
Review of the Synthesis and Anticancer Properties of Pyrazolo[4,3-e][1,2,4]triazine Derivatives

 , ,
, , 

Abstract
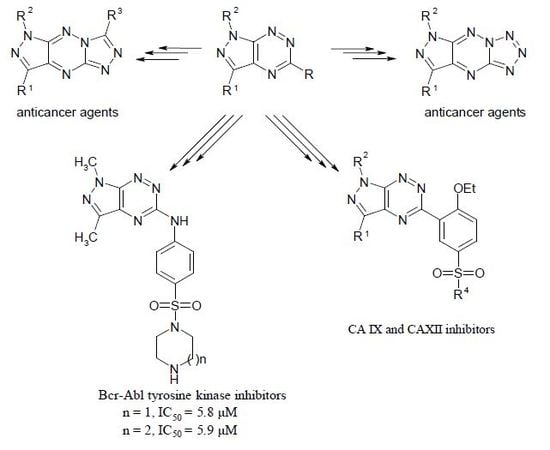
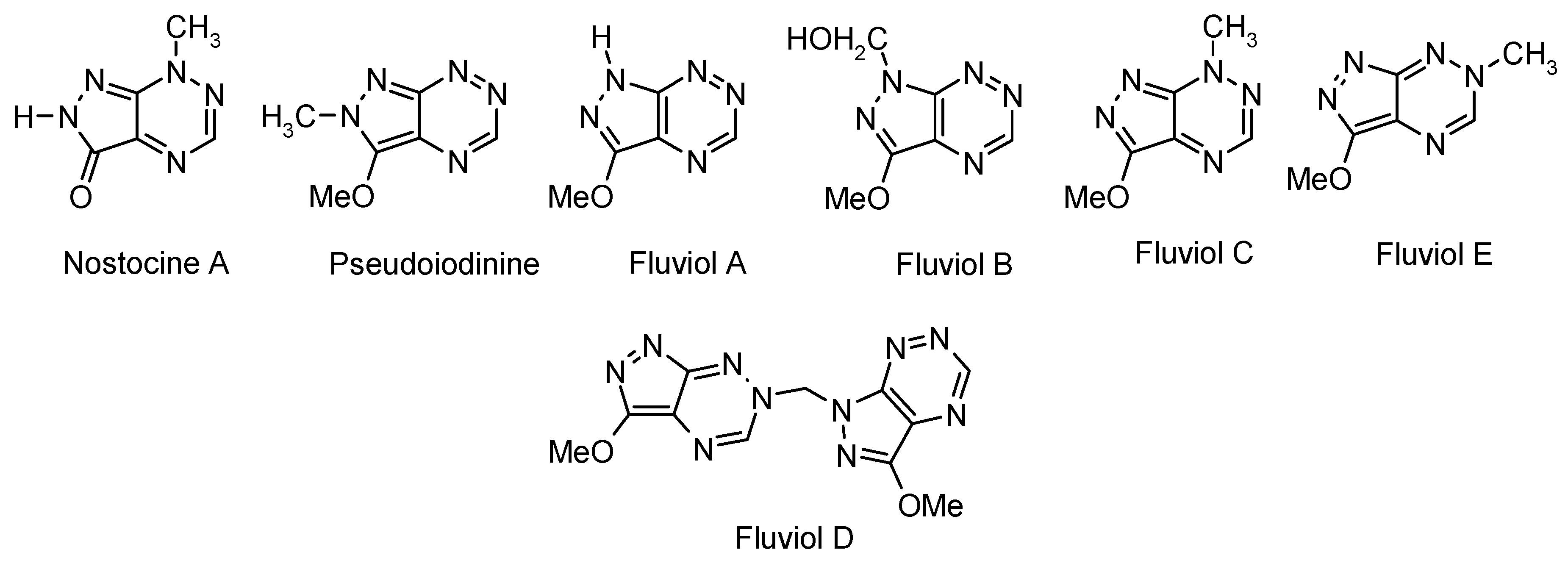
1. Introduction
2. Approach to the Construction of Pyrazolo[4,3-e][1,2,4]triazine Ring System
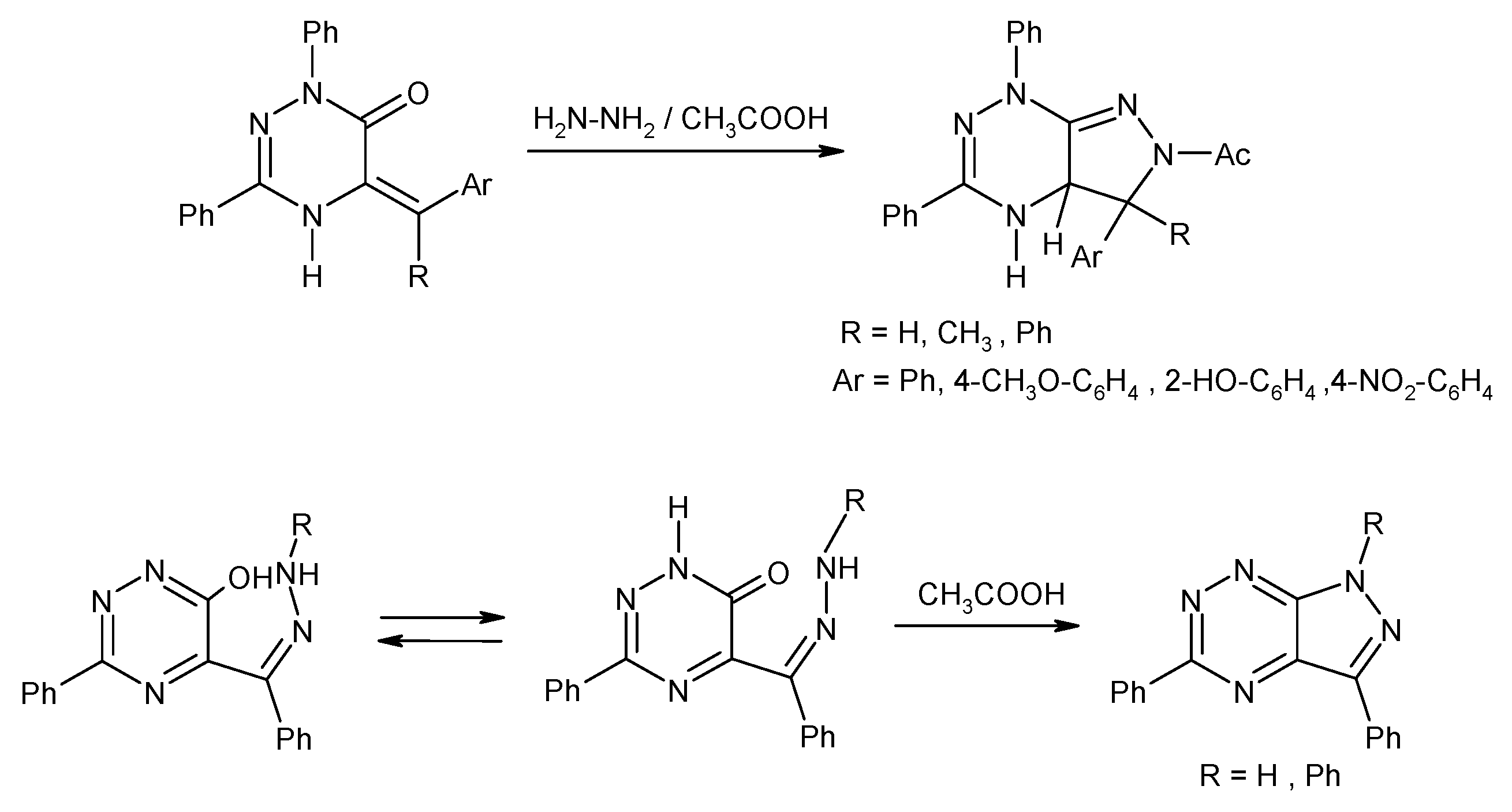
2.1. Synthesis from 4-Amino-5-acylhydrazinopyrazole
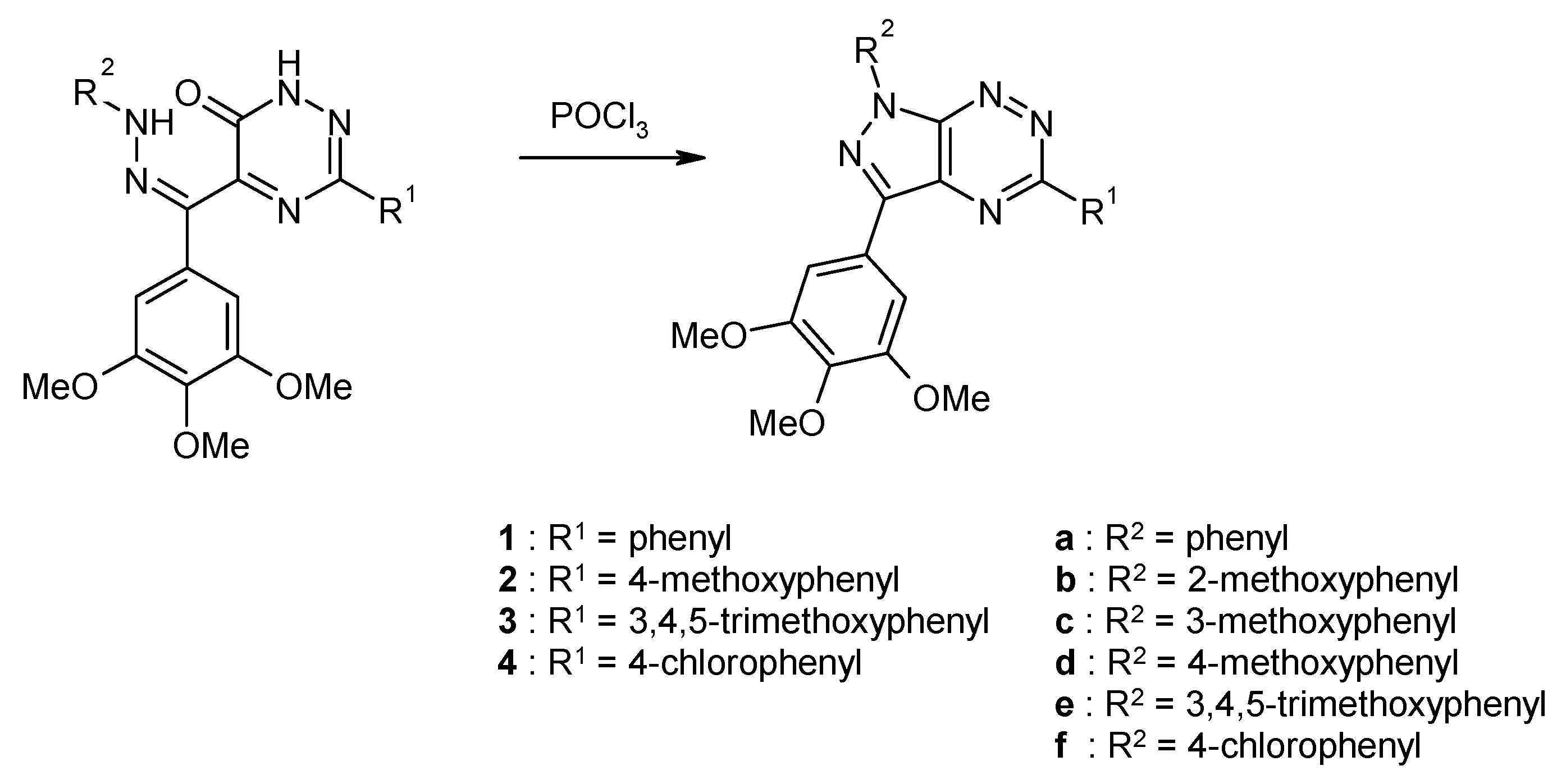
2.2. Synthesis from 3,5-Disubstituted-1,2,4-triazin-6(1H)-one with Cytotoxic Activity
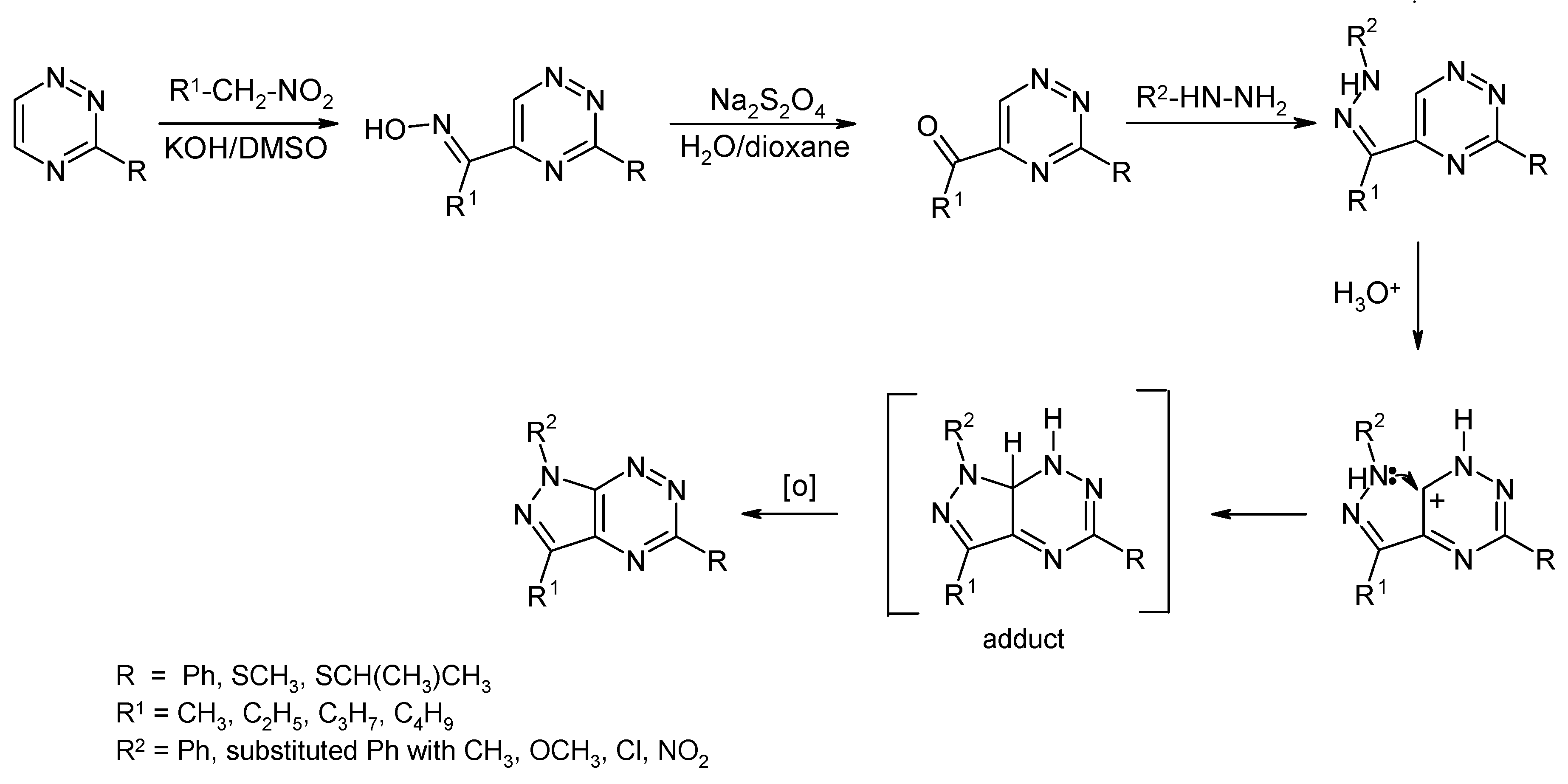
2.3. Synthesis from 3-Substituted-1,2,4-triazine with Cytotoxic Activity
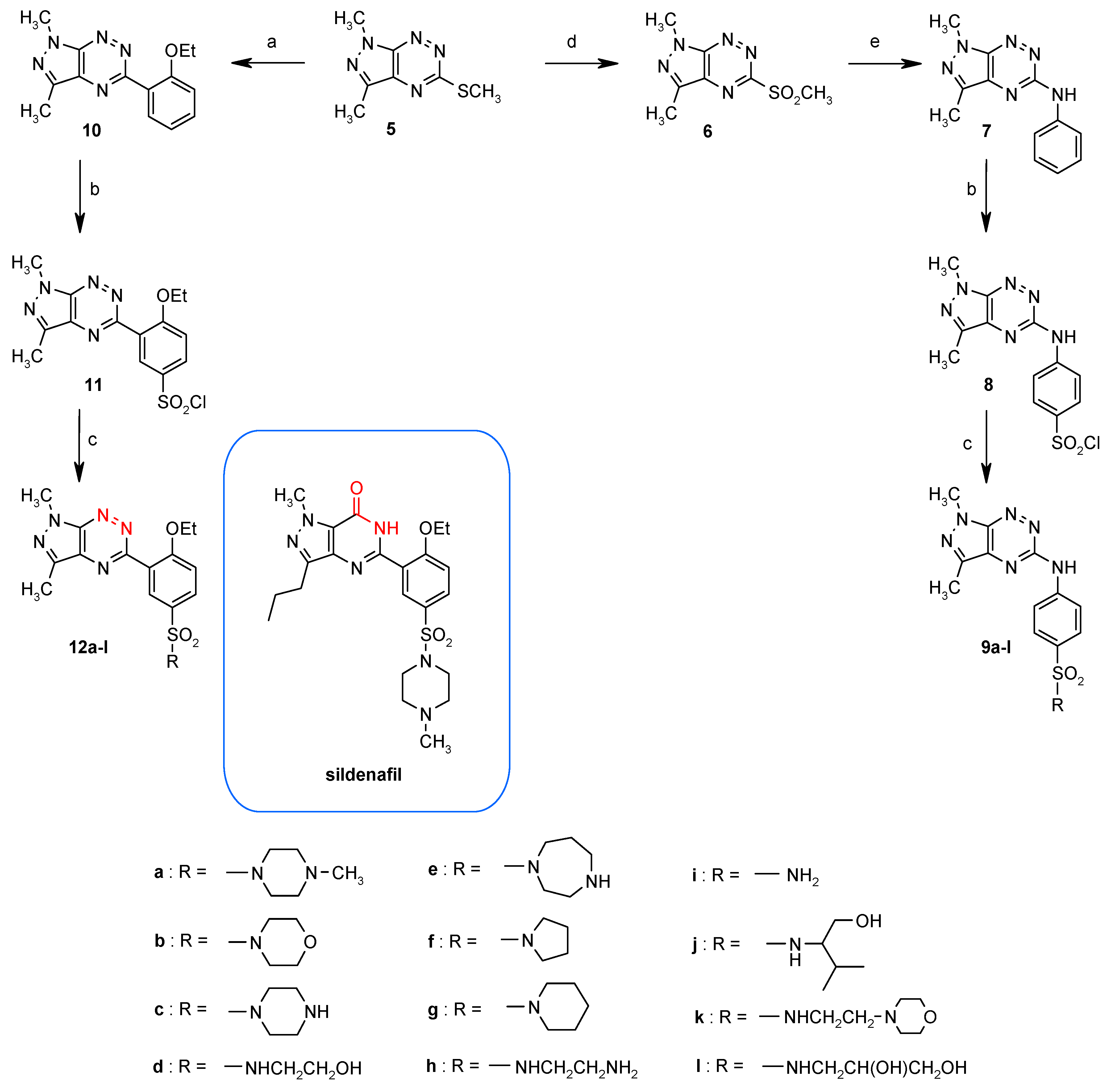
3. Synthesis and Anticancer Activity of Pyrazolo[4,3-e][1,2,4]triazine Sulfonamides
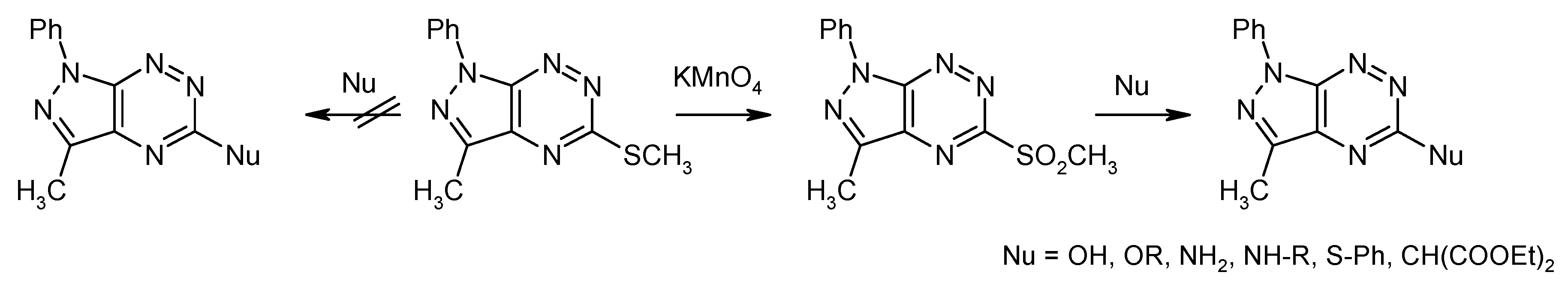
3.1. Synthesis from 5-Methylsulfanylpyrazolo[4,3-e][1,2,4]triazine
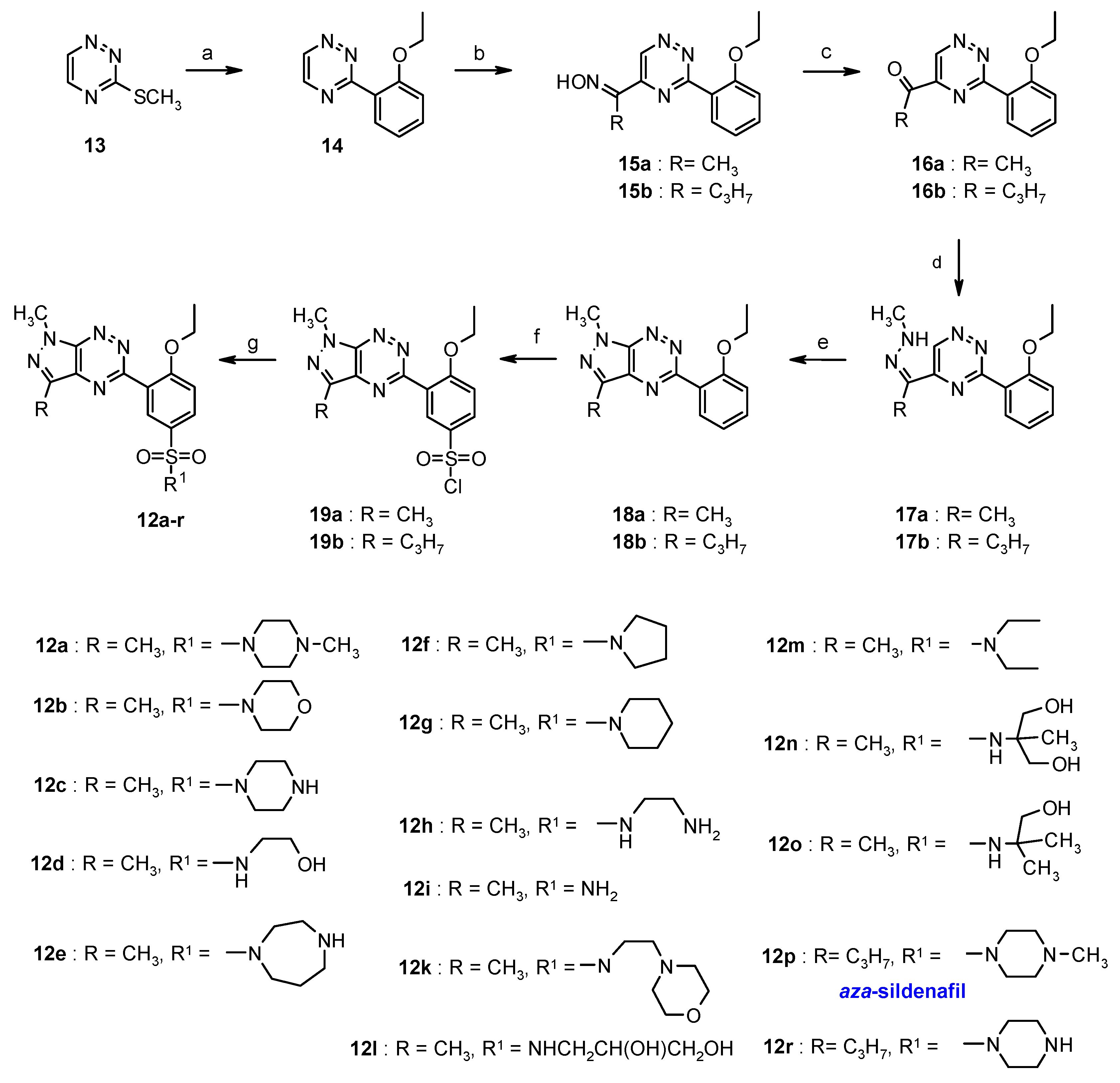
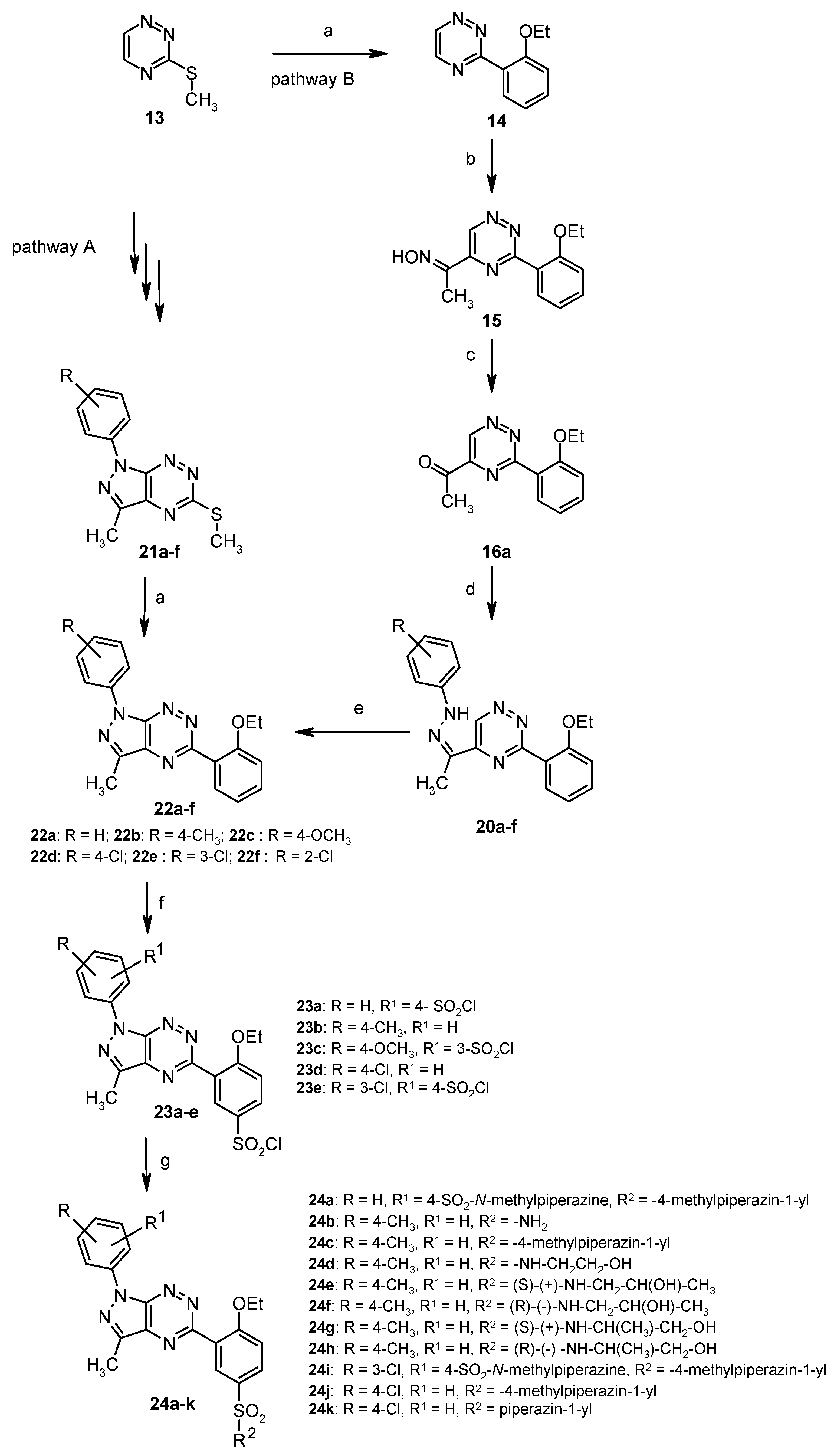
3.2. Synthesis from 3-Methylsulfanyl-1,2,4-triazine
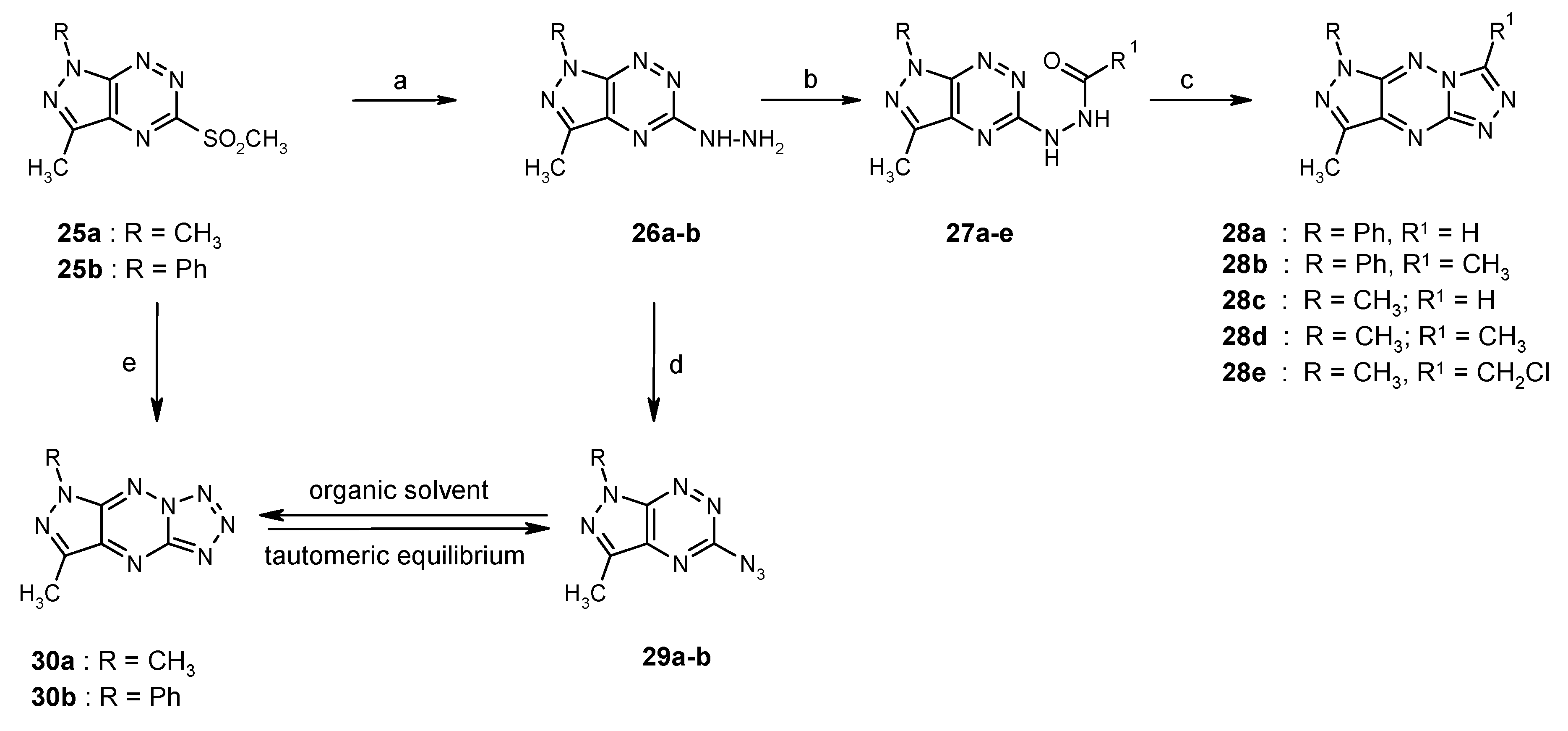
4. Synthesis and Anticancer Activity of Annulated Pyrazolo[4,3-e][1,2,4]triazines: Pyrazolo[4,3-e][1,2,4]triazolo[4,3-b][1,2,4]triazines and Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazines
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PPA | polyphosphoric acid |
| A549 | lung adenocarcinoma cells |
| CEM | T-lymphoblastic leukemia cells |
| CEM DNR Bulk | T-lymphoblastic leukemia daunorubicin resistant cells |
| K562 | myeloid leukemia cells |
| K562 tax | myeloid leukemia, paclitaxel resistant cells |
| SAR | structure–activity relationship |
| SNAr | reaction of nucleophillic aromatic substitution |
| PC-3 | prostate cancer cells |
| MCF-7 | estrogen receptor positive breast cancer cells |
| H460 | non-small-cell lung cancer |
| Colo205 | colorectal adenocarcinoma cells |
| CDK | cyclin dependent kinase |
| CA, EC 4.2.1.1 | carbonic anhydrase |
| RAF kinase | rapidly accelerated fibrosarcoma |
| CA IX | carbonic anhydrase IX |
| CA XII | carbonic anhydrase XII |
| CDK2 | cyclin-dependent kinase 2 |
| MDA-MB-231 | estrogen receptor negative breast cancer cells |
| BV173 | leukemia cells |
| HL60 | leukemia cells |
| CCRF-CEM | leukemia cells |
| KI | inhibitory constant |
| hCA IX | human carbonic anhydrase IX |
| hCA XII | human carbonic anhydrase XII |
| LN229 | glioblastoma tumor cells |
| LS180 | colon adenocarcinoma cells |
References
- Lindner, H.J.; Schaden, G. Pyrazolo(4.3-e)as-triazine, a new heterocyclic system from Pseudomonas fluorescens var. Pseudoidinum. Chem. Ber. 1972, 105, 1949–1955. [Google Scholar] [CrossRef]
- Hirata, K.; Nakagami, H.; Takashina, J.; Mahmud, T.; Kobayashi, M.; In, Y.; Ishida, T.; Miyamoto, K. Novel violet pigment, nostocine A, an extracellular metabolite from Cyanobacterium Nostoc spongiaeforme. Heterocycles 1996, 43, 1513–1519. [Google Scholar]
- Smirnov, V.V.; Kiprianova, E.A.; Garagulya, A.D.; Esipov, S.E.; Dovjenko, S.A. Fluviols, bicyclic nitrogen-rich antibiotics produced by Pseudomonas fluorescens. Fems Microbiol. Lett. 1997, 153, 357–361. [Google Scholar] [CrossRef]
- Kelly, T.R.; Elliott, E.L.; Lebedev, R.; Pagalday, J. Synthesis of the pyrazolo[4,3-e][1,2,4]triazine family of natural products: nostocine A, fluviol A, and pseudoiodinine. J. Am. Chem. Soc. 2006, 128, 5646–5647. [Google Scholar] [CrossRef] [PubMed]
- Badr, M.Z.A.; Aly, M.M.; Khalil, Z.H.; Attalla, A.A. ChemInform Abstract: Synthesis and Reactions of 1,3-Diphenyl-As-Triazin-6-One. Indian J. Chem. Sect. B 1982, 21B, 115. [Google Scholar] [CrossRef]
- Nálepa, K.; Slouka, J. Cyclisierungsreaktionen von Hydrazonen. XVI: Eine einfache Synthese einiger Pyrazolo[4,3-e]-1,2,4-triazinen. Pharmazie 1984, 39, 504–505. [Google Scholar]
- Nálepa, K.; Gucký, T. Synthesis of some new pyrazolo[4,3-e]1,2,4-triazines. Acta Univ. Palacki Olomuc Fac Rer. Nat. Chem. 2001, 49. [Google Scholar]
- Rykowski, A.; Mojzych, M.; Karczmarzyk, Z. A new synthesis of pyrazolo[4,3-e][l,2,4]triazines via acid promoted ring closure of the phenylhydrazones of 5-acyl-l,2,4-triazines. Heterocycles 2000, 53, 2175–2181. [Google Scholar] [CrossRef]
- Mojzych, M.; Rykowski, A. Thermal transformations of phenylhydrazones of 5-acyl-1,2,4-triazines towards pyrazolo[4,3-e][1,2,4]triazine and 4-cyanopyrazole derivative under acidic and no acidic conditions. J. Heterocycl. Chem. 2007, 44, 1003–1007. [Google Scholar] [CrossRef]
- Youssef, M.S.K.; Hassan, K.M.; Atta, F.M.; Abbady, M.S. Synthesis of 1H-pyrazolo[3,4-e]-s-triazolo[3,4-c]-as-triazines, 8H-pyrazolo[3,4-e]tetrazolo[5,1-c]-as-triazine and 1,7-dihydro-8H-pyrazolo[3,4-e]-as-triazine derivatives. J Heterocycl. Chem. 1984, 21, 1565–1568. [Google Scholar] [CrossRef]
- Abu Safieh, A.K.; Abu Mahthieh, A.M.; El-Abadelah, M.M.; Ayoub, M.T.; Voelter, W. Synthesis of 5-substituted 1,3-dimethylpyrazolo[4,3-e][1,2,4]triazines. Monatsh. Chem. 2007, 138, 157–160. [Google Scholar] [CrossRef]
- Gucký, T.; Frysová, I.; Slouka, J.; Hajdúch, M.; Dzubák, P. Cyclocondensation reaction of heterocyclic carbonyl compounds. Part XIII: Synthesis and cytotoxic activity of some 3,7-diaryl-5-(3,4,5-trimethoxyphenyl)pyrazolo[4,3-e][1,2,4]triazines. Eur. J. Med. Chem. 2009, 44, 891–900. [Google Scholar]
- Rykowski, A.; Mąkosza, M. Reaction of 1,2,4-triazines with nitronate anions, direct nucleophilic acylation of 1,2,4-triazines. Tetrahedron Lett. 1984, 25, 4795–4796. [Google Scholar] [CrossRef]
- Rykowski, A.; Lipińska, T. A concise route to a key intermediate in the total synthesis of sempervirine. Synth. Commun. 1996, 26, 4409–4414. [Google Scholar] [CrossRef]
- Mojzych, M.; Rykowski, A. Direct synthesis of pyrazolo[4,3-e][1,2,4]triazine derivatives from oximes of 5-acyl- and 5-formyl-1,2,4-triazines. Heterocycl. Commun. 2006, 12, 191–194. [Google Scholar] [CrossRef]
- Mojzych, M.; Rykowski, A. One-step synthesis and regioselective alkylation of substituted 1H-pyrazolo[4,3-e][l,2,4]triazine. Pol. J. Chem. 2003, 77, 1797–1803. [Google Scholar]
- Shepherd, R.G.; Federic, J.L. Reactivity of azine, benzoazine, and azinoazine derivatives with simple nucleophiles. In Advances in Heterocyclic Chemistry, 1st ed.; Katritzky, A.R., Boulton, A.J., Lagowski, J.M., Eds.; Academic Press Inc.: London, UK, 1965; Volume 4, pp. 145–423. [Google Scholar]
- Oae, S.; Furukawa, N. Heteroaromatic sulfoxides and sulfones: Ligand exchange and coupling in sulfuranes and ipso-substitutions. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press Limited: London, UK, 1990; Volume 48, pp. 1–63. [Google Scholar]
- Mojzych, M.; Rykowski, A. Synthesis of functionalized 1H-pyrazolo[4,3-e][l,2,4]-triazines and their fused derivatives via ipso-substitution of methylsulfonyl group with O-, N-, S- and C-nucleophiles. Heterocycles 2004, 63, 1829–1838. [Google Scholar]
- Mojzych, M.; Rykowski, A. Synthesis of aza-analog of N-methylated acycloformycine A. Heterocycles 2007, 71, 2449–2456. [Google Scholar] [CrossRef]
- Mojzych, M. Cytotoxic activity of some pyrazolo[4,3-e][1,2,4]triazines against human cancer cell lines. J Chem. Soc. Pak. 2011, 33, 123–128. [Google Scholar]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev. 2003, 5, 535–558. [Google Scholar] [CrossRef]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and antiviral sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, A.; Engel, J. Pharmaceutical Substances, Syntheses, Patents, Applications, 3rd ed.; Thieme: Stuttgart, Germany, 1999. [Google Scholar]
- Byth, K.F.; Culshaw, J.D.; Green, S.; Oakes, S.E.; Thomas, A.P. Imidazo[1,2-b]pyridazines: A potent and selective class of cyclin-dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 2249–2252. [Google Scholar] [CrossRef]
- Shah, S.S.A.; Rivera, G.; Ashfaq, M. Recent advances in medicinal chemistry of sulfonamides. Rational design as anti-tumoral, anti-bacterial and anti-inflammatory agents. Mini Rev. Med. Chem. 2013, 13, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Catalytic and inhibition mechanisms, distribution and physiological roles. In Carbonic Anhydrase: Its Inhibitors and Activators; Supuran, C.T., Scozzafava, A., Conway, J., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 1–23. [Google Scholar]
- Supuran, C.T.; Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg. Med. Chem. 2007, 15, 4336–4350. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Enzyme Structure and Function. In xPharm: The Comprehensive Pharmacology Reference; Byland, D.B., Enna, S.J., Eds.; Elsevier Science Inc.: New York, NY, USA, 2007. [Google Scholar]
- Roskoski, R., Jr. An anticancer protein-tyrosine kinase inhibitor. Biochem. Biophys. Res. Commun. 2003, 303, 709–717. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Kryštof, V.; Uldrijan, S. Cyclin-dependent kinase inhibitors as anticancer drugs. Curr. Drug Targets 2010, 11, 291–302. [Google Scholar] [CrossRef]
- Pastorek, J.; Pastoreková, S. Cancer-related carbonic anhydrase Isozymes and their Inhibition. In Carbonic anhydrase: Its inhibitors and activators; Supuran, C.T., Scozzafava, A., Conway, J., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 255–281. [Google Scholar]
- Supuran, C.T. Carbonic anhydrases as drug targets-an overview. Curr Top Med Chem. 2007, 7, 825–833. [Google Scholar] [CrossRef]
- Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic anhydrase inhibitors and activators and their use in therapy. Expert Opin. Pat. 2006, 16, 1627–1664. [Google Scholar] [CrossRef]
- Pastoreková, S.; Kopacek, J.; Pastorek, J. Carbonic anhydrase inhibitors and the management of cancer. Curr. Top. Med. Chem. 2007, 7, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastoreková, S.; Callebaut, I.; Mornon, J.-P.; Zelník, V.; Opavský, R.; Zat’Ovicová, M.; Liao, S.; Portetelle, D.; Stanbridge, E.J. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994, 9, 2788–2888. [Google Scholar]
- Opavský, R.; Pastoreková, S.; Zelník, V.; Gibadulinová, A.; Stanbridge, E.J.; Závada, J.; Kettmann, R.; Pastorek, J. Human MN/CA9 Gene, a Novel Member of the Carbonic Anhydrase Family: Structure and Exon to Protein Domain Relationships. Genomics 1996, 33, 480–487. [Google Scholar] [CrossRef]
- Türeci, Ö.; Şahin, U.; Vollmar, E.; Siemer, S.; Göttert, E.; Seitz, G.; Parkkila, A.-K.; Shah, G.N.; Grubb, J.H.; Pfreundschuh, M.; et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 7608–7613. [Google Scholar] [CrossRef]
- Winum, J.-Y.; Scozzafava, A.; Montero, J.-L.; Supuran, C.T. Inhibition of carbonic anhydrase IX: A new strategy against cancer. Anti-Cancer Agents Med. Chem. 2009, 9, 693–702. [Google Scholar] [CrossRef]
- Winum, J.-Y.; Rami, M.; Scozzafava, A.; Montero, J.-L.; Supuran, C.T. Carbonic anhydrase IX: A new druggable target for the design of antitumor agents. Med. Res. Rev. 2008, 28, 445–463. [Google Scholar] [CrossRef]
- Mojzych, M.; Šubertová, V.; Bielawska, A.; Bielawski, K.; Bazgier, V.; Berka, K.; Gucký, T.; Fornal, E.; Kryštof, V. Synthesis and kinase inhibitory activity of new sulfonamide derivatives of pyrazolo[4,3-e][1,2,4]triazines. Eur. J. Med. Chem. 2014, 78, 217–224. [Google Scholar] [CrossRef]
- Mojzych, M.; Bielawska, A.; Bielawski, K.; Ceruso, M.; Supuran, C.T. Pyrazolo[4,3-e][1,2,4]triazine sulfonamides as carbonic anhydrase inhibitors with antitumor activity. Bioorganic Med. Chem. 2014, 22, 2643–2647. [Google Scholar] [CrossRef]
- Mojzych, M.; Karczmarzyk, Z.; Wysocki, W.; Ceruso, M.; Supuran, C.T.; Kryštof, V.; Urbańczyk-Lipkowska, Z.; Kalicki, P. New approaches to the synthesis of sildenafil analogues and their enzyme inhibitory activity. Bioorganic Med. Chem. 2015, 23, 1421–1429. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; Fendrich, G.; Floersheimer, A.; Furet, P.; Liebetanz, J.; Rummel, G.; Rheinberger, P.; Centeleghe, M.; Fabbro, D.; Manley, P.W. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta. Cryst. Sect. D Boil. Cryst. 2006, 63, 80–93. [Google Scholar] [CrossRef]
- Mojzych, M.; Ceruso, M.; Bielawska, A.; Bielawski, K.; Fornal, E.; Supuran, C.T. New pyrazolo[4,3-e][1,2,4]triazine sulfonamides as carbonic anhydrase inhibitors. Bioorg. Med. Chem. 2015, 23, 3674–3680. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, M.; Karczmarzyk, Z.; Wysocki, W.; Urbańczyk-Lipkowska, Z.; Żaczek, N. Valence tautomerism of new pyrazolo[4,3-e]tetrazole[4,5-b][1,2,4]triazines. J. Mol. Struct. 2014, 1067, 147–153. [Google Scholar] [CrossRef]
- Mojzych, M.; Tarasiuk, P.; Karczmarzyk, Z.; Juszczak, M.; Rzeski, W.; Fruziński, A.; Woźny, A. Synthesis, Structure and Antiproliferative Activity of New pyrazolo[4,3-e]triazolo[4,5-b][1,2,4]triazine Derivatives. Med. Chem. 2018, 14, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Cmoch, P. Multinuclear magnetic resonance study of the structure and tautomerism of azide and iminophosphorane derivatives of chloropyridazines. Magn. Reson. Chem. 2002, 40, 507–516. [Google Scholar] [CrossRef]
- Karczmarzyk, Z.; Mojzych, M.; Rykowski, A. Synthesis and structure of a novel mesomeric betaine 6,7-dimethyl-2H-pyrazolo[4,3-e]tetrazolo[4,5-b][1,2,4]triazine. J. Mol. Struct. 2007, 829, 22–28. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
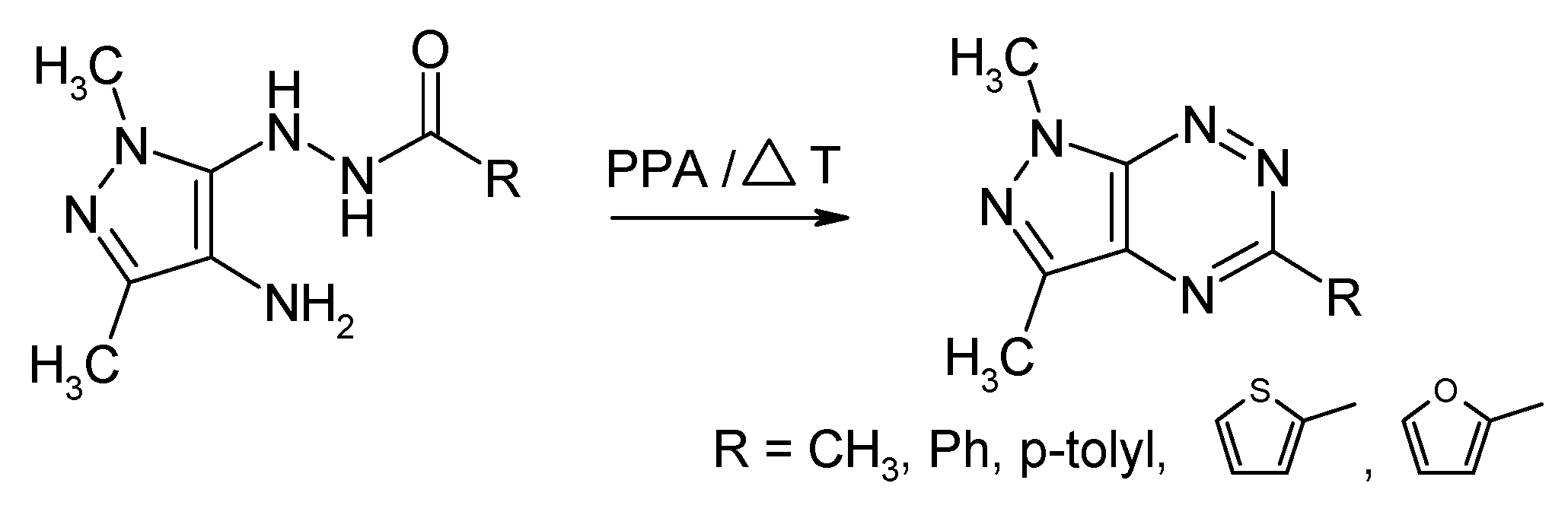{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Compd. | R1 | R2 | MTT Assay, IC50 (µM) 1 | ||||
| CEM | CEM DNR Bulk | K562 | K562 Tax | A549 | |||
| 1b | Ph | 2-OCH3-Ph | 109 | 118 | 182 | 94.3 | 5.41 |
| 1c | Ph | 3-OCH3-Ph | 8.41 | 99.3 | 117 | 159 | 0.61 |
| 1d | Ph | 4-OCH3-Ph | 117 | 100 | 84.4 | 150 | 39.0 |
| 1e | Ph | 3,4,5-(OCH3)3-Ph | 65.2 | 129 | 52.6 | 99.9 | 2.16 |
| 2b | 4-OCH3-Ph | 2-OCH3-Ph | 105 | 119 | 178 | 155 | 6.58 |
| 2c | 4-OCH3-Ph | 3-OCH3-Ph | 54.5 | 184 | 115 | 174 | 124 |
| 3b | 3,4,5-(OCH3)3-Ph | 2-OCH3-Ph | 126 | 125 | 182 | 156 | 15.0 |
| 3c | 3,4,5-(OCH3)3-Ph | 3-OCH3-Ph | 48.3 | 109 | 164 | 141 | 163 |
| 3d | 3,4,5-(OCH3)3-Ph | 4-OCH3-Ph | 36.9 | 118 | 66.2 | 161 | 75.6 |
| 3f | 3,4,5-(OCH3)3-Ph | 4-Cl-Ph | 82.2 | 115 | 158 | 196 | 8.26 |
| 4a | 4-Cl-Ph | Ph | 11.6 | 113 | 220 | 225 | 130 |
| 4b | 4-Cl-Ph | 2-OCH3-Ph | 53.8 | 35.7 | 48.7 | 20.7 | 3.62 |
| 4c | 4-Cl-Ph | 3-OCH3-Ph | 54.7 | 108 | 192 | 219 | 2.86 |
| 4e | 4-Cl-Ph | 3,4,5-(OCH3)3-Ph | 99.9 | 117 | 192 | 210 | 15.0 |
| 4f | 4-Cl-Ph | 4-Cl-Ph | 68.9 | 126 | 190 | 222 | 155 |
 | ||||||
| MTT Assay, IC50 (µM) | ||||||
| R | R1 | R2 | PC-3 | MCF-7 | H460 | Colo205 |
| SCH3 | C2H5 | 4-NO2-Ph | 98 | 78 | 36 | 75 |
| Ph | CH3 | Ph | 98 | NA 1 | NA 1 | NA 1 |
| SO2CH3 | CH3 | Ph | 25 | 50 | 25 | 25 |
| NH-Ph | CH3 | Ph | 81 | 90 | 86 | 4 |
| NH-Bu | CH3 | Ph | 66 | 77 | NA 1 | 25 |
| NH-CH2-Ph | CH3 | Ph | NA 1 | NA 1 | NA 1 | 50 |
| NH2 | CH3 | Ph | NA1 | NA 1 | NA 1 | 72 |
| OCH3 | CH3 | Ph | NA1 | NA 1 | NA 1 | 91 |
 | |||||
| Compd. | R | MTT Assay, IC50 (µM) | |||
| K562 | BV173 | HL60 | CCRF-CEM | ||
| 9a |  | 66 ± 5 | 40 ± 8 | 49 ± 2 | 36 ± 2 |
| 9b |  | 90 ± 8 | 58 ± 5 | 39 ± 1 | 69 ± 8 |
| 9c |  | 27 ± 4 | 22 ± 6 | 55 ± 2 | 20 ± 2 |
| 9d |  | 100 ± 4 | 41 ± 10 | 42 ± 5 | 49 ± 8 |
| 9e |  | 21 ± 5 | 22 ± 4 | 38 ± 1 | 36 ± 12 |
| 9f |  | 102 ± 1 | 47 ± 14 | 56± 6 | 30 ± 2 |
| 9g |  | 98 ± 2 | 36 ± 9 | 24 ± 2 | 30 ± 2 |
| 9h |  | 77 ± 7 | 39 ± 8 | 42 ± 6 | 56 ± 2 |
| 9i |  | 106 ± 8 | 45 ± 11 | 58 ± 3 | 50 ± 1 |
| 9j |  | >200 | 58 ± 9 | 40 ± 2 | 54 ± 8 |
| 9k |  | 96 ± 3 | 39 ± 8 | 41 ± 1 | 64 ± 6 |
| 9l |  | 101 ± 2 | 42 ± 9 | 44 ± 5 | 57 ± 3 |
| Chlorambucil | 84 ± 6 | 34 ± 8 | 38 ± 2 | 21 ± 8 | |
| Imatinib | 13 ± 2 | 20 ± 6 | 55 ± 7 | 45 ± 1 | |
 | |||||
| Compd. | R | MTT Assay, IC50 (µM) | [3H]Thymidine Incorporation, IC50 (µM) | ||
| MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | ||
| 9a |  | 102 ± 2 | 99 ± 2 | 87 ± 2 | 80 ± 2 |
| 9b |  | >200 | >200 | >200 | >200 |
| 9c |  | 150 ± 2 | 130 ± 2 | 170 ± 2 | 103 ± 2 |
| 9d |  | >200 | >200 | >200 | >200 |
| 9e |  | 140 ± 3 | 155 ± 2 | 123 ± 1 | 150 ± 2 |
| 9f |  | >200 | >200 | >200 | >200 |
| 9g |  | 200 ± 2 | 140 ± 1 | 150 ± 2 | 135 ± 1 |
| 9h |  | 126 ± 1 | 120 ± 1 | 85 ± 1 | 90 ± 1 |
| 9i |  | >200 | >200 | nt 1 | nt 1 |
| 9j |  | >200 | >200 | nt 1 | nt 1 |
| 9k |  | 146 ± 1 | 125 ± 2 | 99 ± 1 | 120 ± 2 |
| 9l |  | 200 ± 2 | 140 ± 2 | nt 1 | nt 1 |
| Chlorambucil | 97 ± 2 | 93 ± 2 | 56 ± 2 | 49 ± 2 | |
| Compd. | MTT Assay, IC50 (µM) | [3H]Thymidine Incorporation, IC50 (µM) | ||
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | |
| 12a | 110 ± 2 | 115 ± 2 | 140 ± 1 | 152 ± 2 |
| 12b | 181 ± 2 | 189 ± 2 | 148 ± 1 | 160 ± 1 |
| 12c | 172 ± 2 | 182 ± 2 | 150 ± 2 | 164 ± 2 |
| 12d | >200 | >200 | >200 | >200 |
| 12e | 123 ± 2 | 130 ± 1 | 129 ± 2 | 115 ± 1 |
| 12f | 112 ± 2 | 120 ± 2 | 130 ± 2 | 159 ± 1 |
| 12g | 105 ± 3 | 98 ± 2 | 112 ± 1 | 126 ± 2 |
| 12h | 190 ± 1 | >200 | >200 | 200 ± 1 |
| 12i | nt 1 | nt 1 | nt 1 | nt 1 |
| 12j | nt 1 | nt 1 | nt 1 | nt 1 |
| 12k | 132 ± 2 | 136 ± 2 | 145 ± 1 | 156 ± 2 |
| 12l | nt 1 | nt 1 | nt 1 | nt 1 |
| Chlorambucil | 97 ± 2 | 93 ± 2 | 56 ± 2 | 49 ± 2 |
| Compd. | IC50 (µM) | |
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| 9a | 47 ± 1 | 58 ± 2 |
| 9b | >200 | >200 |
| 9c | 175 ± 1 | 180 ± 2 |
| 9d | >200 | >200 |
| 9e | 133 ± 1 | 135 ± 2 |
| 9f | >200 | >200 |
| 9g | 163 ± 2 | 145 ± 1 |
| 9h | 112 ± 1 | 95 ± 1 |
| 9i | nt 1 | nt 1 |
| 9j | nt 1 | nt 1 |
| 9k | 140 ± 1 | 137 ± 2 |
| Chlorambucil | 52 ± 1 | 72 ± 2 |
| Compd. | KI (nM) | |
|---|---|---|
| hCA IX | hCA XII | |
| 5 | 89 | 6.6 |
| 6 | 82 | 9.0 |
| 9a | >50,000 | 5.3 |
| 9b | nt 1 | nt 1 |
| 9c | >50,000 | 5.7 |
| 9d | 26.5 | 7.5 |
| 9e | >50,000 | 5.9 |
| 9f | 652 | 8.1 |
| 9g | >50,000 | 6.2 |
| 9h | 23.7 | 7.1 |
| 9i | 43.8 | 7.9 |
| 9j | nt 1 | nt 1 |
| 9k | 824 | 6.2 |
| Acetazolamide | 25 | 5.8 |
 | |||||||
| Compd. | R1 | R2 | R3 | MTT Assay, IC50 (µM) | [3H]Thymidine Incorporation, IC50 (µM) | ||
| MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | ||||
| 24a | H | 4-SO2 | 4-methylpiperazin-1-yl | >200 | >200 | >200 | >200 |
| 24b | 4-CH3 | H | NH2 | 154 ± 2 | 171 ± 2 | 155 ± 2 | 148 ± 2 |
| 24c | 4-CH3 | H | 4-methylpiperazin-1-yl | >200 | >200 | >200 | >200 |
| 24d | 4-CH3 | H | NHCH2CH2OH | 126 ± 2 | 147 ± 2 | 132 ± 2 | 136 ± 2 |
| 24e | 4-CH3 | H | (S)-(+)-NHCH2CH(OH)CH3 | >200 | >200 | >200 | >200 |
| 24f | 4-CH3 | H | (R)-(+)-NHCH2CH(OH)CH3 | >200 | >200 | >200 | >200 |
| 24g | 4-CH3 | H | (S)-(+)-NHCH(CH3)CH2OH | >200 | >200 | >200 | >200 |
| 24h | 4-CH3 | H | (R)-(+)-NHCH(CH3)CH2OH | >200 | >200 | >200 | >200 |
| 24i | 3-Cl | 4-SO2-N-methylpiperazine | 4-methylpiperazin-1-yl | 160 ± 2 | 185 ± 2 | 173 ± 2 | 169 ± 2 |
| 24j | 4-Cl | H | 4-methylpiperazin-1-yl | >200 | >200 | >200 | >200 |
| 24k | 4-Cl | H | piperazin-1-yl | >200 | >200 | >200 | >200 |
| Chlorambucil | 97 ± 2 | 93 ± 2 | 56 ± 2 | 49 ± 2 | |||
| Compd. | KI (nM) | |
|---|---|---|
| hCA IX | hCA XII | |
| 24a | 29.6 | 536 |
| 24b | 28.7 | 77.5 |
| 24c | 25.4 | 78.1 |
| 24d | 24.5 | 91.5 |
| 24e | 13.8 | 82.8 |
| 24f | 35.6 | 77.8 |
| 24g | 27.7 | 77.6 |
| 24h | 26.6 | 70.3 |
| 24i | 417 | 79.4 |
| 24j | 403 | 81.5 |
| 24k | 42.3 | 93.1 |
| Acetazolamide | 25 | 5.8 |
| Compd. | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| LS180 | Colo205 | H460 | MCF-7 | PC-3 | LN229 | |
| 30b | 0.131 | <0.400 | <0.400 | 0.500 | <0.400 | 0.270 |
| Compd. | IC50 (µM) | |
| A549 | LS180 | |
| 26 | 17.4 | >25 |
| 27c | >25 | >25 |
| 28a | 2.1 | 5.6 |
| 28b | 9.4 | 37.6 |
| 28c | 2.0 | 2.4 |
| cisplatin | 3.4 | - |
| 5-fluorouracil | - | 19.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernat, Z.; Szymanowska, A.; Kciuk, M.; Kotwica-Mojzych, K.; Mojzych, M. Review of the Synthesis and Anticancer Properties of Pyrazolo[4,3-e][1,2,4]triazine Derivatives. Molecules 2020, 25, 3948. https://doi.org/10.3390/molecules25173948
Bernat Z, Szymanowska A, Kciuk M, Kotwica-Mojzych K, Mojzych M. Review of the Synthesis and Anticancer Properties of Pyrazolo[4,3-e][1,2,4]triazine Derivatives. Molecules. 2020; 25(17):3948. https://doi.org/10.3390/molecules25173948
Chicago/Turabian StyleBernat, Zofia, Anna Szymanowska, Mateusz Kciuk, Katarzyna Kotwica-Mojzych, and Mariusz Mojzych. 2020. "Review of the Synthesis and Anticancer Properties of Pyrazolo[4,3-e][1,2,4]triazine Derivatives" Molecules 25, no. 17: 3948. https://doi.org/10.3390/molecules25173948
APA StyleBernat, Z., Szymanowska, A., Kciuk, M., Kotwica-Mojzych, K., & Mojzych, M. (2020). Review of the Synthesis and Anticancer Properties of Pyrazolo[4,3-e][1,2,4]triazine Derivatives. Molecules, 25(17), 3948. https://doi.org/10.3390/molecules25173948