
Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains

Abstract

1. Introduction
2. Results
2.1. Screening Workflow
2.2. Screening Results
2.3. Hit Follow Up
2.4. Investigating a Possible Orthogonal Binding Mode to PfGCN5
3. Discussion
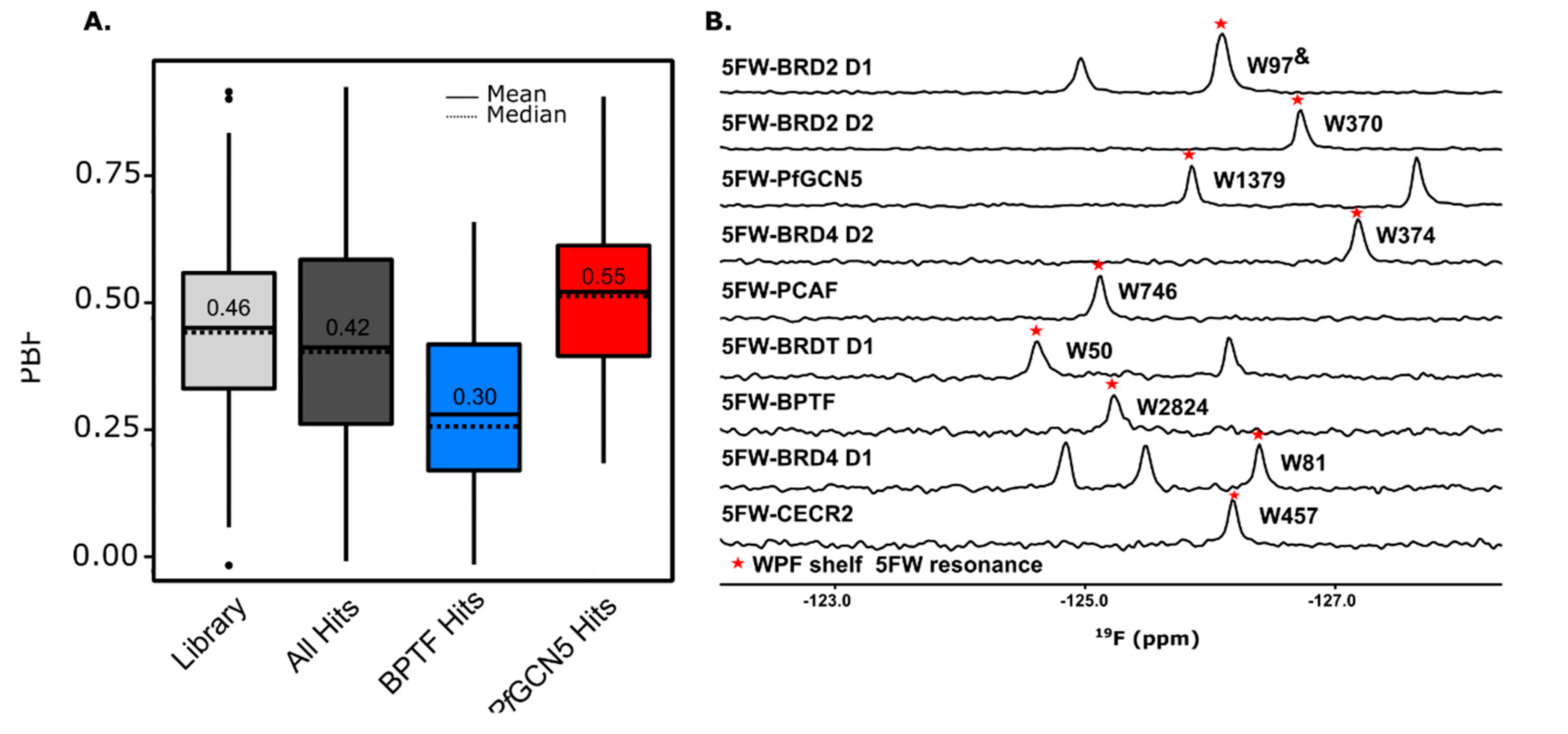
3.1. 3D-Enriched Fragment Analysis
3.2. Initial Assessment of the Broader Applicability of the Screening Workflow
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. NMR Methods
4.4. Peptide Synthesis
4.5. Docking
4.6. Circular Dichroism
4.7. FW PfGCN5 Resonance Assignment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. New Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Newell, D.R.; Angibaud, P. A successful collaboration between academia, biotech and pharma led to discovery of erdafitinib, a selective FGFR inhibitor recently approved by the FDA. MedChemComm 2019, 10, 1509–1511. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.J.; Mortenson, P.N.; Murray, C.W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 2014, 116, 82–91. [Google Scholar] [CrossRef]
- Di Micco, S.; Vitale, R.; Pellecchia, M.; Rega, M.F.; Riva, R.; Basso, A.; Bifulco, G. Identification of Lead Compounds As Antagonists of Protein Bcl-xL with a Diversity-Oriented Multidisciplinary Approach. J. Med. Chem. 2009, 52, 7856–7867. [Google Scholar] [CrossRef]
- Foley, D.J.; Craven, P.G.E.; Collins, P.M.; Doveston, R.G.; Aimon, A.; Talon, R.; Churcher, I.; von Delft, F.; Marsden, S.P.; Nelson, A. Synthesis and Demonstration of the Biological Relevance of sp3-rich Scaffolds Distantly Related to Natural Product Frameworks. Chem. Eur. J. 2017, 23, 15227–15232. [Google Scholar] [CrossRef]
- Grädler, U.; Schwarz, D.; Blaesse, M.; Leuthner, B.; Johnson, T.L.; Bernard, F.; Jiang, X.; Marx, A.; Gilardone, M.; Lemoine, H.; et al. Discovery of novel Cyclophilin D inhibitors starting from three dimensional fragments with millimolar potencies. Bioorg. Med. Chem. Lett. 2019, 29, 126717. [Google Scholar] [CrossRef]
- Vu, H.; Pedro, L.; Mak, T.; McCormick, B.; Rowley, J.; Liu, M.; Di Capua, A.; Williams-Noonan, B.; Pham, N.B.; Pouwer, R.; et al. Fragment-Based Screening of a Natural Product Library against 62 Potential Malaria Drug Targets Employing Native Mass Spectrometry. Acs Infect. Dis. 2018, 4, 431–444. [Google Scholar] [CrossRef]
- Zhang, R.; McIntyre, P.J.; Collins, P.M.; Foley, D.J.; Arter, C.; vonDelft, F.; Bayliss, R.; Warriner, S.; Nelson, A. Construction of a Shape-Diverse Fragment Set: Design, Synthesis and Screen against Aurora-A Kinase. Chem. Euro. J. 2019, 25, 6831–6839. [Google Scholar] [CrossRef]
- Johnson, J.A.; Nicolaou, C.A.; Kirberger, S.E.; Pandey, A.K.; Hu, H.; Pomerantz, W.C.K. Evaluating the Advantages of Using 3D-Enriched Fragments for Targeting BET Bromodomains. Acs Med. Chem. Lett. 2019, 10, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Morley, A.D.; Pugliese, A.; Birchall, K.; Bower, J.; Brennan, P.; Brown, N.; Chapman, T.; Drysdale, M.; Gilbert, I.H.; Hoelder, S.; et al. Fragment-based hit identification: Thinking in 3D. Drug Discov. Today 2013, 18, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Clemons, P.A.; Bodycombe, N.E.; Carrinski, H.A.; Wilson, J.A.; Shamji, A.F.; Wagner, B.K.; Koehler, A.N.; Schreiber, S.L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. USA 2010, 107, 18787–18792. [Google Scholar] [CrossRef]
- Dobson, C.M. Chemical space and biology. Nature 2004, 432, 824. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Sauer, W.H.B.; Schwarz, M.K. Molecular Shape Diversity of Combinatorial Libraries: A Prerequisite for Broad Bioactivity. J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. [Google Scholar] [CrossRef]
- Schuffenhauer, A.; Brown, N.; Selzer, P.; Ertl, P.; Jacoby, E. Relationships between Molecular Complexity, Biological Activity, and Structural Diversity. J. Chem. Inf. Model. 2006, 46, 525–535. [Google Scholar] [CrossRef]
- Erlanson, D. Poll results: Affiliation and fragment-finding methods in 2019. Practicle Fragments. Erlanson, D., Ed.; 2019. Available online: https://practicalfragments.blogspot.com/ (accessed on 25 July 2020).
- Keserű, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef]
- Harner, M.J.; Chauder, B.A.; Phan, J.; Fesik, S.W. Fragment-Based Screening of the Bromodomain of ATAD2. J. Med. Chem. 2014, 57, 9687–9692. [Google Scholar] [CrossRef]
- Mashalidis, E.H.; Śledź, P.; Lang, S.; Abell, C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309–2324. [Google Scholar] [CrossRef]
- Norton, R.S.; Leung, E.W.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of (19)F-NMR in Fragment-Based Drug Discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef] [PubMed]
- Urick, A.K.; Hawk, L.M.L.; Cassel, M.K.; Mishra, N.K.; Liu, S.; Adhikari, N.; Zhang, W.; dos Santos, C.O.; Hall, J.L.; Pomerantz, W.C.K. Dual Screening of BPTF and Brd4 Using Protein-Observed Fluorine NMR Uncovers New Bromodomain Probe Molecules. Acs Chem. Biol. 2015, 10, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Zartler, E.R.; Hanson, J.; Jones, B.E.; Kline, A.D.; Martin, G.; Mo, H.; Shapiro, M.J.; Wang, R.; Wu, H.; Yan, J. RAMPED-UP NMR: Multiplexed NMR-Based Screening for Drug Discovery. J. Am. Chem. Soc. 2003, 125, 10941–10946. [Google Scholar] [CrossRef]
- Urick, A.K.; Calle Jiménez, L.P.; Espinosa, J.F.; Hu, H.; Pomerantz, W.C.K. Protein-Observed Fluorine NMR is a Complementary Ligand Discovery Method to 1H CPMG Ligand-Observed NMR. Acs Chem. Biol. 2016, 11, 3154–3164. [Google Scholar] [CrossRef] [PubMed]
- Stadmiller, S.S.; Aguilar, J.S.; Waudby, C.A.; Pielak, G.J. Rapid Quantification of Protein-Ligand Binding via (19)F NMR Lineshape Analysis. Biophys. J. 2020, 118, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Arntson, K.E.; Pomerantz, W.C.K. Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J. Med. Chem. 2016, 59, 5158–5171. [Google Scholar] [CrossRef] [PubMed]
- Evanics, F.; Kitevski, J.L.; Bezsonova, I.; Forman-Kay, J.; Prosser, R.S. 19F NMR studies of solvent exposure and peptide binding to an SH3 domain. Biochim. Biophys. Acta. (Bba)-Gen. Subj. 2007, 1770, 221–230. [Google Scholar] [CrossRef]
- Boeszoermenyi, A.; Chhabra, S.; Dubey, A.; Radeva, D.L.; Burdzhiev, N.T.; Chanev, C.D.; Petrov, O.I.; Gelev, V.M.; Zhang, M.; Anklin, C.; et al. Aromatic 19F-13C TROSY: A background-free approach to probe biomolecular structure, function, and dynamics. Nat. Methods 2019, 16, 333–340. [Google Scholar] [CrossRef]
- Vidler, L.R.; Brown, N.; Knapp, S.; Hoelder, S. Druggability Analysis and Structural Classification of Bromodomain Acetyl-lysine Binding Sites. J. Med. Chem. 2012, 55, 7346–7359. [Google Scholar] [CrossRef]
- Dar, A.A.; Nosrati, M.; Bezrookove, V.; de Semir, D.; Majid, S.; Thummala, S.; Sun, V.; Tong, S.; Leong, S.P.L.; Minor, D.; et al. The Role of BPTF in Melanoma Progression and in Response to BRAF-Targeted Therapy. J. Natl. Cancer Ins. 2015, 107, djv034. [Google Scholar] [CrossRef]
- Landry, J.; Sharov, A.A.; Piao, Y.; Sharova, L.V.; Xiao, H.; Southon, E.; Matta, J.; Tessarollo, L.; Zhang, Y.E.; Ko, M.S.H.; et al. Essential Role of Chromatin Remodeling Protein Bptf in Early Mouse Embryos and Embryonic Stem Cells. Plos Genet. 2008, 4, e1000241. [Google Scholar] [CrossRef]
- Frey, W.D.; Chaudhry, A.; Slepicka, P.F.; Ouellette, A.M.; Kirberger, S.E.; Pomerantz, W.C.K.; Hannon, G.J.; dos Santos, C.O. BPTF Maintains Chromatin Accessibility and the Self-Renewal Capacity of Mammary Gland Stem Cells. Stem Cell Rep. 2017, 9, 23–31. [Google Scholar] [CrossRef] [PubMed]
- TP-238 A chemical probe for CECR2/BPTF bromodomains. Available online: https://www.thesgc.org/chemical-probes/TP-238 (accessed on 5 September 2018).
- NVS-BPTF-1 A chemical probe for BPTF. Available online: https://www.thesgc.org/chemical-probes/NVS-BPTF-1 (accessed on 19 February 2019).
- Kirberger, S.E.; Ycas, P.D.; Johnson, J.A.; Chen, C.; Ciccone, M.F.; Woo, R.W.L.; Urick, A.K.; Zahid, H.; Shi, K.; Aihara, H.; et al. Selectivity, ligand deconstruction, and cellular activity analysis of a BPTF bromodomain inhibitor. Org. Biomol. Chem. 2019, 17, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Q.; Leung, E.L.H.; Li, Y.; Fan, X.; Wu, Q.; Yao, X.; Liu, L. Compound C620-0696, a new potent inhibitor targeting BPTF, the chromatin-remodeling factor in non-small-cell lung cancer. Front. Med. 2019, 14, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Han, J.; Lu, W.; Lian, F.; Wang, J.; Lu, T.; Tao, H.; Xiao, S.; Zhang, F.; Liu, Y.-C.; et al. Discovery of alkoxy benzamide derivatives as novel BPTF bromodomain inhibitors via structure-based virtual screening. Bioorg. Chem. 2019, 86, 494–500. [Google Scholar] [CrossRef]
- Saraf, A.; Cervantes, S.; Bunnik, E.M.; Ponts, N.; Sardiu, M.E.; Chung, D.-W.D.; Prudhomme, J.; Varberg, J.M.; Wen, Z.; Washburn, M.P.; et al. Dynamic and Combinatorial Landscape of Histone Modifications during the Intraerythrocytic Developmental Cycle of the Malaria Parasite. J. Proteome Res. 2016, 15, 2787–2801. [Google Scholar] [CrossRef] [PubMed]
- Trelle, M.B.; Salcedo-Amaya, A.M.; Cohen, A.M.; Stunnenberg, H.G.; Jensen, O.N. Global Histone Analysis by Mass Spectrometry Reveals a High Content of Acetylated Lysine Residues in the Malaria Parasite Plasmodium falciparum. J. Proteome Res 2009, 8, 3439–3450. [Google Scholar] [CrossRef] [PubMed]
- Josling Gabrielle, A.; Petter, M.; Oehring Sophie, C.; Gupta Archna, P.; Dietz, O.; Wilson Danny, W.; Schubert, T.; Längst, G.; Gilson Paul, R.; Crabb Brendan, S.; et al. A Plasmodium Falciparum Bromodomain Protein Regulates Invasion Gene Expression. Cell Host Microbe. 2015, 17, 741–751. [Google Scholar] [CrossRef]
- Fan, Q.; An, L.; Cui, L. Plasmodium falciparum Histone Acetyltransferase, a Yeast GCN5 Homologue Involved in Chromatin Remodeling. Eukaryot. Cell 2004, 3, 264–276. [Google Scholar] [CrossRef]
- Jeffers, V.; Yang, C.; Huang, S.; Sullivan, W.J. Bromodomains in Protozoan Parasites: Evolution, Function, and Opportunities for Drug Development. Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef]
- Miao, J.; Fan, Q.; Cui, L.; Li, J.; Li, J.; Cui, L. The malaria parasite Plasmodium falciparum histones: Organization, expression, and acetylation. Gene. 2006, 369, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.J.; Robaa, D.; Skinner-Adams, T.S.; Sippl, W.; Andrews, K.T. Activity of bromodomain protein inhibitors/binders against asexual-stage Plasmodium falciparum parasites. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Moustakim, M.; Clark, P.G.K.; Trulli, L.; Fuentes de Arriba, A.L.; Ehebauer, M.T.; Chaikuad, A.; Murphy, E.J.; Mendez-Johnson, J.; Daniels, D.; Hou, C.-F.D.; et al. Discovery of a PCAF Bromodomain Chemical Probe. Angew. Chem. Int. Ed. 2017, 56, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Gee, C.T.; Arntson, K.E.; Urick, A.K.; Mishra, N.K.; Hawk, L.M.; Wisniewski, A.J.; Pomerantz, W.C. Protein-Observed (19)F-NMR for Fragment Screening, Affinity Quantification and Druggability Assessment. Nat. Protc. 2016, 11, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Urick, A.K.; Ember, S.W.J.; Schönbrunn, E.; Pomerantz, W.C. Dual Screening of BPTF and Brd4 Using Protein-Observed Fluorine NMR Uncovers New Bromodomain Probe Molecules. Acs Chem. Biol. 2014, 9, 2755–2760. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Urick, A.K.; Ember, S.W.J.; Schönbrunn, E.; Pomerantz, W.C. Fluorinated Aromatic Amino Acids Are Sensitive 19F NMR Probes for Bromodomain-Ligand Interactions. Acs Chem. Biol. 2014, 9, 2755–2760. [Google Scholar] [CrossRef] [PubMed]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef]
- Ycas, P.D.; Zahid, H.; Chan, A.; Olson, N.M.; Johnson, J.A.; Talluri, S.K.; Schonbrunn, E.; Pomerantz, W.C.K. New inhibitors for the BPTF bromodomain enabled by structural biology and biophysical assay. Org. Biomol. Chem. 2020, 18, 5174–5182. [Google Scholar] [CrossRef]
- Olp, M.D.; Sprague, D.J.; Goetz, C.J.; Kathman, S.G.; Wynia-Smith, S.L.; Shishodia, S.; Summers, S.B.; Xu, Z.; Statsyuk, A.V.; Smith, B.C. Covalent-Fragment Screening of BRD4 Identifies a Ligandable Site Orthogonal to the Acetyl-Lysine Binding Sites. Acs Chem. Biol. 2020, 15, 1036–1049. [Google Scholar] [CrossRef]
- Humphreys, P.G.; Bamborough, P.; Chung, C.W.; Craggs, P.D.; Gordon, L.; Grandi, P.; Hayhow, T.G.; Hussain, J.; Jones, K.L.; Lindon, M.; et al. Discovery of a Potent, Cell Penetrant, and Selective p300/CBP-Associated Factor (PCAF)/General Control Nonderepressible 5 (GCN5) Bromodomain Chemical Probe. J. Med. Chem. 2017, 60, 695–709. [Google Scholar] [CrossRef]
- Structural Genomics Consortium. Available online: https://www.thesgc.org/chemical-probes/TP-238 (accessed on 15 May 2004).
- Olson, N.M.; Kroc, S.; Johnson, J.A.; Zahid, H.; Ycas, P.D.; Chan, A.; Kimbrough, J.R.; Kalra, P.; Schönbrunn, E.; Pomerantz, W.C.K. NMR Analyses of Acetylated H2A.Z Isoforms Identify Differential Binding Interactions with the Bromodomain of the NURF Nucleosome Remodeling Complex. Biochemistry 2020, 59, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Morcos, F.; Cheng, R.R.; Jiang, H.; Onuchic, J.N. Elucidating the druggable interface of protein−protein interactions using fragment docking and coevolutionary analysis. Proc. Natl. Acad. Sci. USA 2016, 113, E8051–E8058. [Google Scholar] [CrossRef] [PubMed]
- Feher, M.; Schmidt, J.M. Property Distributions: Differences between Drugs, Natural Products, and Molecules from Combinatorial Chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Garner, P.; Cox, P.B.; Rathnayake, U.; Holloran, N.; Erdman, P. Design and Synthesis of Pyrrolidine-based Fragments That Sample Three-dimensional Molecular Space. Acs Med. Chem. Lett. 2019, 10, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Hanby, A.R.; Troelsen, N.S.; Osberger, T.J.; Kidd, S.L.; Mortensen, K.T.; Spring, D.R. Fsp(3)-rich and diverse fragments inspired by natural products as a collection to enhance fragment-based drug discovery. Chemcomm (Camb. Engl. ) 2020, 56, 2280–2283. [Google Scholar] [CrossRef]
- Liu, M.; Quinn, R.J. Fragment-based screening with natural products for novel anti-parasitic disease drug discovery. Expert Opin. Drug Discov. 2019, 14, 1283–1295. [Google Scholar] [CrossRef]
- Firth, N.C.; Brown, N.; Blagg, J. Plane of Best Fit: A Novel Method to Characterize the Three-Dimensionality of Molecules. J. Chem. Inf. Model. 2012, 52, 2516–2525. [Google Scholar] [CrossRef]
- Fuller, N.; Spadola, L.; Cowen, S.; Patel, J.; Schonherr, H.; Cao, Q.; McKenzie, A.; Edfeldt, F.; Rabow, A.; Goodnow, R. An Improved Model for Fragment-Based Lead Generation at AstraZeneca. Drug Discov Today 2016. [Google Scholar] [CrossRef]
- Divakaran, A.; Kirberger, S.E.; Pomerantz, W.C.K. SAR by (Protein-Observed) 19F NMR. Acc. Chem. Res. 2019, 52, 3407–3418. [Google Scholar] [CrossRef]
- Ye, L.; Larda, S.T.; Frank Li, Y.F.; Manglik, A.; Prosser, R.S. A comparison of chemical shift sensitivity of trifluoromethyl tags: Optimizing resolution in ¹⁹F NMR studies of proteins. J. Biomol Nmr 2015, 62, 97–103. [Google Scholar] [CrossRef]
- Lu, M.; Ishima, R.; Polenova, T.; Gronenborn, A.M. 19F NMR relaxation studies of fluorosubstituted tryptophans. J. Biomol. Nmr 2019, 73, 401–409. [Google Scholar] [CrossRef] [PubMed]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P., Jr. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front Chem 2020, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ranganathan, A.; Ijzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in Silico and Biophysical Screening Approaches in Fragment-Based Lead Discovery against the A2A Adenosine Receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Spiliotopoulos, D.; Wamhoff, E.-C.; Lolli, G.; Rademacher, C.; Caflisch, A. Discovery of BAZ2A bromodomain ligands. Eur. J. Med. Chem. 2017, 139, 564–572. [Google Scholar] [CrossRef]
- Zhao, H.; Gartenmann, L.; Dong, J.; Spiliotopoulos, D.; Caflisch, A. Discovery of BRD4 bromodomain inhibitors by fragment-based high-throughput docking. Bioorganic Med. Chem. Lett. 2014, 24, 2493–2496. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Divers. 2019, 23, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Unzue, A.; Dong, J.; Spiliotopoulos, D.; Nevado, C.; Caflisch, A. Discovery of CREBBP Bromodomain Inhibitors by High-Throughput Docking and Hit Optimization Guided by Molecular Dynamics. J. Med. Chem. 2016, 59, 1340–1349. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
Sample Availability: All small molecule fragments are commercially available and can be purchased from Life Chemicals. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein(s) | Number of Hit Mixtures from PrOF NMR | Number of Hits after 1H CPMG NMR Deconvolution |
|---|---|---|
| BPTF | 14 (14.2%) | 27 (5.7%), (9.8%*) |
| PfGCN5 | 16 (16.3%) | 24 (5.1%), (9.2%*) |
| BPTF and PfGCN5 | 19 (19.4%) | 19 (4.1%) |
| ID | Structure | Initial Mixture BPTF PrOF NMR Δδ | Initial Mixture PfGCN5 PrOF NMR Δδ | 1H CPMG NMR % Drop in Resonance Intensity BPTF | 1H CPMG NMR % Drop in Resonance Intensity PfGCN5 | BPTF Kd (µM) | PfGCN5 Kd (µM) | BRD4 D1 Kd (µM) b |
|---|---|---|---|---|---|---|---|---|
| 1 |  | - | ++ | NA | +++++ | NA | NS | NB |
| 2 |  | ++ | +++ | NA | +++++ | NA | 360 | 450 |
| 3 |  | - | ++++ | NA | ++++ | NA | NS | NB |
| 4 |  | + | - | +++++ | NA | NB | NA | NB |
| 5 |  | ++ | - | +++ | NA | NS | NA | NB |
| 6 |  | + | - | + | NA | 540 | NA | NB |
| 7 |  | +++ | +++ | ++ | +++++ | >1 mM | 150 | NB |
| 8 |  | +++ | ++ | ++ | ++ | 720 | >1 mM | 24 |
| 9 |  | ++++ | ++++ | ++++ a | ++++ a | 180 | 16 & (8.3, s = 7.0 *) | 50 |
| 2 1H CPMG NMR Screen | 2 PrOF NMR Screens | Dual-Protein PrOF NMR Screen | This Method | |
|---|---|---|---|---|
| Screen Time (h) a | 30 | 21.7 | 10.9 | 10.8 |
| Deconvolution Time a | 0 | 36.7 | 36.7 | 10.8 |
| Amount of Protein Used for Screen (mg) b | 8.5 (BPTF), 7.4(PfGCN5) | 42 (BPTF), 37 (PfGCN5) | 85 (BPTF), 74 (PfGCN5) | 85 (BPTF), 74 (PfGCN5) |
| Amount of Protein Used for Deconvolution (mg) b | 0 | 74 (BPTF), 61 (PfGCN5) | 74 (BPTF),61 (PfGCN5) | 2.6 (BPTF), 2.1 (PfGCN5) |
| Amount of Fragment Used for Screen (mg) c | 0.03 | 0.12 | 0.06 | 0.06 |
| Amount of Fragment Used for Deconvolution (mg) d | 0 | 0.06 (bind 1 protein), 0.12 (bind both proteins) | 0.06 (bind 1 protein), 0.12 (bind both proteins) | 0.015 (bind 1 protein), 0.03 (bind both proteins) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, J.A.; Olson, N.M.; Tooker, M.J.; Bur, S.K.; Pomerantz, W.C.K. Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains. Molecules 2020, 25, 3949. https://doi.org/10.3390/molecules25173949
Johnson JA, Olson NM, Tooker MJ, Bur SK, Pomerantz WCK. Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains. Molecules. 2020; 25(17):3949. https://doi.org/10.3390/molecules25173949
Chicago/Turabian StyleJohnson, Jorden A., Noelle M. Olson, Madison J. Tooker, Scott K. Bur, and William C.K. Pomerantz. 2020. "Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains" Molecules 25, no. 17: 3949. https://doi.org/10.3390/molecules25173949
APA StyleJohnson, J. A., Olson, N. M., Tooker, M. J., Bur, S. K., & Pomerantz, W. C. K. (2020). Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains. Molecules, 25(17), 3949. https://doi.org/10.3390/molecules25173949