin, R., Ed.; New Developments in NMR, Royal Society of Chemistry: Cambridge, UK, 2016. [Google Scholar] [CrossRef]
- Peschier, L.J.C.; Bouwstra, J.A.; de Bleyser, J.; Junginger, H.E.; Leyte, J.C. Cross-Relaxation Effects in Pulsed-Field-Gradient Stimulated-Echo Measurements on Water in a Macromolecular Matrix. J. Magn. Res. B 1996, 110, 150–157. [Google Scholar] [CrossRef]
- Topgaard, D.; Söderman, O. Self-Diffusion of Nonfreezing Water in Porous Carbohydrate Polymer Systems Studied with Nuclear Magnetic Resonance. Biophys. J. 2002, 83, 3596–3606. [Google Scholar] [CrossRef][Green Version]
- Rosenstihl, M.; Vogel, M. Static and pulsed field gradient nuclear magnetic resonance studies of water diffusion in protein matrices. J. Chem. Phys. 2011, 135, 164503. [Google Scholar] [CrossRef]
- Edzes, H.T.; Samulski, E.T. The measurement of cross-relaxation effects in the proton NMR spin-lattice relaxation of water in biological systems: Hydrated collagen and muscle. J. Magn. Reson. (1969) 1978, 31, 207–229. [Google Scholar] [CrossRef]
- Hills, B.P.; Wright, K.M.; Belton, P.S. Proton N.M.R. studies of chemical and diffusive exchange in carbohydrate systems. Mol. Phys. 1989, 67, 1309–1326. [Google Scholar] [CrossRef]
- Palit, S.; Yethiraj, A. Dynamics and cluster formation in charged and uncharged Ficoll70 solutions. J. Chem. Phys. 2017, 147, 074901. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, D.; Leisen, J.; Spiess, H.W. Experimental Aspects of Multidimensional Exchange Solid-State NMR. J. Magn. Reson. A 1995, 115, 60–79. [Google Scholar] [CrossRef]
- Chang, I.; Fujara, F.; Geil, B.; Hinze, G.; Sillescu, H.; Tölle, A. New perspectives of NMR in ultrahigh static magnetic field gradients. J. Non-Cryst. Solids 1994, 172, 674–681. [Google Scholar] [CrossRef]
Sample Availability: Most NMR samples are available from the corresponding author upon reasonable request. |











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demuth, D.; Reuhl, M.; Hopfenmüller, M.; Karabas, N.; Schoner, S.; Vogel, M. Confinement Effects on Glass-Forming Aqueous Dimethyl Sulfoxide Solutions. Molecules 2020, 25, 4127. https://doi.org/10.3390/molecules25184127
Demuth D, Reuhl M, Hopfenmüller M, Karabas N, Schoner S, Vogel M. Confinement Effects on Glass-Forming Aqueous Dimethyl Sulfoxide Solutions. Molecules. 2020; 25(18):4127. https://doi.org/10.3390/molecules25184127
Chicago/Turabian StyleDemuth, Dominik, Melanie Reuhl, Moritz Hopfenmüller, Nail Karabas, Simon Schoner, and Michael Vogel. 2020. "Confinement Effects on Glass-Forming Aqueous Dimethyl Sulfoxide Solutions" Molecules 25, no. 18: 4127. https://doi.org/10.3390/molecules25184127
APA StyleDemuth, D., Reuhl, M., Hopfenmüller, M., Karabas, N., Schoner, S., & Vogel, M. (2020). Confinement Effects on Glass-Forming Aqueous Dimethyl Sulfoxide Solutions. Molecules, 25(18), 4127. https://doi.org/10.3390/molecules25184127