
The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations




Abstract
{kind=link}
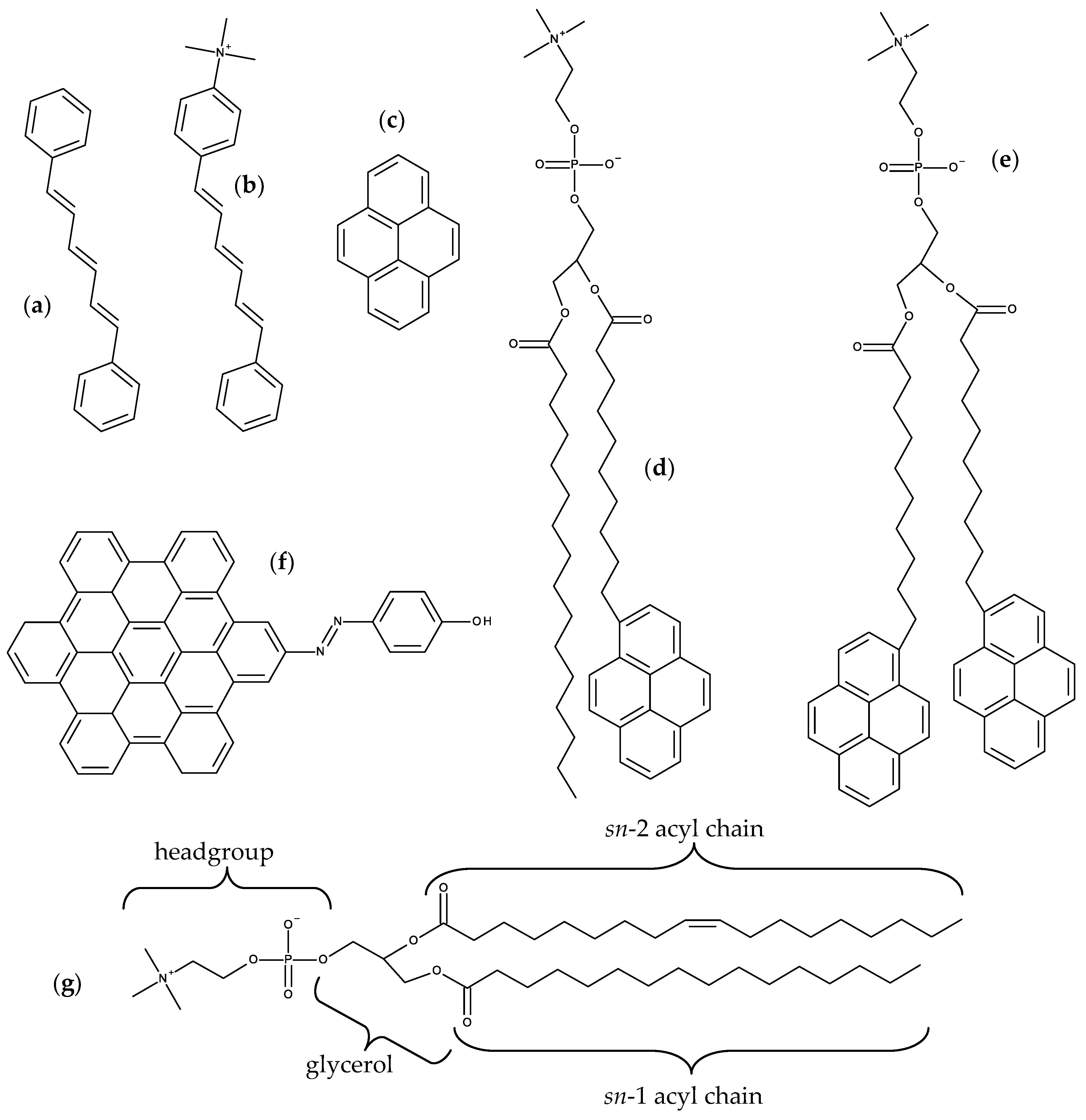{kind=link}
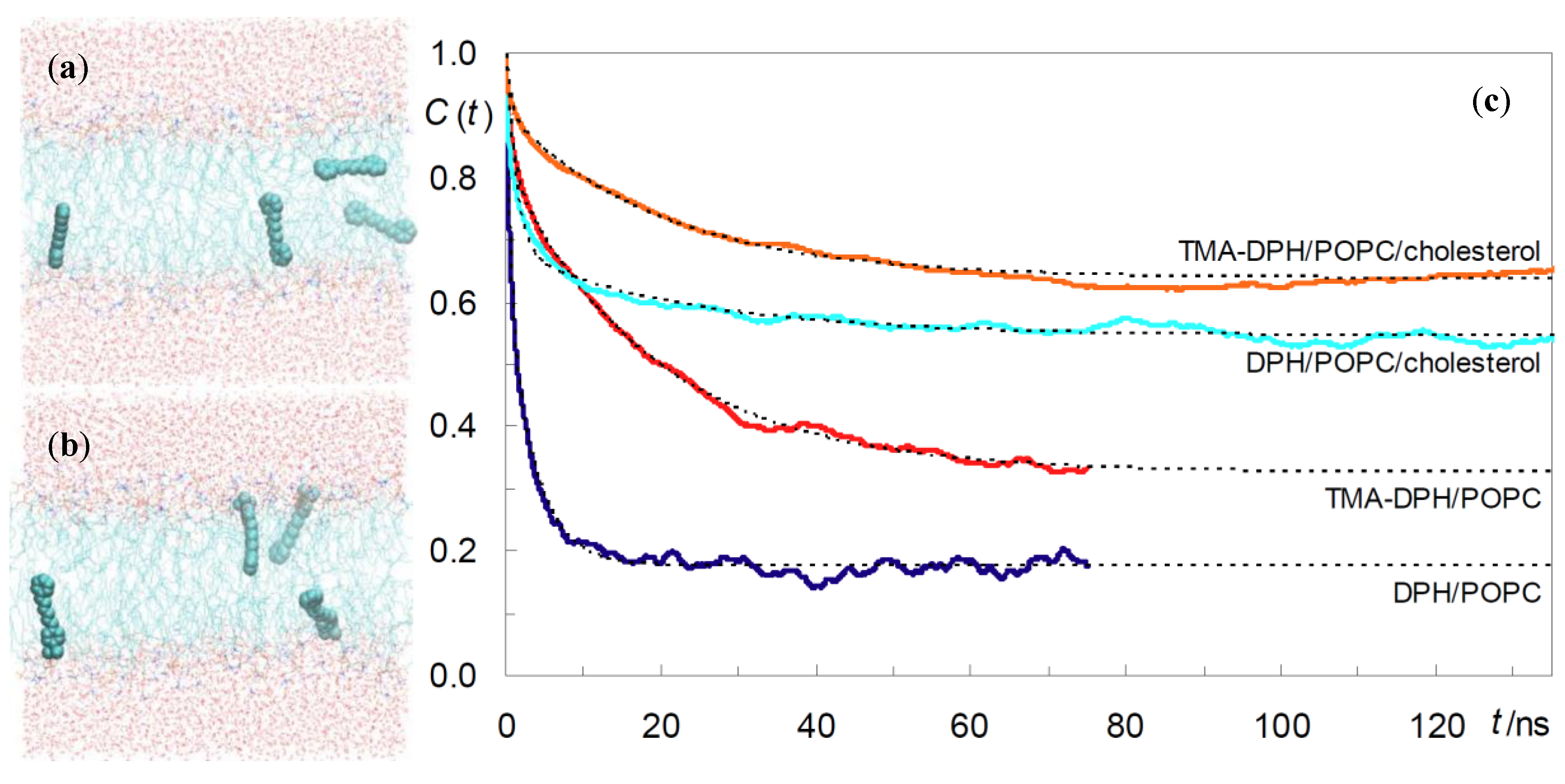{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Aromatic Hydrocarbon Fluorophores
2.1. DPH and TMA-DPH
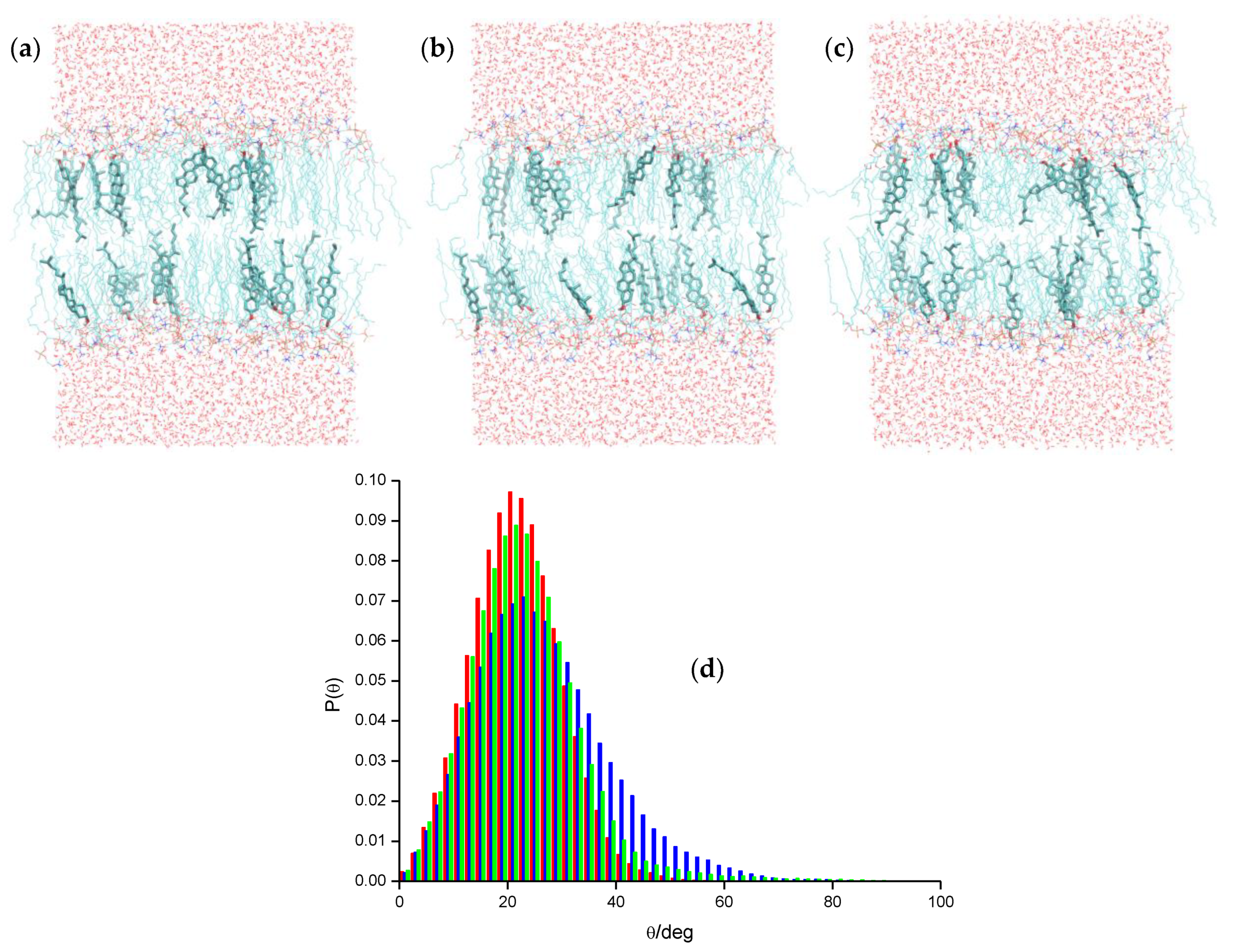
2.2. Pyrene and Its Derivatives
2.3. Hexabenzocoronene Photoswitchable Probes
3. Charge-Transfer-Based and Other Solvatochromic Probes
3.1. NBD Probes
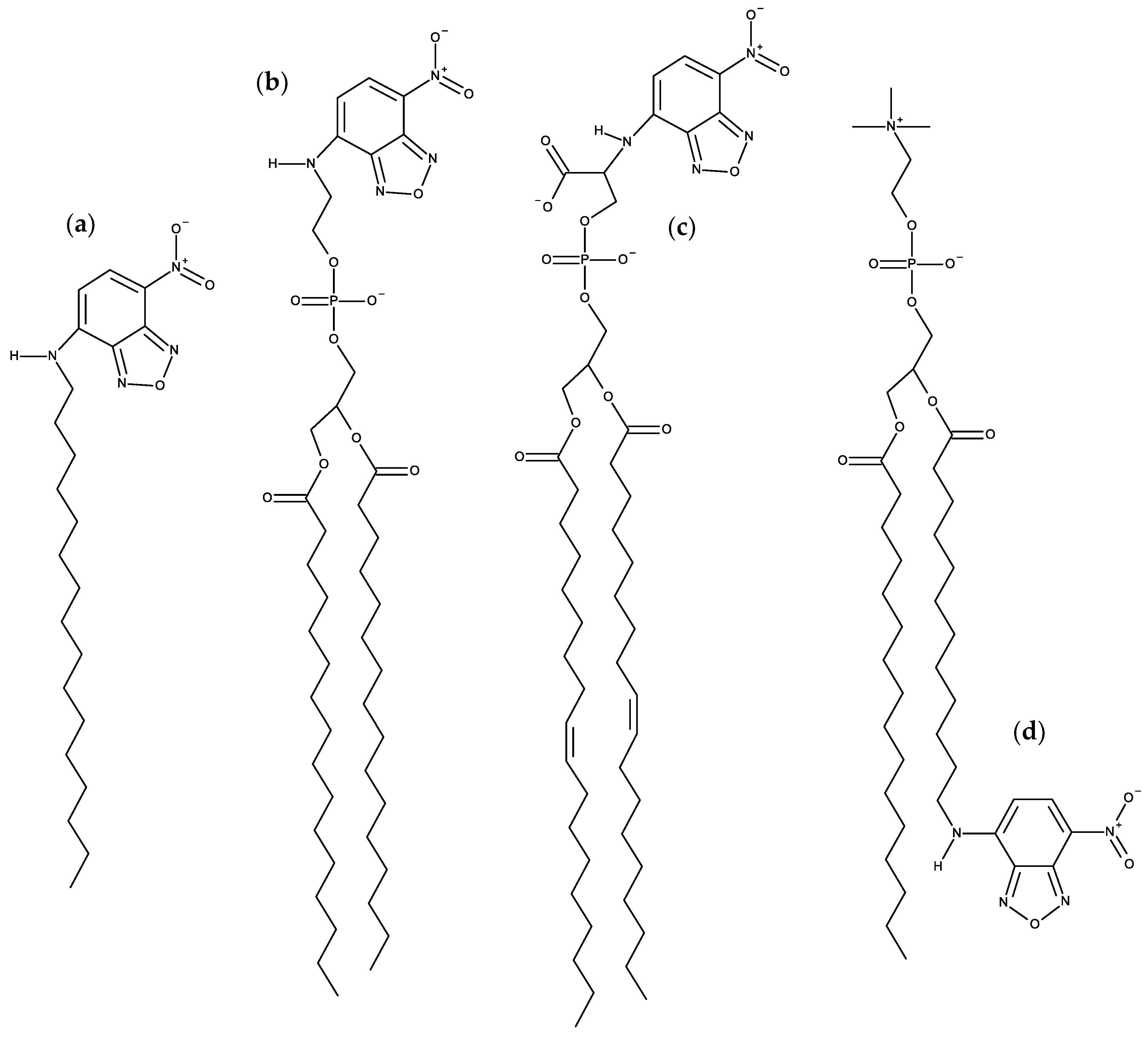
3.1.1. NBD Fatty Amines (NBD-Cn)
3.1.2. NBD Phospholipid Probes
3.2. Prodan and Laurdan
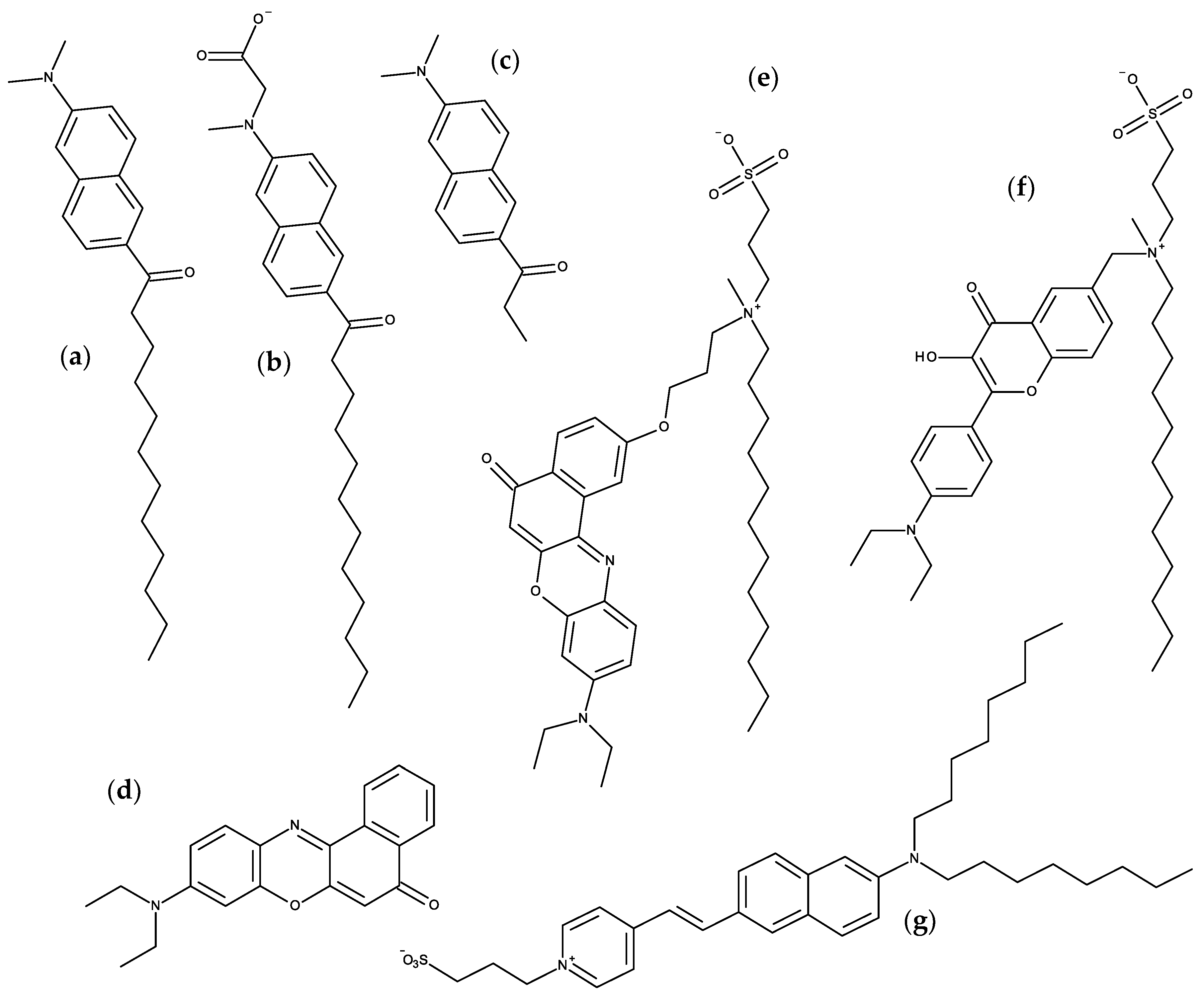
3.3. Nile Red
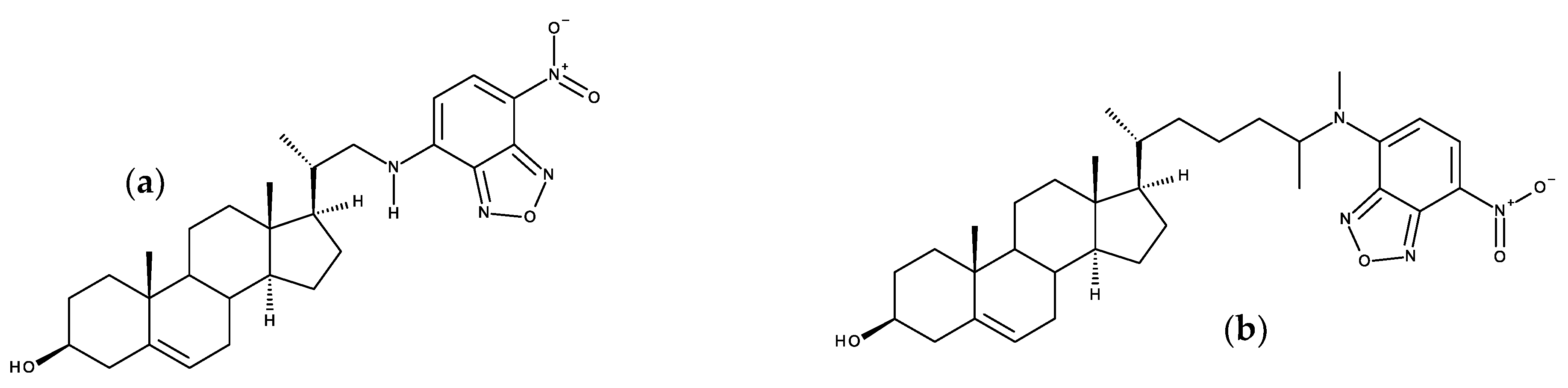
3.4. Voltage and pH-Sensitive Probes
3.5. Other Solvatochromic Probes
4. BODIPY Probes
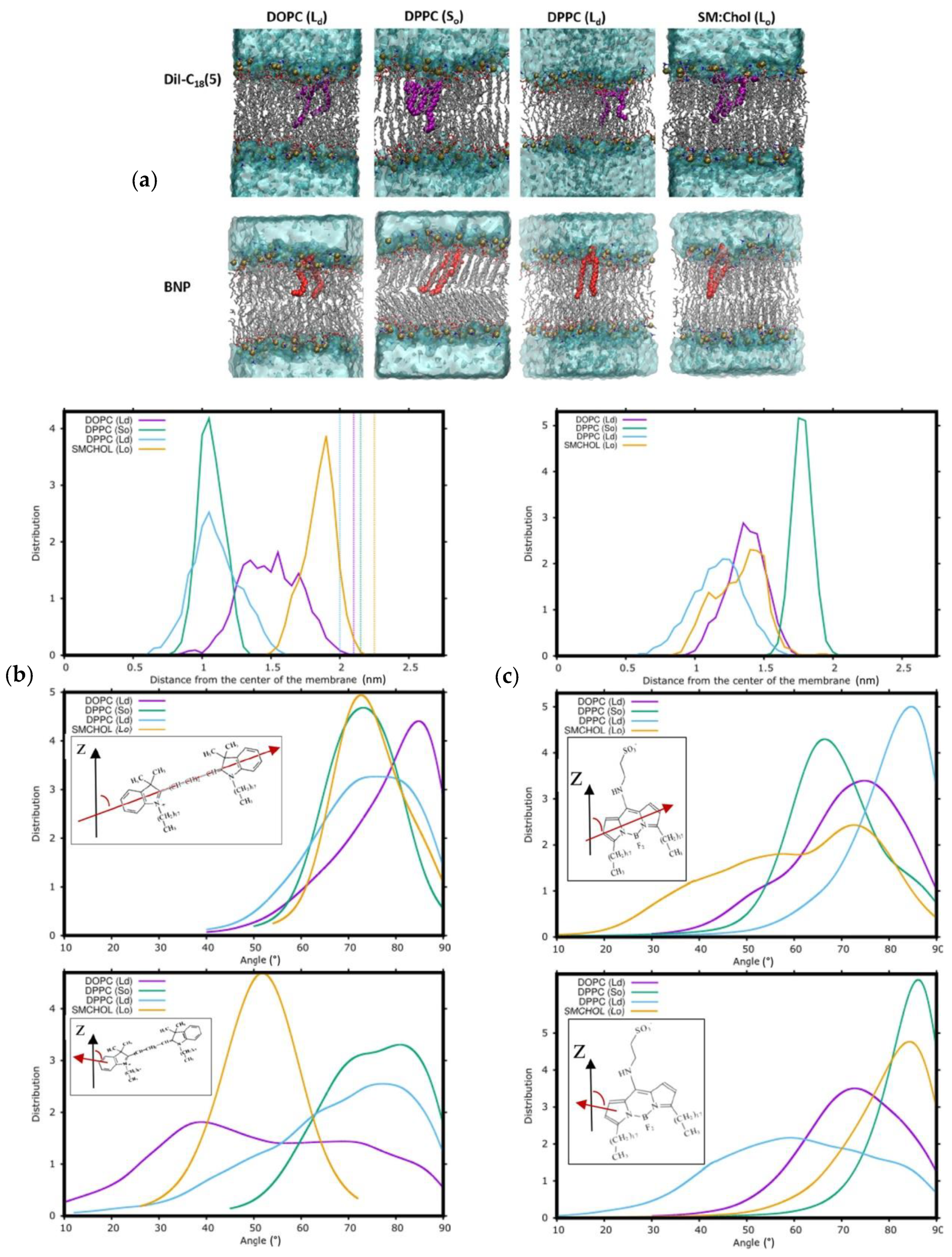
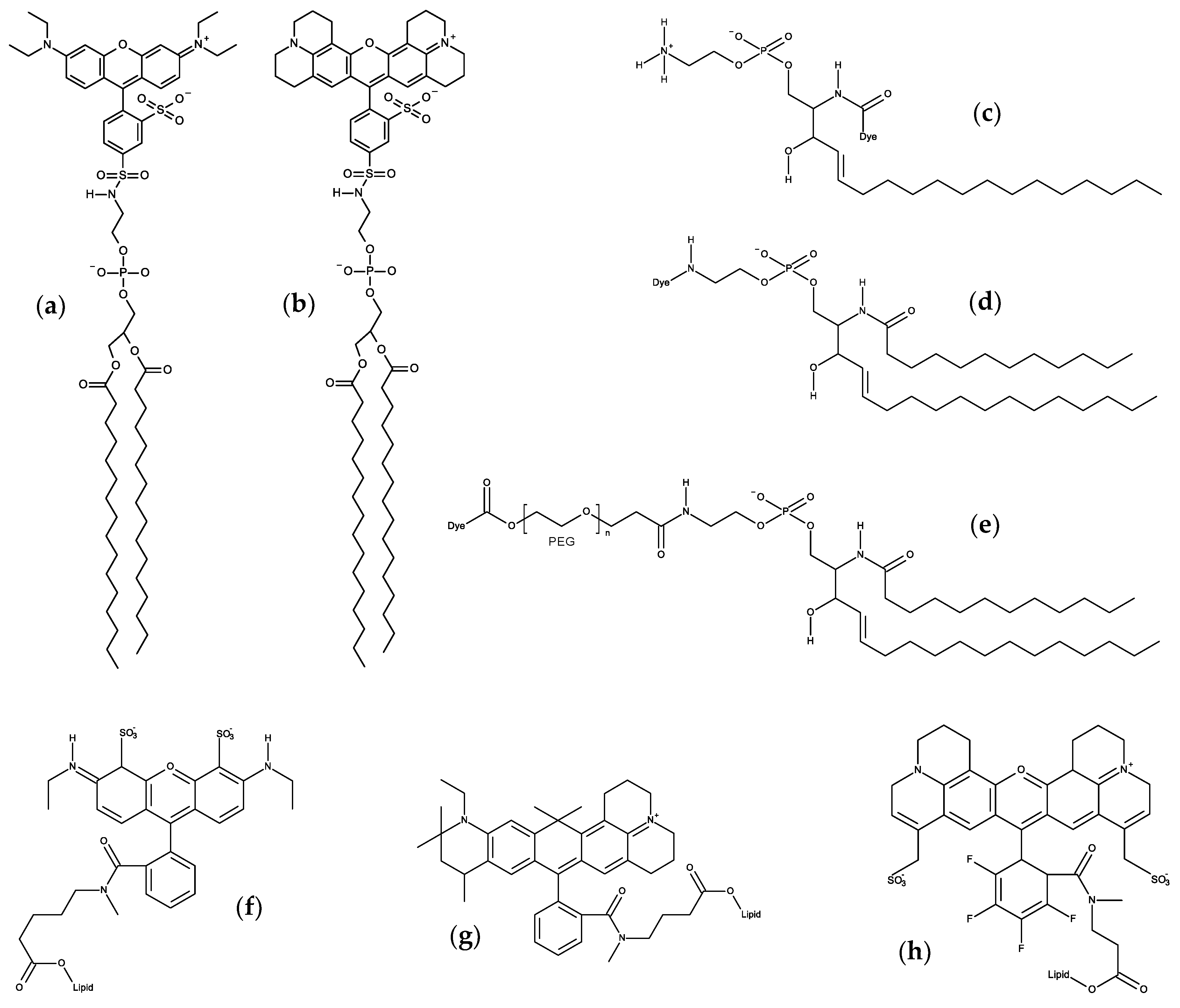
5. Carbocyanine Probes
6. Xanthene Probes
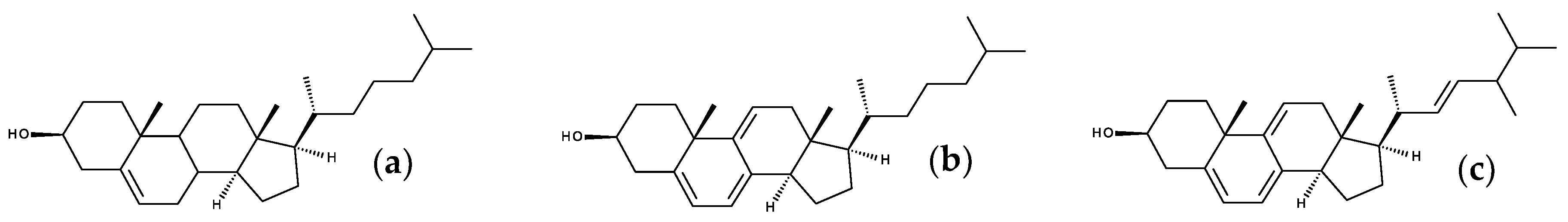
7. Fluorescent Sterols
7.1. Intriniscally Fluorescent Sterols
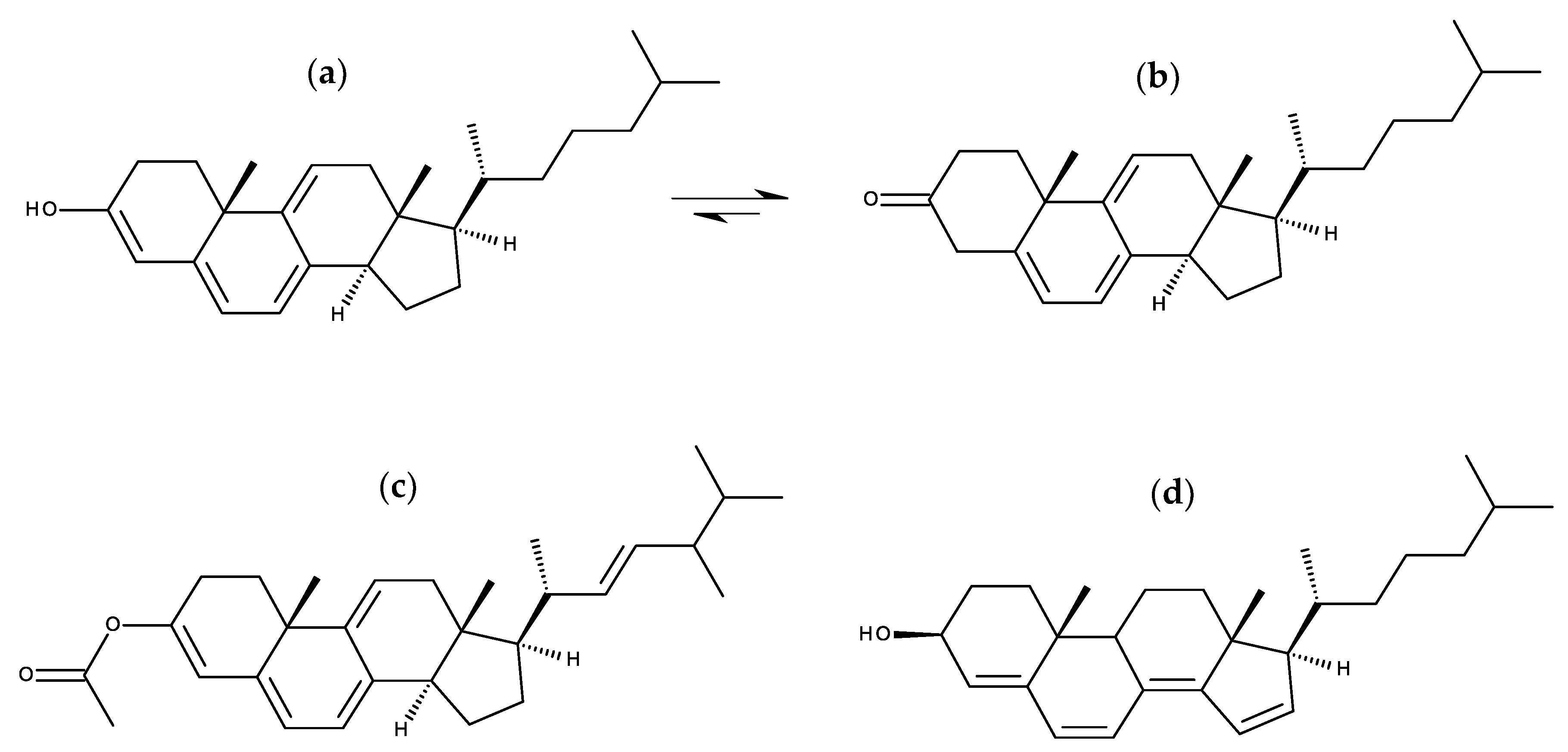
7.1.1. Cholestatrienol and Dehydroergosterol
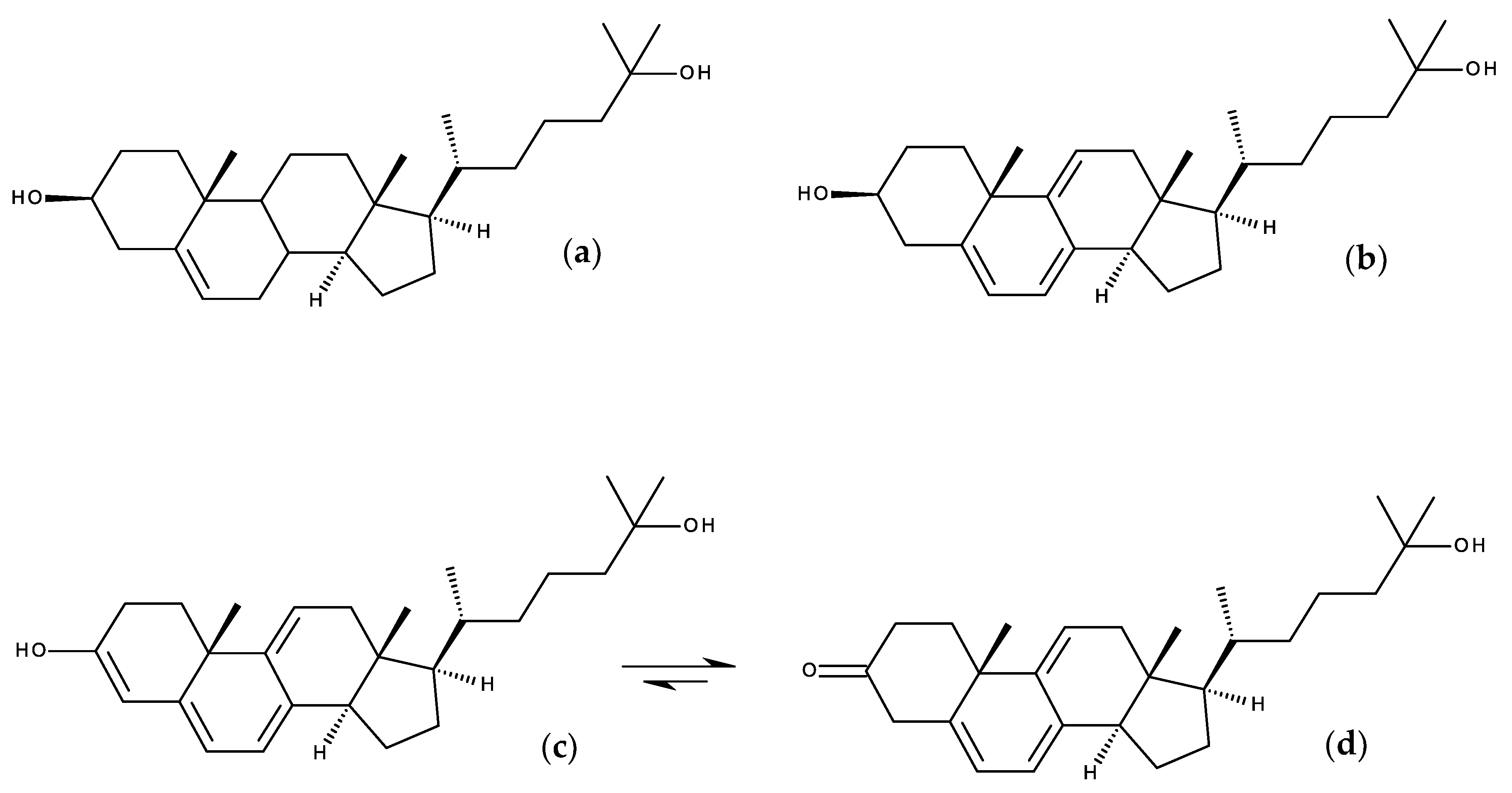
7.1.2. Design Probes with Extended Double Bond Conjugation in the Ring System
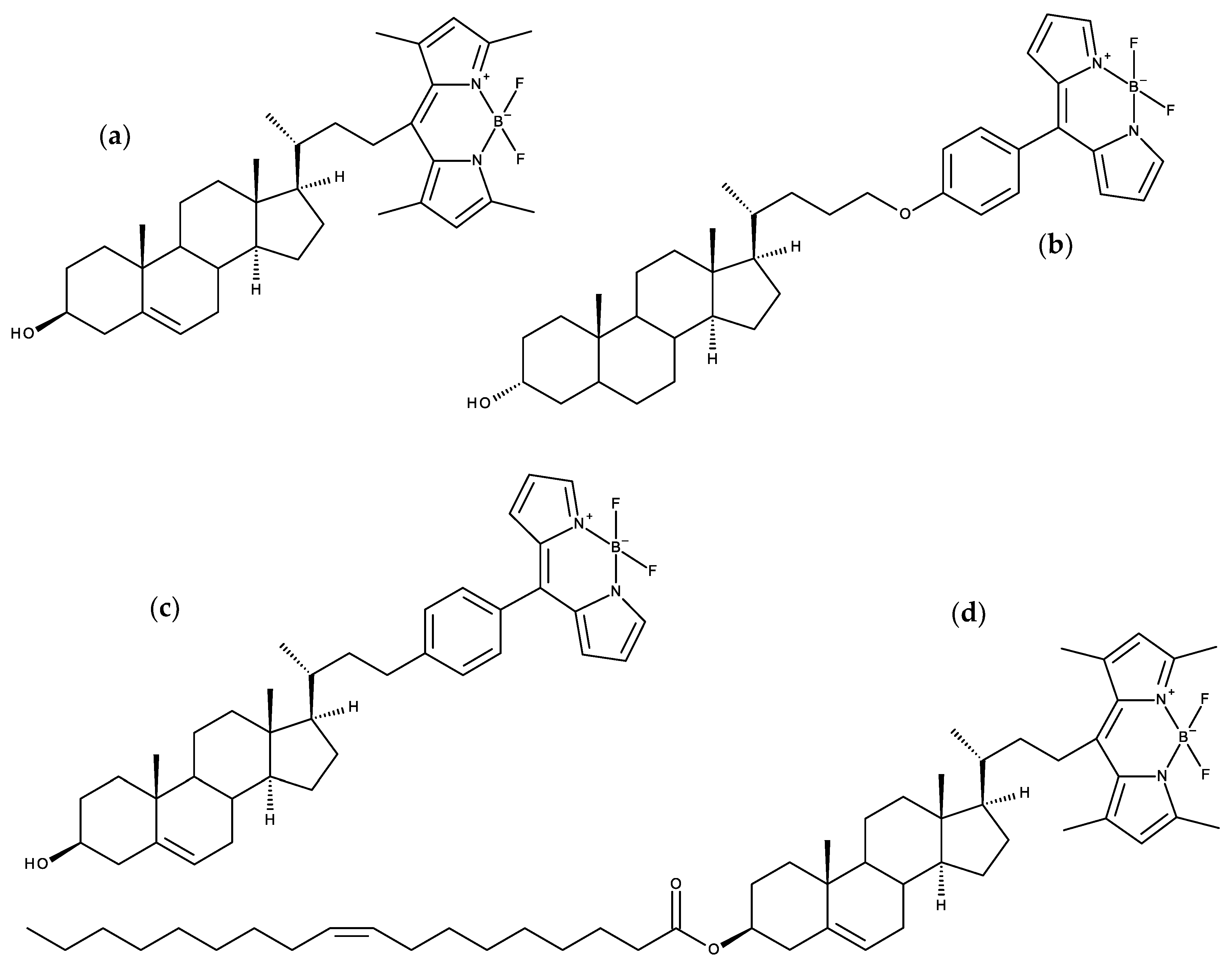
7.2. Side-Chain Labeled Sterols
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Waggoner, A.S.; Stryer, L. Fluorescent probes of biological membranes. Proc. Natl. Acad. Sci. USA 1970, 67, 579–589. [Google Scholar] [CrossRef]
- Radda, G.K. Fluorescent probes in membrane studies. In Biophysical Approaches; Korn, E.D., Ed.; Springer: Boston, MA, USA, 1975; pp. 97–188. ISBN 978-1-4684-2907-7. [Google Scholar]
- Demchenko, A.P.; Mély, Y.; Duportail, G.; Klymchenko, A.S. Monitoring biophysical properties of lipid membranes by environment-sensitive fluorescent probes. Biophys. J. 2009, 96, 3461–3470. [Google Scholar] [CrossRef]
- Demchenko, A.P.; Duportail, G.; Oncul, S.; Klymchenko, A.S.; Mély, Y. Introduction to fluorescence probing of biological membranes. Methods Mol. Biol. 2015, 1232, 19–43. [Google Scholar] [CrossRef]
- Kyrychenko, A. Using fluorescence for studies of biological membranes: A review. Methods Appl. Fluoresc. 2015, 3, 042003. [Google Scholar] [CrossRef] [PubMed]
- Klymchenko, A.S. Solvatochromic and fluorogenic dyes as environment-sensitive probes: Design and biological applications. Acc. Chem. Res. 2017, 50, 366–375. [Google Scholar] [CrossRef]
- Loura, L.M.S.; de Almeida, R.F.M.; Silva, L.C.; Prieto, M. FRET analysis of domain formation and properties in complex membrane systems. Biochim. Biophys. Acta Biomembr. 2009, 1788, 209–224. [Google Scholar] [CrossRef]
- Lesslauer, W.; Cain, J.E.; Blasie, J.K. X-ray diffraction studies of lecithin bimolecular leaflets with incorporated fluorescent probes. Proc. Natl. Acad. Sci. USA 1972, 69, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, A.L.; Lyubartsev, A.P. Computer simulation of lipid membranes: Methodology and achievements. Polym. Sci. Ser. C 2013, 55, 162–180. [Google Scholar] [CrossRef]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale simulations of biological membranes: The challenge to understand biological phenomena in a living substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.C.; Filipe, H.A.L.; Reis, R.L.; Prates Ramalho, J.P.; Coreta-Gomes, F.; Moreno, M.J.; Loura, L.M.S. Interaction of bile salts with lipid bilayers: An atomistic molecular dynamics study. Front. Physiol. 2019, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Loura, L.M.S.; Prates Ramalho, J.P. Fluorescent membrane probes’ behavior in lipid bilayers: Insights from molecular dynamics simulations. Biophys. Rev. 2009, 1, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Loura, L.M.S.; Ramalho, J.P.P. Recent developments in molecular dynamics simulations of fluorescent membrane probes. Molecules 2011, 16, 5437–5452. [Google Scholar] [CrossRef]
- Faller, R. Molecular modeling of lipid probes and their influence on the membrane. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2353–2361. [Google Scholar] [CrossRef]
- Repáková, J.; Čapková, P.; Holopainen, J.M.; Vattulainen, I. Distribution, orientation, and dynamics of DPH probes in DPPC bilayer. J. Phys. Chem. B 2004, 108, 13438–13448. [Google Scholar] [CrossRef]
- Repáková, J.; Holopainen, J.M.; Karttunen, M.; Vattulainen, I. Influence of pyrene-labeling on fluid lipid membranes. J. Phys. Chem. B 2006, 110, 15403–15410. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and validation of the automated topology builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput. Aided. Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Javanainen, M.; Salvador, A.; Galvão, A.M.; Vattulainen, I.; Loura, L.M.S.; Moreno, M.J. Quantitative assessment of methods used to obtain rate constants from molecular dynamics simulations–Translocation of cholesterol across lipid bilayers. J. Chem. Theory Comput. 2018, 14, 3840–3848. [Google Scholar] [CrossRef] [PubMed]
- Juračka, J.; Šrejber, M.; Melíková, M.; Bazgier, V.; Berka, K. MolMeDB: Molecules on membranes database. Database 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Lomize, A.L.; Hage, J.M.; Schnitzer, K.; Golobokov, K.; Lafaive, M.B.; Forsyth, A.C.; Pogozheva, I.D. PerMM: A web tool and database for analysis of passive membrane permeability and translocation pathways of bioactive molecules. J. Chem. Inf. Model. 2019, 59, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Lentz, B.R. Membrane “fluidity” as detected by diphenylhexatriene probes. Chem. Phys. Lipids 1989, 50, 171–190. [Google Scholar] [CrossRef]
- Mitchell, D.C.; Litman, B.J. Molecular order and dynamics in bilayers consisting of highly polyunsaturated phospholipids. Biophys. J. 1998, 74, 879–891. [Google Scholar] [CrossRef]
- López Cascales, J.J.; Huertas, M.L.; García De La Torre, J. Molecular dynamics simulation of a dye molecule in the interior of a bilayer: 1,6-diphenyl-1,3,5-hexatriene in dipalmitoylphosphatidylcholine. Biophys. Chem. 1997, 69, 1–8. [Google Scholar] [CrossRef]
- Repáková, J.; Holopainen, J.M.; Morrow, M.R.; McDonald, M.C.; Čapková, P.; Vattulainen, I. Influence of DPH on the structure and dynamics of a DPPC bilayer. Biophys. J. 2005, 88, 3398–3410. [Google Scholar] [CrossRef]
- Fraňová, M.; Repáková, J.; Čapková, P.; Holopainen, J.M.; Vattulainen, L. Effects of DPH on DPPC-cholesterol membranes with varying concentrations of cholesterol: From local perturbations to limitations in fluorescence anisotropy experiments. J. Phys. Chem. B 2010, 114, 2704–2711. [Google Scholar] [CrossRef]
- Hurjui, I.; Neamtu, A.; Dorohoi, D.O. The interaction of fluorescent DPH probes with unsaturated phospholipid membranes: A molecular dynamics study. J. Mol. Struct. 2013, 1044, 134–139. [Google Scholar] [CrossRef]
- Do Canto, A.M.T.M.; Robalo, J.R.; Santos, P.D.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Diphenylhexatriene membrane probes DPH and TMA-DPH: A comparative molecular dynamics simulation study. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2647–2661. [Google Scholar] [CrossRef] [PubMed]
- Paloncýová, M.; Ameloot, M.; Knippenberg, S. Orientational distribution of DPH in lipid membranes: A comparison of molecular dynamics calculations and experimental time-resolved anisotropy experiments. Phys. Chem. Chem. Phys. 2019, 21, 7594–7604. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, W.; Pottel, H.; Herreman, W.; Ameloot, M.; Hendrickx, H.; Schröder, H. Effect of orientational order on the decay of the fluorescence anisotropy in membrane suspensions. A new approximate solution of the rotational diffusion equation. Biophys. J. 1984, 46, 515–523. [Google Scholar] [CrossRef]
- Poojari, C.; Wilkosz, N.; Lira, R.B.; Dimova, R.; Jurkiewicz, P.; Petka, R.; Kepczynski, M.; Róg, T. Behavior of the DPH fluorescence probe in membranes perturbed by drugs. Chem. Phys. Lipids 2019, 223, 104784. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.C.; Neeley, S.K.; Melonakos, E.D.; Bell, J.D.; Busath, D.D. Fluorescence anisotropy of diphenylhexatriene and its cationic Trimethylamino derivative in liquid dipalmitoylphosphatidylcholine liposomes: Opposing responses to isoflurane. BMC Biophys. 2012, 5, 5. [Google Scholar] [CrossRef]
- Somerharju, P. Pyrene-labeled lipids as tools in membrane biophysics and cell biology. Chem. Phys. Lipids 2002, 116, 57–74. [Google Scholar] [CrossRef]
- Hoff, B.; Strandberg, E.; Ulrich, A.S.; Tieleman, D.P.; Posten, C. 2H-NMR study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in POPC bilayers. Biophys. J. 2005, 88, 1818–1827. [Google Scholar] [CrossRef]
- Čurdová, J.; Čapková, P.; Plášek, J.; Repáková, J.; Vattulainen, I. Free pyrene probes in gel and fluid membranes: Perspective through atomistic simulations. J. Phys. Chem. B 2007, 111, 3640–3650. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Do Canto, A.M.T.M.; Martins, J. Sensing hydration and behavior of pyrene in POPC and POPC/cholesterol bilayers: A molecular dynamics study. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1094–1101. [Google Scholar] [CrossRef]
- Arrais, D.; Martins, J. Bilayer polarity and its thermal dependency in the ℓo and ℓd phases of binary phosphatidylcholine/cholesterol mixtures. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2914–2922. [Google Scholar] [CrossRef][Green Version]
- Do Canto, A.M.T.M.; Santos, P.D.; Martins, J.; Loura, L.M.S. Behavior of pyrene as a polarity probe in palmitoylsphingomyelin and palmitoylsphingomyelin/cholesterol bilayers: A molecular dynamics simulation study. Colloids Surf. Physicochem. Eng. Asp. 2015, 480, 296–306. [Google Scholar] [CrossRef]
- Fraňová, M.D.; Repáková, J.; Holopainen, J.M.; Vattulainen, I. How to link pyrene to its host lipid to minimize the extent of membrane perturbations and to optimize pyrene dimer formation. Chem. Phys. Lipids 2014, 177, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.; Vaz, W.L.C.; Melo, E. Long-range diffusion coefficients in two-dimensional fluid media measured by the pyrene excimer reaction. J. Phys. Chem. 1996, 100, 1889–1895. [Google Scholar] [CrossRef]
- Templer, R.H.; Castle, S.J.; Curran, A.R.; Rumbles, G.; Klug, D.R. Sensing isothermal changes in the lateral pressure in model membranes using di-pyrenyl phosphatidylcholine. Faraday Discuss. 1999, 111, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Dékány Fraňová, M.; Vattulainen, I.; Samuli Ollila, O.H. Can pyrene probes be used to measure lateral pressure profiles of lipid membranes? Perspective through atomistic simulations. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1406–1411. [Google Scholar] [CrossRef]
- Osella, S.; Knippenberg, S. Triggering on/off states of photoswitchable probes in biological environments. J. Am. Chem. Soc. 2017, 139, 4418–4428. [Google Scholar] [CrossRef]
- Chattopadhyay, A. Chemistry and biology of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: Fluorescent probes of biological and model membranes. Chem. Phys. Lipids 1990, 53, 1–15. [Google Scholar] [CrossRef]
- Mukherjee, S.; Raghuraman, H.; Dasgupta, S.; Chattopadhyay, A. Organization and dynamics of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: A fluorescence approach. Chem. Phys. Lipids 2004, 127, 91–101. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Ramalho, J.P.P. Location and dynamics of acyl chain NBD-labeled phosphatidylcholine (NBD-PC) in DPPC bilayers. A molecular dynamics and time-resolved fluorescence anisotropy study. Biochim. Biophys. Acta Biomembr. 2007, 1768, 467–478. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Fernandes, F.; Fernandes, A.C.; Ramalho, J.P.P. Effects of fluorescent probe NBD-PC on the structure, dynamics and phase transition of DPPC. A molecular dynamics and differential scanning calorimetry study. Biochim. Biophys. Acta Biomembr. 2008, 1778, 491–501. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Carvalho, A.J.P.; Ramalho, J.P.P. Direct calculation of Förster orientation factor of membrane probes by molecular simulation. J. Mol. Struct. Theochem. 2010, 946, 107–112. [Google Scholar] [CrossRef]
- Loura, L.M.S. Simple estimation of förster resonance energy transfer (FRET) orientation factor distribution in membranes. Int. J. Mol. Sci. 2012, 13, 15252–15270. [Google Scholar] [CrossRef] [PubMed]
- Loura, L.M.S. Lateral distribution of NBD-PC fluorescent lipid analogs in membranes probed by molecular dynamics-assisted analysis of förster resonance energy transfer (FRET) and fluorescence quenching. Int. J. Mol. Sci. 2012, 13, 14545–14564. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, R.M.S.; Filipe, H.A.L.; Gomes, F.; Moreira, N.D.; Vaz, W.L.C.; Moreno, M.J. Chain length effect on the binding of amphiphiles to serum albumin and to POPC bilayers. J. Phys. Chem. B 2010, 114, 16337–16346. [Google Scholar] [CrossRef] [PubMed]
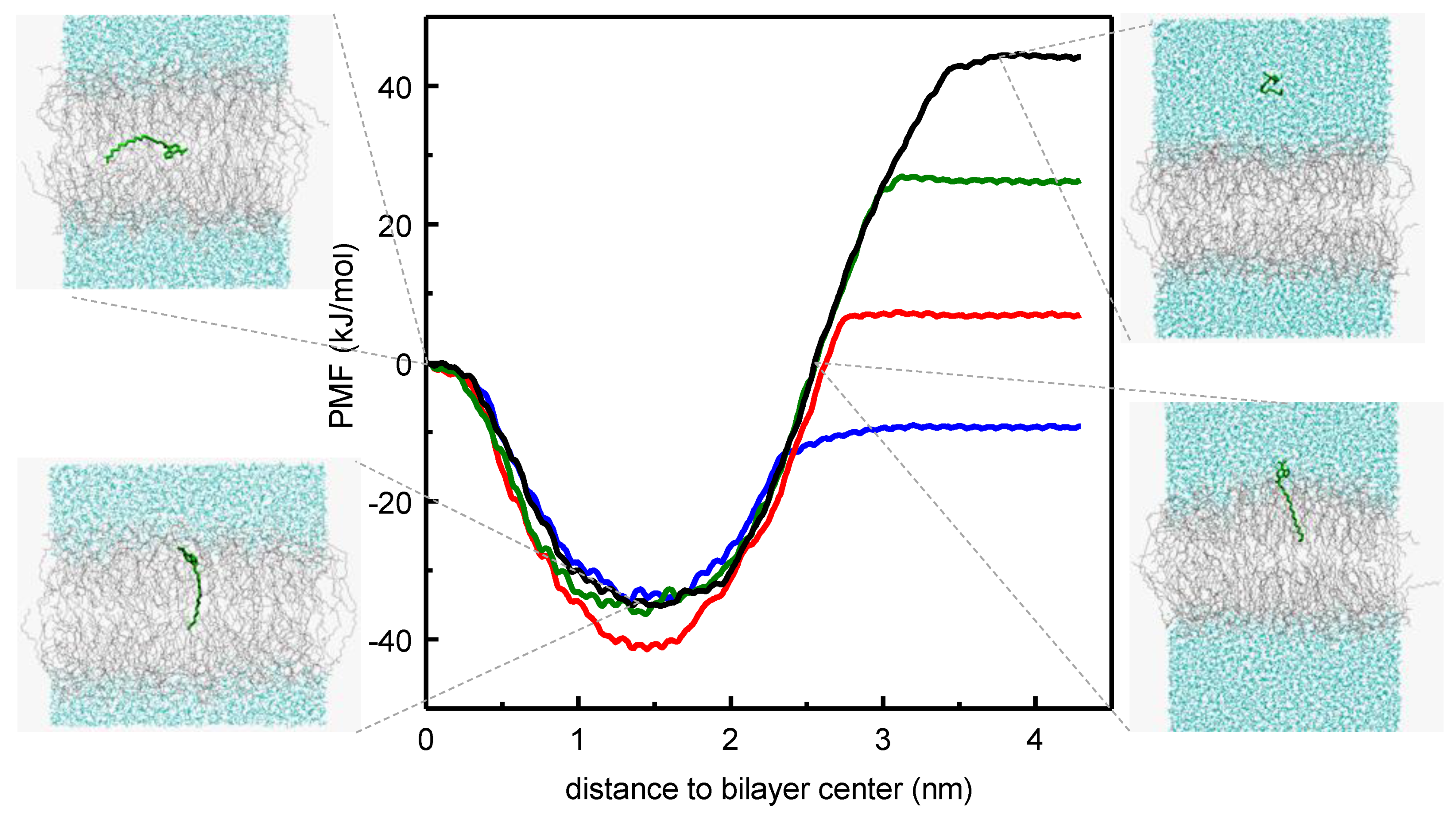
- Cardoso, R.M.S.; Martins, P.A.T.; Gomes, F.; Doktorovova, S.; Vaz, W.L.C.; Moreno, M.J. Chain-length dependence of insertion, desorption, and translocation of a homologous series of 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled aliphatic amines in membranes. J. Phys. Chem. B 2011, 115, 10098–10108. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Moreno, M.J.; Loura, L.M.S. Interaction of 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled fatty amines with 1-palmitoyl, 2-oleoyl-sn-glycero-3-phosphocholine bilayers: A molecular dynamics study. J. Phys. Chem. B 2011, 115, 10109–10119. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Moreno, M.J.; Róg, T.; Vattulainen, I.; Loura, L.M.S. How to tackle the issues in free energy simulations of long amphiphiles interacting with lipid membranes: Convergence and local membrane deformations. J. Phys. Chem. B 2014, 118, 3572–3581. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Bowman, D.; Palmeira, T.; Cardoso, R.M.S.; Loura, L.M.S.; Moreno, M.J. Interaction of NBD-labelled fatty amines with liquid-ordered membranes: A combined molecular dynamics simulation and fluorescence spectroscopy study. Phys. Chem. Chem. Phys. 2015, 17, 27534–27547. [Google Scholar] [CrossRef]
- Hinner, M.J.; Marrink, S.J.; De Vries, A.H. Location, tilt, and binding: A molecular dynamics study of voltage-sensitive dyes in biomembranes. J. Phys. Chem. B 2009, 113, 15807–15819. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Rodnin, M.V.; Ladokhin, A.S. Calibration of distribution analysis of the depth of membrane penetration using simulations and depth-dependent fluorescence quenching. J. Membr. Biol. 2015, 248, 583–594. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Santos, L.S.; Prates Ramalho, J.P.; Moreno, M.J.; Loura, L.M.S. Behaviour of NBD-head group labelled phosphatidylethanolamines in POPC bilayers: A molecular dynamics study. Phys. Chem. Chem. Phys. 2015, 17, 20066–20079. [Google Scholar] [CrossRef]
- London, E.; Chattopadhyay, A. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 1987, 26, 39–45. [Google Scholar] [CrossRef]
- Amaro, M.; Filipe, H.A.L.; Prates Ramalho, J.P.; Hof, M.; Loura, L.M.S. Fluorescence of nitrobenzoxadiazole (NBD)-labeled lipids in model membranes is connected not to lipid mobility but to probe location. Phys. Chem. Chem. Phys. 2016, 18, 7042–7054. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Chattopadhyay, A. Application of NBD-labeled lipids in membrane and cell biology. In Fluorescent Methods to Study Biological Membranes; Mély, Y., Duportail, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 37–50. ISBN 978-3-642-33128-2. [Google Scholar]
- Fery-Forgues, S.; Fayet, J.P.; Lopez, A. Drastic changes in the fluorescence properties of NBD probes with the polarity of the medium: Involvement of a TICT state? J. Photochem. Photobiol. A Chem. 1993, 70, 229–243. [Google Scholar] [CrossRef]
- Paprica, P.A.; Baird, N.C.; Petersen, N.O. Theoretical and experimental analyses of optical transitions of nitrobenzoxadiazole (NBD) derivatives. J. Photochem. Photobiol. A Chem. 1993, 70, 51–57. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chattopadhyay, A.; Samanta, A.; Soujanya, T. Dipole moment change of NBD group upon excitation studied using solvatochromic and quantum chemical approaches: Implications in membrane research. J. Phys. Chem. 1994, 98, 2809–2812. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Pokorná, Š.; Hof, M.; Amaro, M.; Loura, L.M.S. Orientation of nitro-group governs the fluorescence lifetime of nitrobenzoxadiazole (NBD)-labeled lipids in lipid bilayers. Phys. Chem. Chem. Phys. 2019, 21, 1682–1688. [Google Scholar] [CrossRef]
- Bagatolli, L.A. To see or not to see: Lateral organization of biological membranes and fluorescence microscopy. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1541–1556. [Google Scholar] [CrossRef]
- Parasassi, T.; De Stasio, G.; Ravagnan, G.; Rusch, R.M.; Gratton, E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys. J. 1991, 60, 179–189. [Google Scholar] [CrossRef]
- Barucha-Kraszewska, J.; Kraszewski, S.; Jurkiewicz, P.; Ramseyer, C.; Hof, M. Numerical studies of the membrane fluorescent dyes dynamics in ground and excited states. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1724–1734. [Google Scholar] [CrossRef]
- Cwiklik, L.; Aquino, A.J.A.; Vazdar, M.; Jurkiewicz, P.; Pittner, J.; Hof, M.; Lischka, H. Absorption and fluorescence of PRODAN in phospholipid bilayers: A combined quantum mechanics and classical molecular dynamics study. J. Phys. Chem. A 2011, 115, 11428–11437. [Google Scholar] [CrossRef] [PubMed]
- Barucha-Kraszewska, J.; Kraszewski, S.; Ramseyer, C. Will C-Laurdan dethrone Laurdan in fluorescent solvent relaxation techniques for lipid membrane studies? Langmuir 2013, 29, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Osella, S.; Murugan, N.A.; Jena, N.K.; Knippenberg, S. Investigation into biological environments through (non)linear optics: A multiscale study of laurdan derivatives. J. Chem. Theory Comput. 2016, 12, 6169–6181. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.W.; Pederzoli, M.; Jurkiewicz, P.; Cwiklik, L.; Pittner, J. Orientation of Laurdan in phospholipid bilayers influences its fluorescence: Quantum mechanics and classical molecular dynamics study. Molecules 2018, 23, 1707. [Google Scholar] [CrossRef] [PubMed]
- Osella, S.; Smisdom, N.; Ameloot, M.; Knippenberg, S. Conformational changes as driving force for phase recognition: The case of Laurdan. Langmuir 2019, 35, 11471–11481. [Google Scholar] [CrossRef]
- Osella, S.; Knippenberg, S. Laurdan as a molecular rotor in biological environments. ACS Appl. Bio Mater. 2019, 2, 5769–5778. [Google Scholar] [CrossRef]
- Suhaj, A.; Le Marois, A.; Williamson, D.J.; Suhling, K.; Lorenz, C.D.; Owen, D.M. PRODAN differentially influences its local environment. Phys. Chem. Chem. Phys. 2018, 20, 16060–16066. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef]
- Martinez, V.; Henary, M. Nile red and Nile blue: Applications and syntheses of structural analogues. Chem. A Eur. J. 2016, 22, 13764–13782. [Google Scholar] [CrossRef]
- Singh, G.; Chamberlin, A.C.; Zhekova, H.R.; Noskov, S.Y.; Tieleman, D.P. Two-dimensional potentials of mean force of Nile red in intact and damaged model bilayers. Application to calculations of fluorescence spectra. J. Chem. Theory Comput. 2016, 12, 364–371. [Google Scholar] [CrossRef]
- Ray, A.; Das, S.; Chattopadhyay, N. Aggregation of Nile red in water: Prevention through encapsulation in β-cyclodextrin. ACS Omega 2019, 4, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Pyrshev, K.A.; Yesylevskyy, S.O.; Mély, Y.; Demchenko, A.P.; Klymchenko, A.S. Caspase-3 activation decreases lipid order in the outer plasma membrane leaflet during apoptosis: A fluorescent probe study. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Prioli, S.; Reinholdt, P.; Hornum, M.; Kongsted, J. Rational design of Nile red analogs for sensing in membranes. J. Phys. Chem. B 2019, 123, 10424–10432. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Measurements and implications of the membrane dipole potential. Annu. Rev. Biochem. 2012, 81, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Loew, L.M. Design and use of organic voltage sensitive dyes. Adv. Exp. Med. Biol. 2015, 859, 27–53. [Google Scholar] [CrossRef]
- Warshaviak, D.T.; Muellner, M.J.; Chachisvilis, M. Effect of membrane tension on the electric field and dipole potential of lipid bilayer membrane. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2608–2617. [Google Scholar] [CrossRef]
- Bouquiaux, C.; Tonnelé, C.; Castet, F.; Champagne, B. Second-order nonlinear optical properties of an amphiphilic dye embedded in a lipid bilayer. A combined molecular dynamics-quantum chemistry study. J. Phys. Chem. B 2020. [Google Scholar] [CrossRef]
- Kulkarni, R.U.; Yin, H.; Pourmandi, N.; James, F.; Adil, M.M.; Schaffer, D.V.; Wang, Y.; Miller, E.W. A rationally designed, general strategy for membrane orientation of photoinduced electron transfer-based voltage-sensitive dyes. ACS Chem. Biol. 2017, 12, 407–413. [Google Scholar] [CrossRef]
- Yue, Y.; Huo, F.; Lee, S.; Yin, C.; Yoon, J. A review: The trend of progress about pH probes in cell application in recent years. Analyst 2017, 142, 30–41. [Google Scholar] [CrossRef]
- Diana, R.; Panunzi, B.; Tuzi, A.; Piotto, S.; Concilio, S.; Caruso, U. An amphiphilic pyridinoyl-hydrazone probe for colorimetric and fluorescence pH sensing. Molecules 2019, 24, 3833. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, P.V.; Koehorst, R.B.M.; Vos, W.L.; Apanasovich, V.V.; Hemminga, M.A. FRET study of membrane proteins: Determination of the tilt and orientation of the N-terminal domain of M13 major coat protein. Biophys. J. 2007, 92, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Vos, W.L.; Schor, M.; Baumgaertner, A.; Tieleman, D.P.; Hemminga, M.A. Molecular dynamics simulations reveal that AEDANS is an inert fluorescent probe for the study of membrane proteins. Eur. Biophys. J. 2010, 39, 229–239. [Google Scholar] [CrossRef] [PubMed]
- García-Beltrán, O.; Mena, N.; Yañez, O.; Caballero, J.; Vargas, V.; Nuñez, M.T.; Cassels, B.K. Design, synthesis and cellular dynamics studies in membranes of a new coumarin-based “turn-off” fluorescent probe selective for Fe2+. Eur. J. Med. Chem. 2013, 67, 60–63. [Google Scholar] [CrossRef]
- García-Beltrán, O.; Cassels, B.K.; Mena, N.; Nuñez, M.T.; Yañez, O.; Caballero, J. A coumarinylaldoxime as a specific sensor for Cu2+ and its biological application. Tetrahedron Lett. 2014, 55, 873–876. [Google Scholar] [CrossRef]
- García-Beltrán, O.; Yañez, O.; Caballero, J.; Galdámez, A.; Mena, N.; Nuñez, M.T.; Cassels, B.K. Synthesis of coumarin derivatives as fluorescent probes for membrane and cell dynamics studies. Eur. J. Med. Chem. 2014, 76, 79–86. [Google Scholar] [CrossRef]
- Paloncýová, M.; Berka, K.; Otyepka, M. Convergence of free energy profile of coumarin in lipid bilayer. J. Chem. Theory Comput. 2012, 8, 1200–1211. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Wu, F.; Thummel, R.P.; Waluk, J.; Ladokhin, A.S. Partitioning and localization of environment-sensitive 2-(2′-pyridyl)- and 2-(2′-pyrimidyl)-indoles in lipid membranes: A joint refinement using fluorescence measurements and molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 13574–13584. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Sevriukov, I.Y.; Syzova, Z.A.; Ladokhin, A.S.; Doroshenko, A.O. Partitioning of 2,6-Bis(1H-Benzimidazol-2-yl)pyridine fluorophore into a phospholipid bilayer: Complementary use of fluorescence quenching studies and molecular dynamics simulations. Biophys. Chem. 2011, 154, 8–17. [Google Scholar] [CrossRef]
- Posokhov, Y.; Kyrychenko, A. Location of fluorescent probes (2′-hydroxy derivatives of 2,5-diaryl-1,3-oxazole) in lipid membrane studied by fluorescence spectroscopy and molecular dynamics simulation. Biophys. Chem. 2018, 235, 9–18. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Karpushina, G.V.; Svechkarev, D.; Kolodezny, D.; Bogatyrenko, S.I.; Kryshtal, A.P.; Doroshenko, A.O. Fluorescence probing of thiol-functionalized gold nanoparticles: Is alkylthiol coating of a nanoparticle as hydrophobic as expected? J. Phys. Chem. C 2012, 116, 21059–21068. [Google Scholar] [CrossRef]
- Ladokhin, A.S. Distribution analysis of depth-dependent fluorescence quenching in membranes: A practical guide. Methods Enzymol. 1997, 278, 462–473. [Google Scholar] [CrossRef] [PubMed]
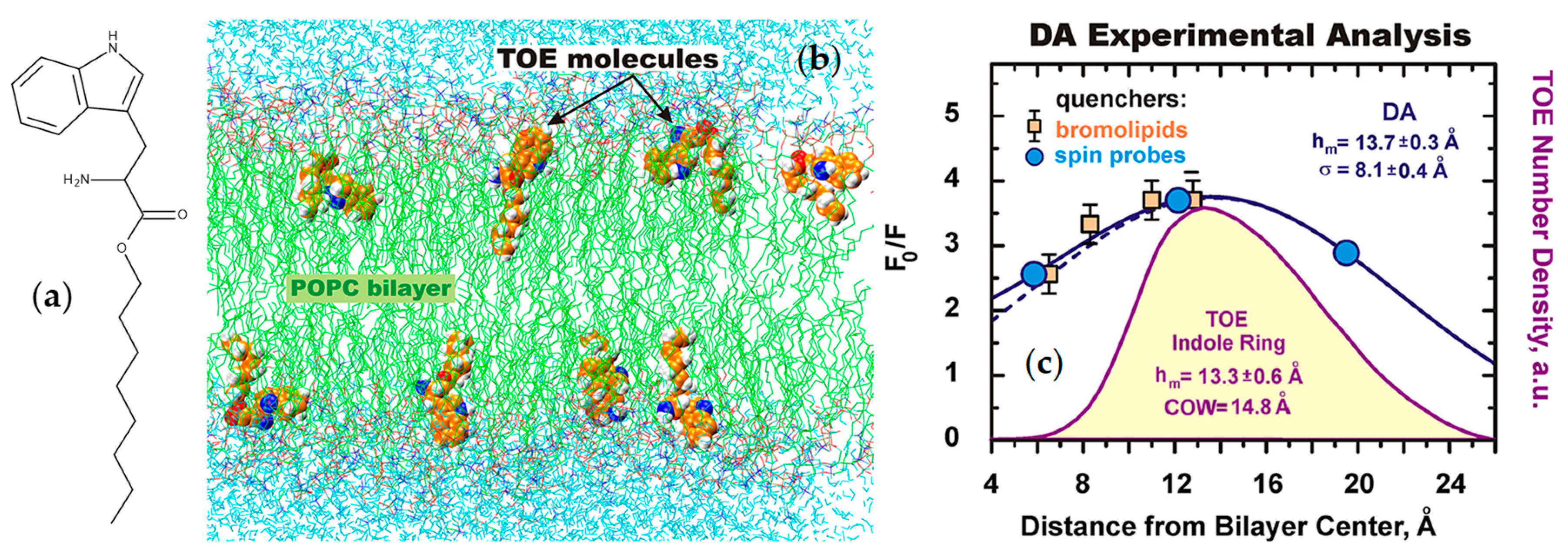
- Kyrychenko, A.; Tobias, D.J.; Ladokhin, A.S. Validation of depth-dependent fluorescence quenching in membranes by molecular dynamics simulation of tryptophan octyl ester in POPC bilayer. J. Phys. Chem. B 2013, 117, 4770–4778. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S.; Holloway, P.W. Fluorescence of membrane-bound tryptophan octyl ester: A model for studying intrinsic fluorescence of protein-membrane interactions. Biophys. J. 1995, 69, 506–517. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Ladokhin, A.S. Molecular dynamics simulations of depth distribution of spin-labeled phospholipids within lipid bilayer. J. Phys. Chem. B 2013, 117, 5875–5885. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Ladokhin, A.S. Location of TEMPO-PC in lipid bilayers: Implications for fluorescence quenching. J. Membr. Biol. 2020, 253, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, A.; Lim, N.M.; Vasquez-Montes, V.; Rodnin, M.V.; Freites, J.A.; Nguyen, L.P.; Tobias, D.J.; Mobley, D.L.; Ladokhin, A.S. Refining protein penetration into the lipid bilayer using fluorescence quenching and molecular dynamics simulations: The case of diphtheria toxin translocation domain. J. Membr. Biol. 2018, 251, 379–391. [Google Scholar] [CrossRef]
- Timr, Š.; Bondar, A.; Cwiklik, L.; Štefl, M.; Hof, M.; Vazdar, M.; Lazar, J.; Jungwirth, P. Accurate determination of the orientational distribution of a fluorescent molecule in a phospholipid membrane. J. Phys. Chem. B 2014, 118, 855–863. [Google Scholar] [CrossRef]
- Timr, Š.; Brabec, J.; Bondar, A.; Ryba, T.; Železný, M.; Lazar, J.; Jungwirth, P. Nonlinear optical properties of fluorescent dyes allow for accurate determination of their molecular orientations in phospholipid membranes. J. Phys. Chem. B 2015, 119, 9706–9716. [Google Scholar] [CrossRef]
- Singh, M.K.; Shweta, H.; Khan, M.F.; Sen, S. New insight into probe-location dependent polarity and hydration at lipid/water interfaces: Comparison between gel- and fluid-phases of lipid bilayers. Phys. Chem. Chem. Phys. 2016, 18, 24185–24197. [Google Scholar] [CrossRef]
- Singh, M.K.; Khan, M.F.; Shweta, H.; Sen, S. Probe-location dependent resonance energy transfer at lipid/water interfaces: Comparison between the gel- and fluid-phase of lipid bilayer. Phys. Chem. Chem. Phys. 2017, 19, 25870–25885. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Bottari, G.; Brancato, G.; Barone, V.; Guldi, D.M.; Torres, T. Unraveling the peculiar modus operandi of a new class of solvatochromic fluorescent molecular rotors by spectroscopic and quantum mechanical methods. Chem. Sci. 2013, 4, 2502–2511. [Google Scholar] [CrossRef]
- Koenig, M.; Storti, B.; Bizzarri, R.; Guldi, D.M.; Brancato, G.; Bottari, G. A fluorescent molecular rotor showing vapochromism, aggregation-induced emission, and environmental sensing in living cells. J. Mater. Chem. C 2016, 4, 3018–3027. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Del Frate, G.; Brancato, G.; Chandramouli, B.; Mancini, G.; Barone, V. Computational study of the DPAP molecular rotor in various environments: From force field development to molecular dynamics simulations and spectroscopic calculations. Phys. Chem. Chem. Phys. 2017, 19, 30590–30602. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lee, M.M.S.; Shan, G.; Kwok, R.T.K.; Lam, J.W.Y.; Su, H.; Cai, Y.; Tang, B.Z. Highly efficient photosensitizers with far-red/near-infrared aggregation-induced emission for in vitro and in vivo cancer theranostics. Adv. Mater. 2018, 30, e1802105. [Google Scholar] [CrossRef]
- Wang, D.; Su, H.; Kwok, R.T.K.; Hu, X.; Zou, H.; Luo, Q.; Lee, M.M.S.; Xu, W.; Lam, J.W.Y.; Tang, B.Z. Rational design of a water-soluble NIR AIEgen, and its application in ultrafast wash-free cellular imaging and photodynamic cancer cell ablation. Chem. Sci. 2018, 9, 3685–3693. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, D.; Xu, W.; Cao, S.; Peng, Q.; Tang, B.Z. Charge control of fluorescent probes to selectively target the cell membrane or mitochondria: Theoretical prediction and experimental validation. Mater. Horiz. 2019, 6, 2016–2023. [Google Scholar] [CrossRef]
- Marrink, S.J.; Berendsen, H.J.C. Simulation of water transport through a lipid membrane. J. Phys. Chem. 1994, 98, 4155–4168. [Google Scholar] [CrossRef]
- Kowada, T.; Maeda, H.; Kikuchi, K. BODIPY-based probes for the fluorescence imaging of biomolecules in living cells. Chem. Soc. Rev. 2015, 44, 4953–4972. [Google Scholar] [CrossRef]
- Hölttä-Vuori, M.; Uronen, R.L.; Repakova, J.; Salonen, E.; Vattulainen, I.; Panula, P.; Li, Z.; Bittman, R.; Ikonen, E. BODIPY-cholesterol: A new tool to visualize sterol trafficking in living cells and organisms. Traffic 2008, 9, 1839–1849. [Google Scholar] [CrossRef]
- Song, K.C.; Livanec, P.W.; Klauda, J.B.; Kuczera, K.; Dunn, R.C.; Im, W. Orientation of fluorescent lipid analogue BODIPY-PC to probe lipid membrane properties: Insights from molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 6157–6165. [Google Scholar] [CrossRef] [PubMed]
- Dent, M.R.; López-Duarte, I.; Dickson, C.J.; Geoghegan, N.D.; Cooper, J.M.; Gould, I.R.; Krams, R.; Bull, J.A.; Brooks, N.J.; Kuimova, M.K. Imaging phase separation in model lipid membranes through the use of BODIPY based molecular rotors. Phys. Chem. Chem. Phys. 2015, 17, 18393–18402. [Google Scholar] [CrossRef] [PubMed]
- Steinmark, I.E.; James, A.L.; Chung, P.-H.; Morton, P.E.; Parsons, M.; Dreiss, C.A.; Lorenz, C.D.; Yahioglu, G.; Suhling, K. Targeted fluorescence lifetime probes reveal responsive organelle viscosity and membrane fluidity. PLoS ONE 2019, 14, e0211165. [Google Scholar] [CrossRef] [PubMed]
- Steinmark, I.E.; Chung, P.; Ziolek, R.M.; Cornell, B.; Smith, P.; Levitt, J.A.; Tregidgo, C.; Molteni, C.; Yahioglu, G.; Lorenz, C.D.; et al. Time-resolved fluorescence anisotropy of a molecular rotor resolves microscopic viscosity parameters in complex environments. Small 2020, 16, e1907139. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, S.; Grzybek, M.; Orłowski, A.; Róg, T.; Cramariuc, O.; Levental, I.; Eggeling, C.; Sezgin, E.; Vattulainen, I. Phase partitioning of GM1 and its bodipy-labeled analog determine their different binding to cholera toxin. Front. Physiol. 2017, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Bacalum, M.; Wang, L.; Boodts, S.; Yuan, P.; Leen, V.; Smisdom, N.; Fron, E.; Knippenberg, S.; Fabre, G.; Trouillas, P.; et al. A blue-light-emitting BODIPY probe for lipid membranes. Langmuir 2016, 32, 3495–3505. [Google Scholar] [CrossRef]
- Knippenberg, S.; Fabre, G.; Osella, S.; Di Meo, F.; Paloncýová, M.; Ameloot, M.; Trouillas, P. Atomistic picture of fluorescent probes with hydrocarbon tails in lipid bilayer membranes: An investigation of selective affinities and fluorescent anisotropies in different environmental phases. Langmuir 2018, 34, 9072–9084. [Google Scholar] [CrossRef]
- Gullapalli, R.R.; Demirel, M.C.; Butler, P.J. Molecular dynamics simulations of DiI-C18(3) in a DPPC lipid bilayer. Phys. Chem. Chem. Phys. 2008, 10, 3548–3560. [Google Scholar] [CrossRef]
- Muddana, H.S.; Gullapalli, R.R.; Manias, E.; Butler, P.J. Atomistic simulation of lipid and DiI dynamics in membrane bilayers under tension. Phys. Chem. Chem. Phys. 2011, 13, 1368–1378. [Google Scholar] [CrossRef]
- Ackerman, D.G.; Heberle, F.A.; Feigenson, G.W. Limited perturbation of a DPPC bilayer by fluorescent lipid probes: A molecular dynamics study. J. Phys. Chem. B 2013, 117, 4844–4852. [Google Scholar] [CrossRef]
- Osella, S.; Di Meo, F.; Murugan, N.A.; Fabre, G.; Ameloot, M.; Trouillas, P.; Knippenberg, S. Combining (non)linear optical and fluorescence analysis of DiD to enhance lipid phase recognition. J. Chem. Theory Comput. 2018, 14, 5350–5359. [Google Scholar] [CrossRef] [PubMed]
- Paloncýová, M.; Aniander, G.; Larsson, E.; Knippenberg, S. Cyanine dyes with tail length asymmetry enhance photoselection: A multiscale study on DiD probes in a liquid disordered membrane. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2020, 224, 117329. [Google Scholar] [CrossRef] [PubMed]
- Ardizzone, A.; Kurhuzenkau, S.; Illa-Tuset, S.; Faraudo, J.; Bondar, M.; Hagan, D.; Van Stryland, E.W.; Painelli, A.; Sissa, C.; Feiner, N.; et al. Nanostructuring lipophilic dyes in water using stable vesicles, quatsomes, as scaffolds and their use as probes for bioimaging. Small 2018, 14, e1703851. [Google Scholar] [CrossRef] [PubMed]
- Beija, M.; Afonso, C.A.M.; Martinho, J.M.G. Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. [Google Scholar] [CrossRef]
- Yan, F.; Fan, K.; Bai, Z.; Zhang, R.; Zu, F.; Xu, J.; Li, X. Fluorescein applications as fluorescent probes for the detection of analytes. Trends Anal. Chem. 2017, 97, 15–35. [Google Scholar] [CrossRef]
- Skaug, M.J.; Longo, M.L.; Faller, R. Computational studies of Texas Red-1,2-dihexadecanoyl-sn-glycero-3- phosphoethanolamine-model building and applications. J. Phys. Chem. B 2009, 113, 8758–8766. [Google Scholar] [CrossRef]
- Kyrychenko, A. A molecular dynamics model of rhodamine-labeled phospholipid incorporated into a lipid bilayer. Chem. Phys. Lett. 2010, 485, 95–99. [Google Scholar] [CrossRef]
- Skaug, M.J.; Longo, M.L.; Faller, R. The impact of texas red on lipid bilayer properties. J. Phys. Chem. B 2011, 115, 8500–8505. [Google Scholar] [CrossRef]
- Putzel, G.G.; Schick, M. Theory of raft formation by the cross-linking of saturated or unsaturated lipids in model lipid bilayers. Biophys. J. 2009, 96, 4935–4940. [Google Scholar] [CrossRef]
- Jan Akhunzada, M.; D’Autilia, F.; Chandramouli, B.; Bhattacharjee, N.; Catte, A.; Di Rienzo, R.; Cardarelli, F.; Brancato, G. Interplay between lipid lateral diffusion, dye concentration and membrane permeability unveiled by a combined spectroscopic and computational study of a model lipid bilayer. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Akhunzada, M.J.; Sagresti, L.; Catte, A.; Bhattacharjee, N.; D’Agostino, T.; Brancato, G. Temperature dependence of the structure and dynamics of a dye-labeled lipid in a planar phospholipid bilayer: A computational study. J. Membr. Biol. 2019, 252, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Mobarak, E.; Javanainen, M.; Kulig, W.; Honigmann, A.; Sezgin, E.; Aho, N.; Eggeling, C.; Rog, T.; Vattulainen, I. How to minimize dye-induced perturbations while studying biomembrane structure and dynamics: PEG linkers as a rational alternative. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, S.; Lee, E.; Manna, M.; Steinert, S.; Kumar, G.S.; Wenk, M.; Wohland, T.; Kraut, R. A fluorescent sphingolipid binding domain peptide probe interacts with sphingolipids and cholesterol-dependent raft domains[s]. J. Lipid Res. 2008, 49, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kraut, R.; Mu, Y. Aβ1-25-derived sphingolipid-domain tracer peptide sbd interacts with membrane ganglioside clusters via a coil-helix-coil motif. Int. J. Mol. Sci. 2015, 16, 26318–26322. [Google Scholar] [CrossRef]
- Gimpl, G.; Gehrig-Burger, K. Probes for studying cholesterol binding and cell biology. Steroids 2011, 76, 216–231. [Google Scholar] [CrossRef]
- Wüstner, D.; Modzel, M.; Lund, F.W.; Lomholt, M.A. Imaging approaches for analysis of cholesterol distribution and dynamics in the plasma membrane. Chem. Phys. Lipids 2016, 199, 106–135. [Google Scholar] [CrossRef]
- Robalo, J.R.; Do Canto, A.M.T.M.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Behavior of fluorescent cholesterol analogues dehydroergosterol and cholestatrienol in lipid bilayers: A molecular dynamics study. J. Phys. Chem. B 2013, 117, 5806–5819. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Müller, P.; Herrmann, A.; Huster, D. The potential of fluorescent and spin-labeled steroid analogs to mimic natural cholesterol. J. Biol. Chem. 2003, 278, 45563–45569. [Google Scholar] [CrossRef]
- Pourmousa, M.; Róg, T.; Mikkeli, R.; Vattulainen, L.; Solanko, L.M.; Wüstner, D.; List, N.H.; Kongsted, J.; Karttunen, M. Dehydroergosterol as an analogue for cholesterol: Why it mimics cholesterol so well-or does it? J. Phys. Chem. B 2014, 118, 7345–7357. [Google Scholar] [CrossRef]
- Nåbo, L.J.; List, N.H.; Witzke, S.; Wüstner, D.; Khandelia, H.; Kongsted, J. Design of new fluorescent cholesterol and ergosterol analogs: Insights from theory. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2188–2199. [Google Scholar] [CrossRef]
- Nåbo, L.J.; List, N.H.; Steinmann, C.; Kongsted, J. Computational approach to evaluation of optical properties of membrane probes. J. Chem. Theory Comput. 2017, 13, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Bonvicini, A.; Reinholdt, P.; Tognetti, V.; Joubert, L.; Wüstner, D.; Kongsted, J. Rational design of novel fluorescent analogues of cholesterol: A “step-by-step” computational study. Phys. Chem. Chem. Phys. 2019, 21, 15487–15503. [Google Scholar] [CrossRef] [PubMed]
- Modzel, M.; Solanko, K.A.; Szomek, M.; Hansen, S.K.; Dupont, A.; Nåbo, L.J.; Kongsted, J.; Wüstner, D. Live-cell imaging of new polyene sterols for improved analysis of intracellular cholesterol transport. J. Microsc. 2018, 271, 36–48. [Google Scholar] [CrossRef]
- Nåbo, L.J.; Modzel, M.; Krishnan, K.; Covey, D.F.; Fujiwara, H.; Ory, D.S.; Szomek, M.; Khandelia, H.; Wüstner, D.; Kongsted, J. Structural design of intrinsically fluorescent oxysterols. Chem. Phys. Lipids 2018, 212, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Thacker, S.G.; Fernandez-Castillejo, S.; Neufeld, E.B.; Remaley, A.T.; Bittman, R. Synthesis of cholesterol analogues bearing BODIPY fluorophores by suzuki or liebeskind-srogl cross-coupling and evaluation of their potential for visualization of cholesterol pools. ChemBioChem 2014, 15, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Reinholdt, P.; Wind, S.; Wüstner, D.; Kongsted, J. Computational characterization of a cholesterol-based molecular rotor in lipid membranes. J. Phys. Chem. B 2019, 123, 7313–7326. [Google Scholar] [CrossRef]
- Solanko, L.M.; Honigmann, A.; Midtiby, H.S.; Lund, F.W.; Brewer, J.R.; Dekaris, V.; Bittman, R.; Eggeling, C.; Wüstner, D. Membrane orientation and lateral diffusion of BODIPY-cholesterol as a function of probe structure. Biophys. J. 2013, 105, 2082–2092. [Google Scholar] [CrossRef]
- Karilainen, T.; Vuorela, T.; Vattulainen, I. How well does BODIPY-cholesteryl ester mimic unlabeled cholesteryl esters in high density lipoprotein particles? J. Phys. Chem. B 2015, 119, 15848–15856. [Google Scholar] [CrossRef]
- Robalo, J.R.; Ramalho, J.P.P.; Loura, L.M.S. NBD-labeled cholesterol analogues in phospholipid bilayers: Insights from molecular dynamics. J. Phys. Chem. B 2013, 117, 13731–13742. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Fedorov, A.; Prieto, M. Exclusion of a cholesterol analog from the cholesterol-rich phase in model membranes. Biochim. Biophys. Acta Biomembr. 2001, 1511, 236–243. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chattopadhyay, A. Membrane organization at low cholesterol concentrations: A study using 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled cholesterol. Biochemistry 1996, 35, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Loura, L.M.S.; Prieto, M. Dehydroergosterol structural organization in aqueous medium and in a model system of membranes. Biophys. J. 1997, 72, 2226–2236. [Google Scholar] [CrossRef]
















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipe, H.A.L.; Moreno, M.J.; Loura, L.M.S. The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations. Molecules 2020, 25, 3424. https://doi.org/10.3390/molecules25153424
Filipe HAL, Moreno MJ, Loura LMS. The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations. Molecules. 2020; 25(15):3424. https://doi.org/10.3390/molecules25153424
Chicago/Turabian StyleFilipe, Hugo A. L., Maria João Moreno, and Luís M. S. Loura. 2020. "The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations" Molecules 25, no. 15: 3424. https://doi.org/10.3390/molecules25153424
APA StyleFilipe, H. A. L., Moreno, M. J., & Loura, L. M. S. (2020). The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations. Molecules, 25(15), 3424. https://doi.org/10.3390/molecules25153424