
Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer′s Disease Therapy



Abstract
1. Introduction
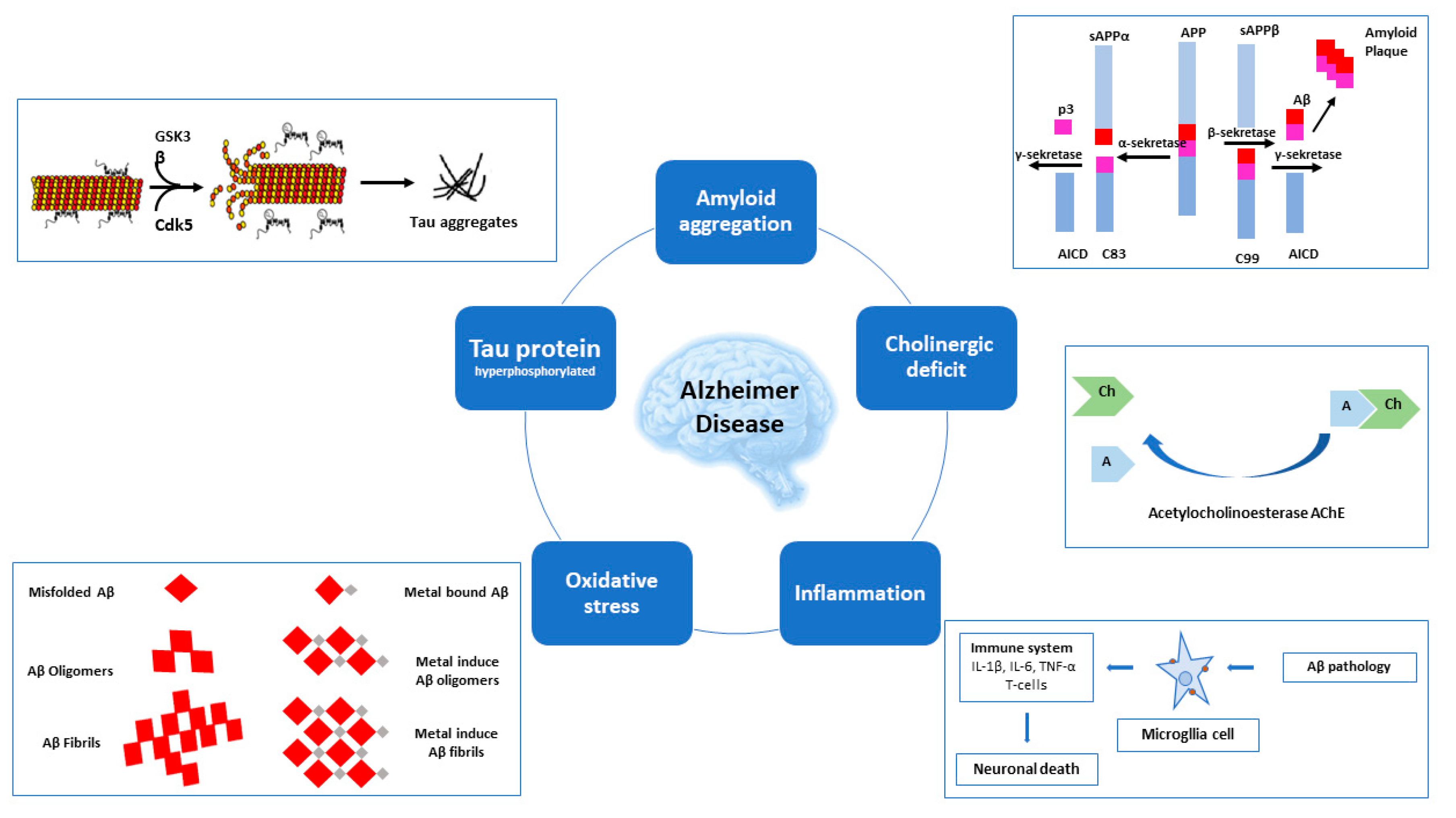
2. AD-Related Targets
3. Multi-Target Strategy for AD
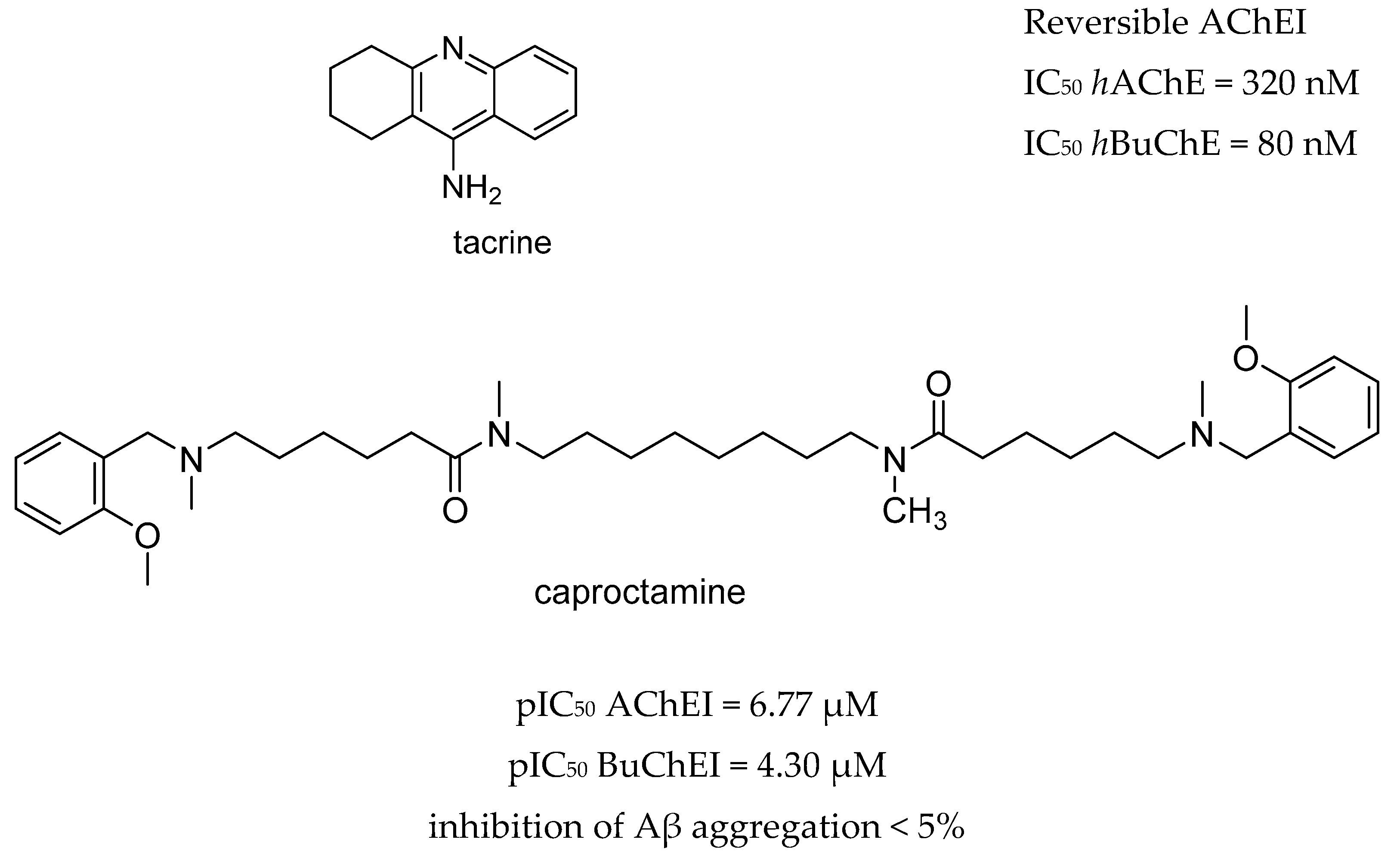
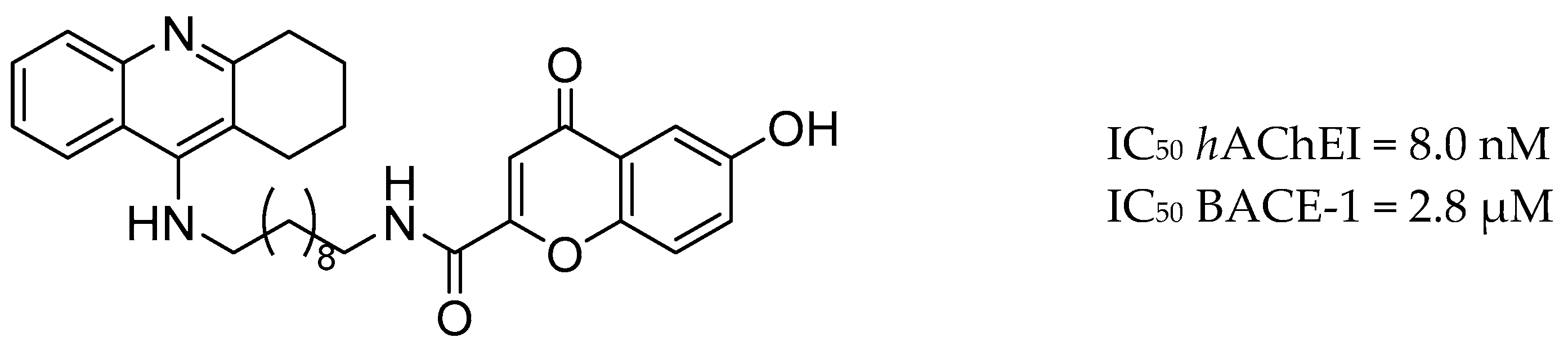
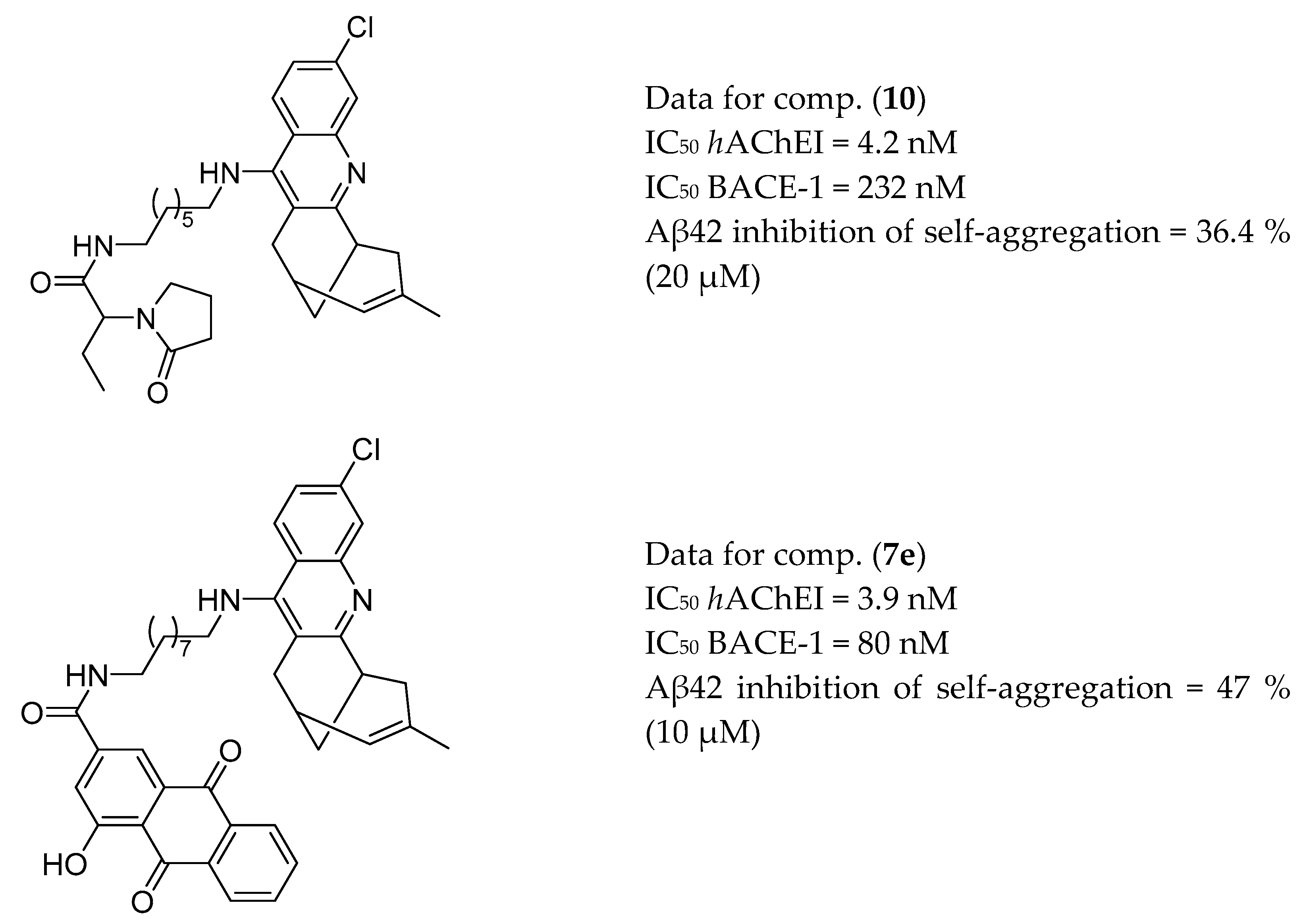
3.1. AChE and BACE-1 Inhibitors
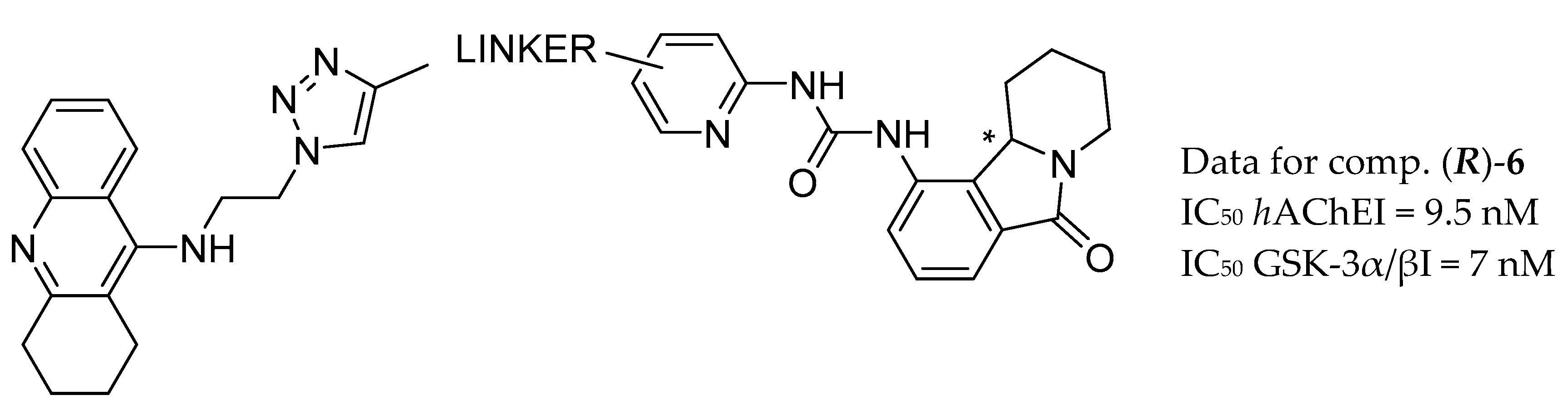
3.2. AChE and GSK-3β Inhibitors
3.3. AChE and MAO Inhibitors
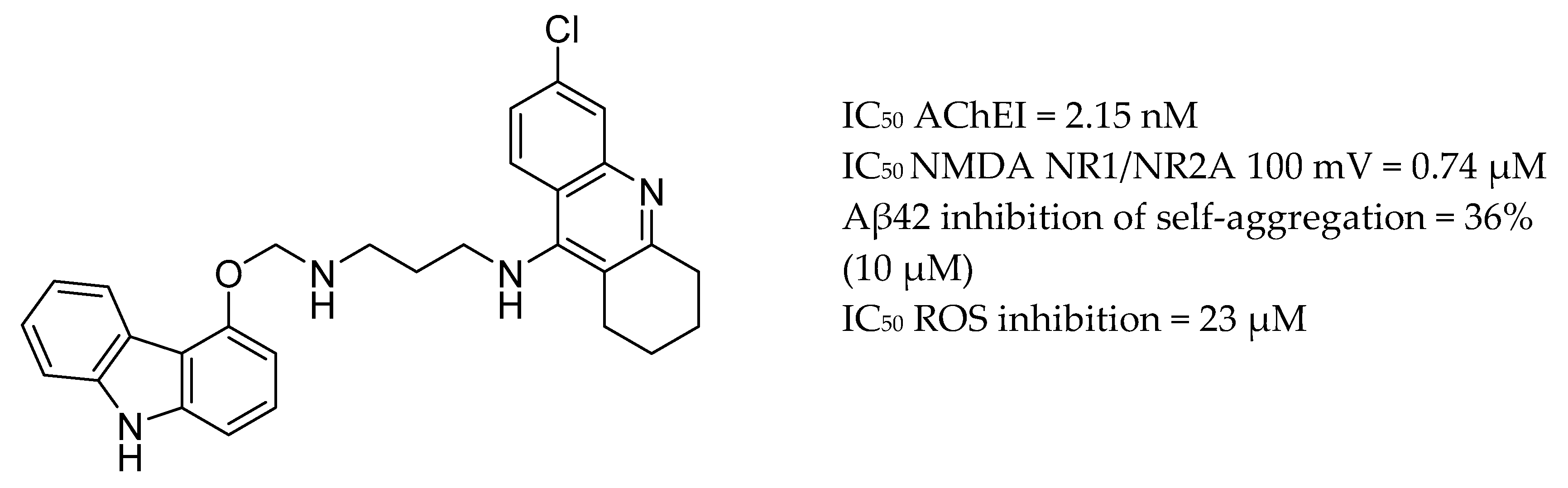
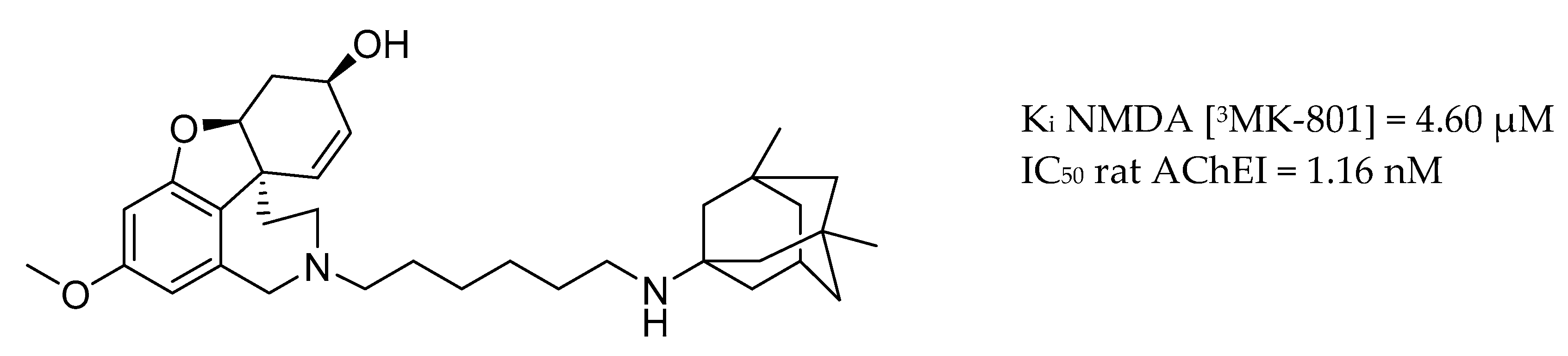
3.4. AChE Inhibitors and NMDA Antagonists
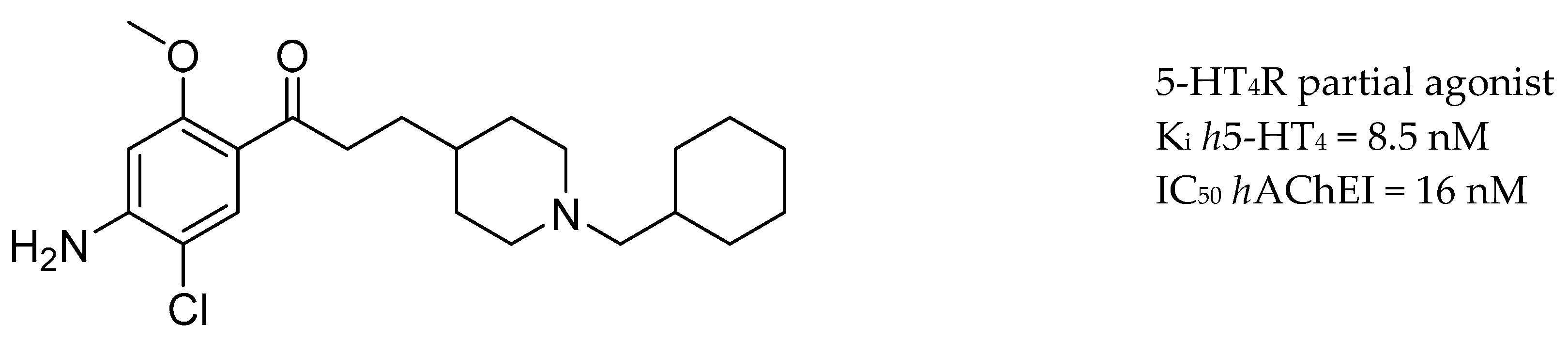
3.5. AChE Inhibitors and 5-HTR
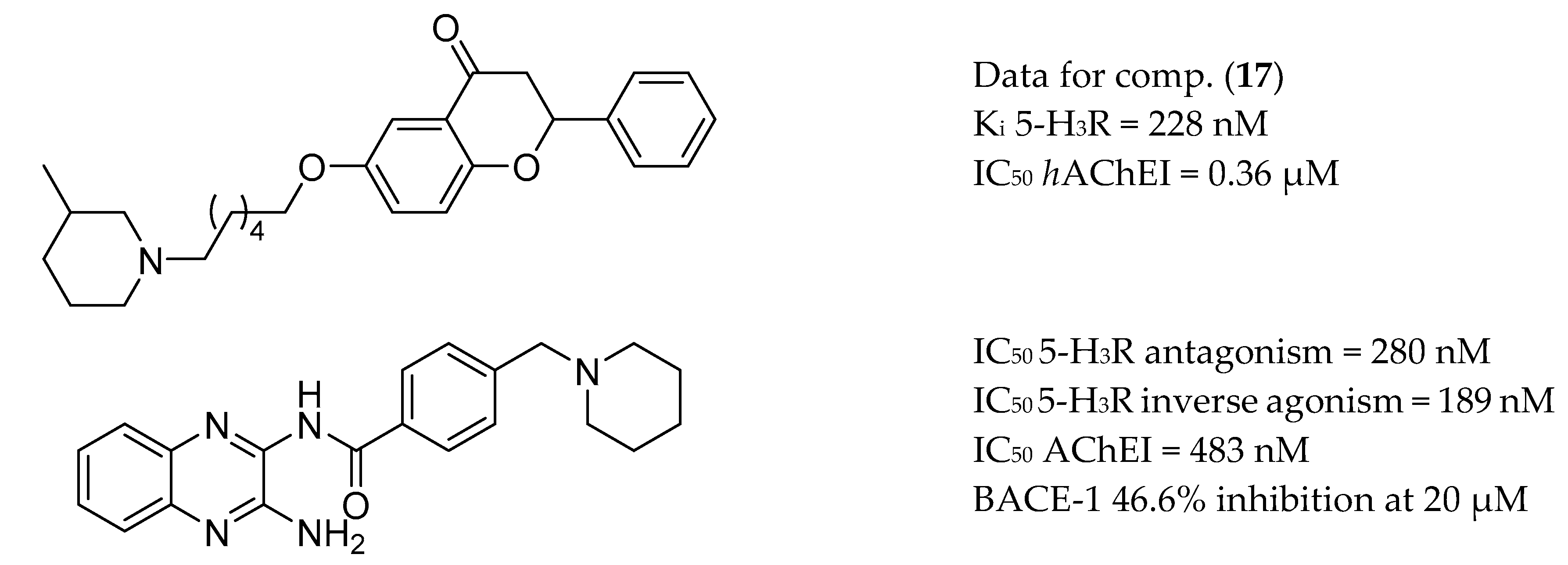
3.6. AChE Inhibitors and H3R
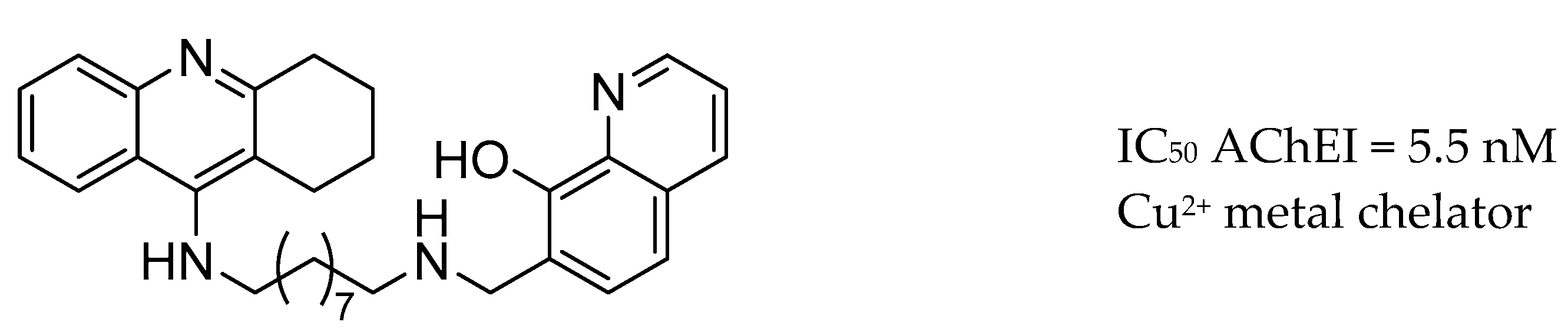
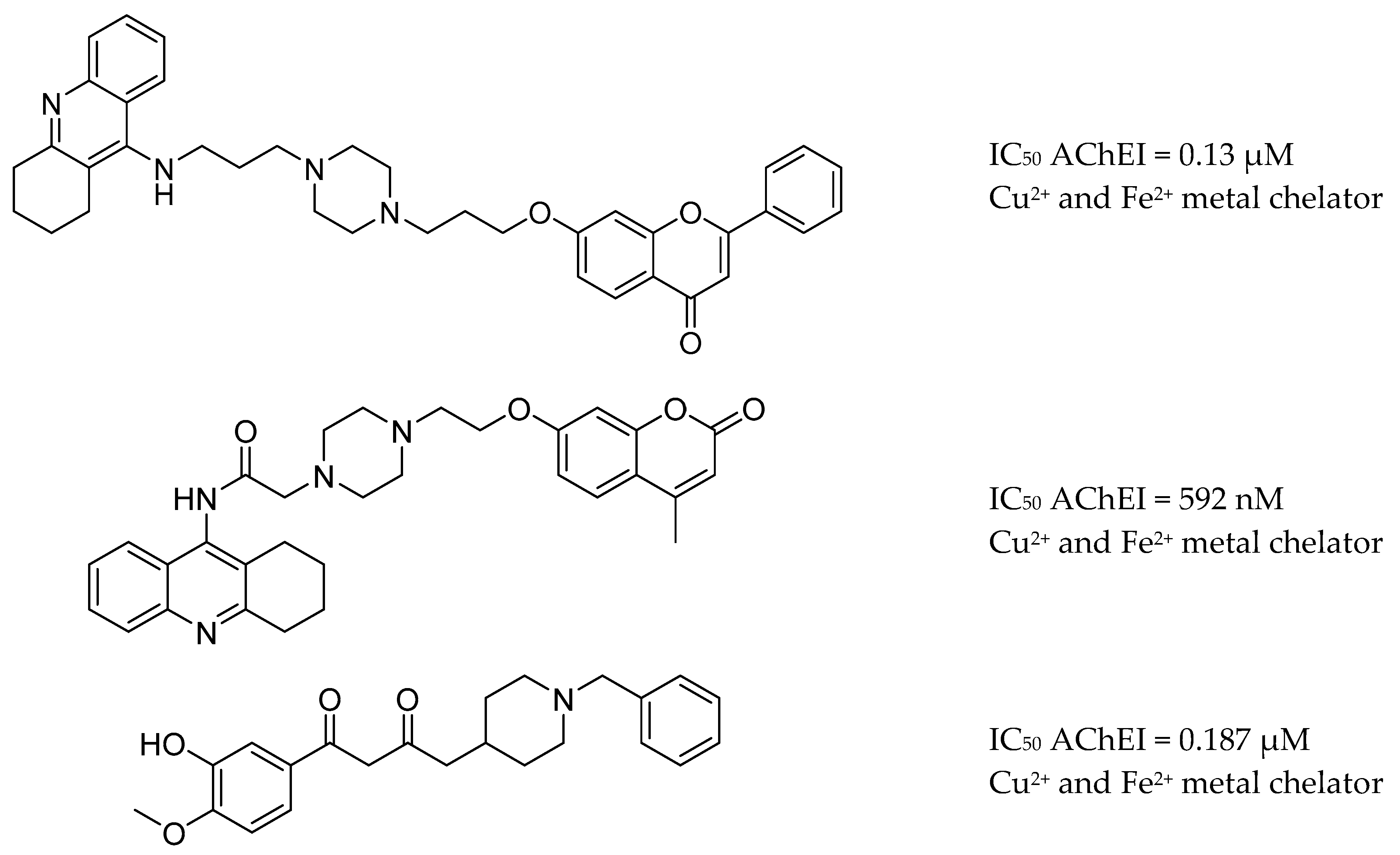
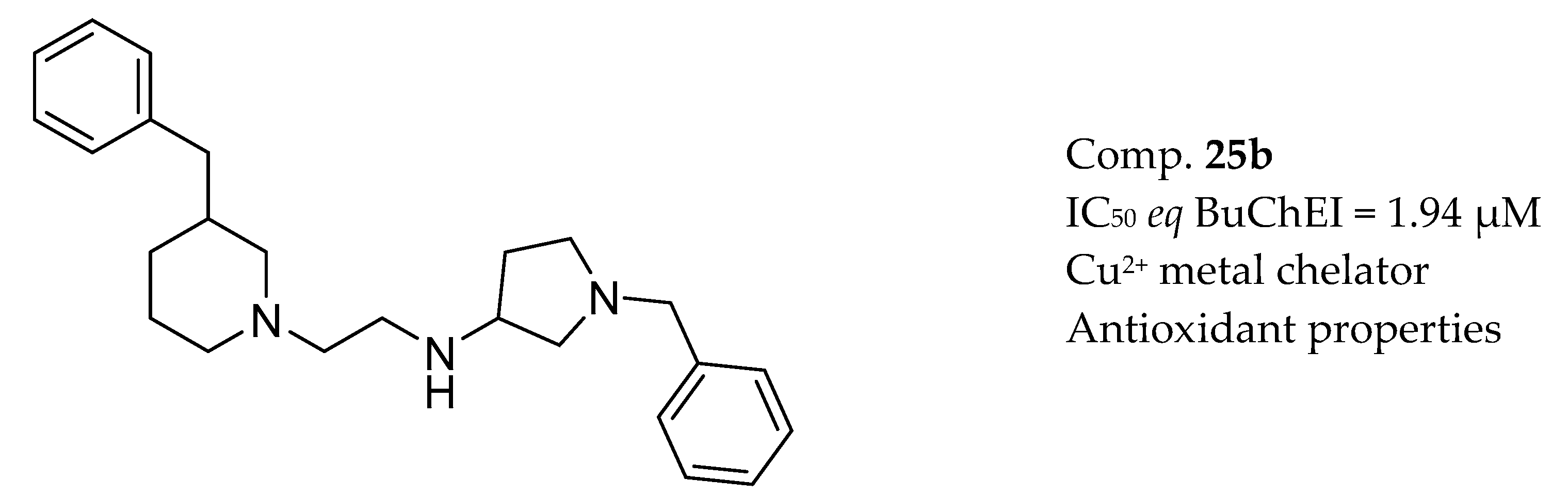
3.7. AChE Inhibitors and Biometal Chelators
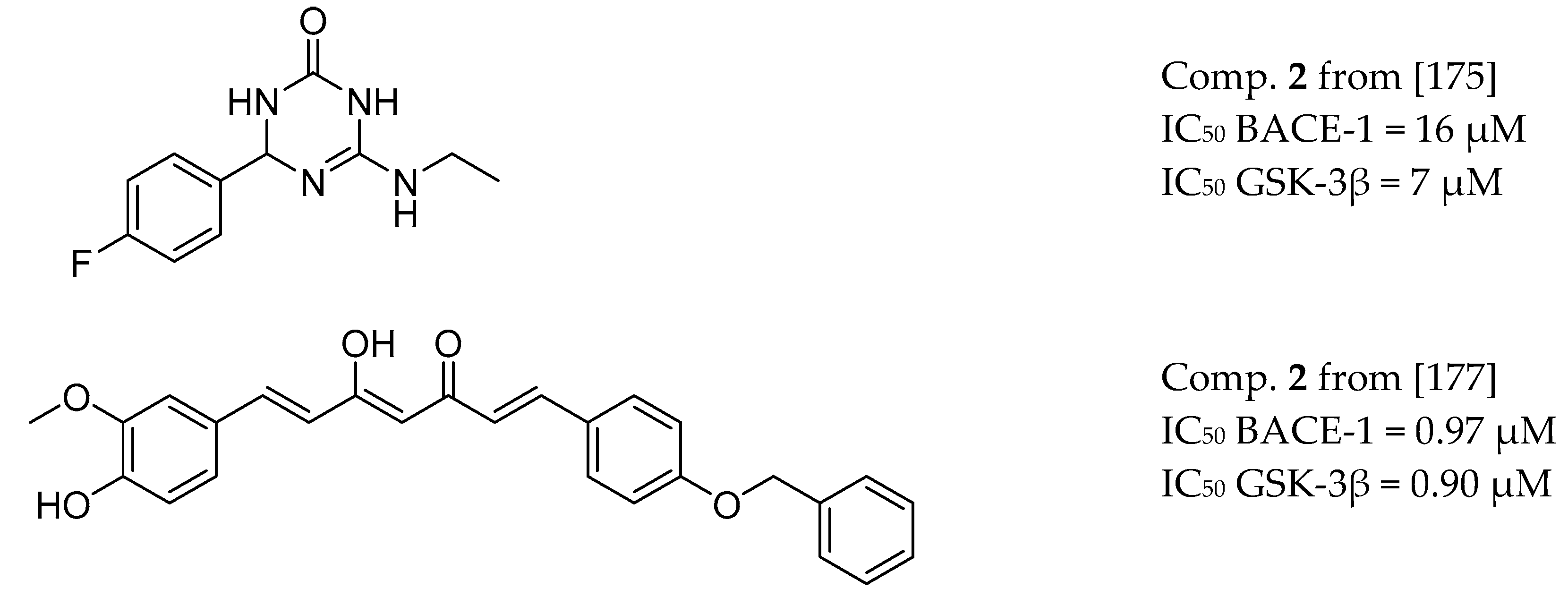
3.8. BACE-1 and GSK-3β Inhibitors
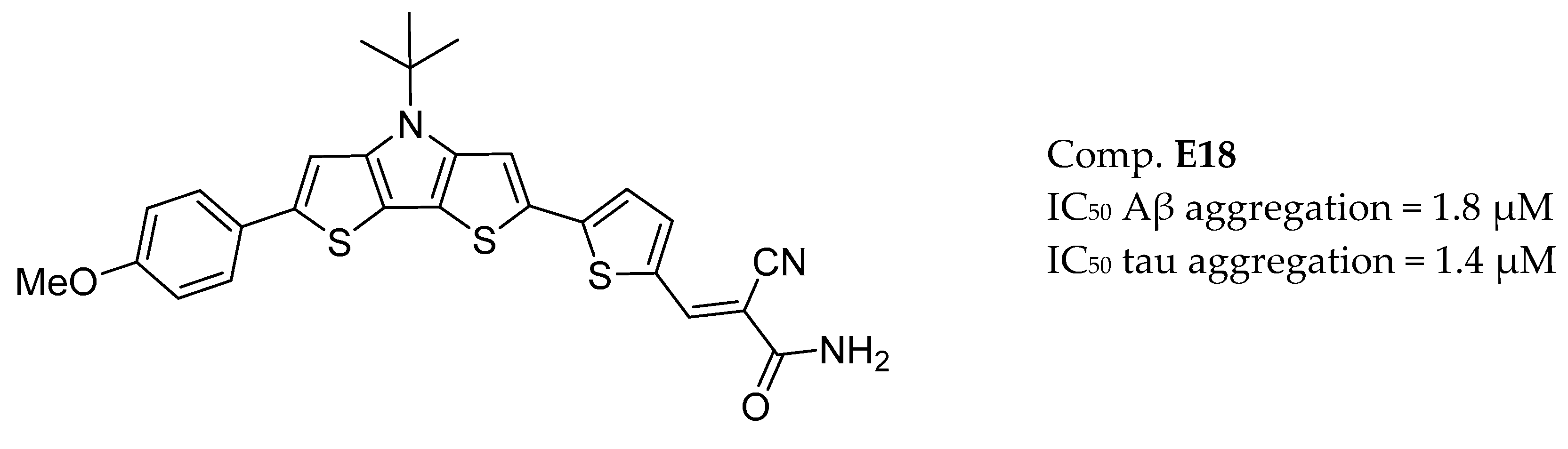
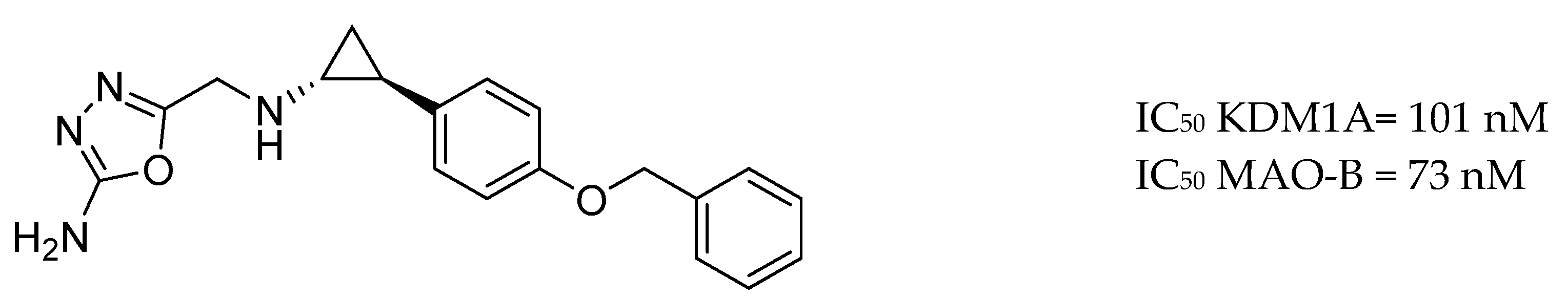
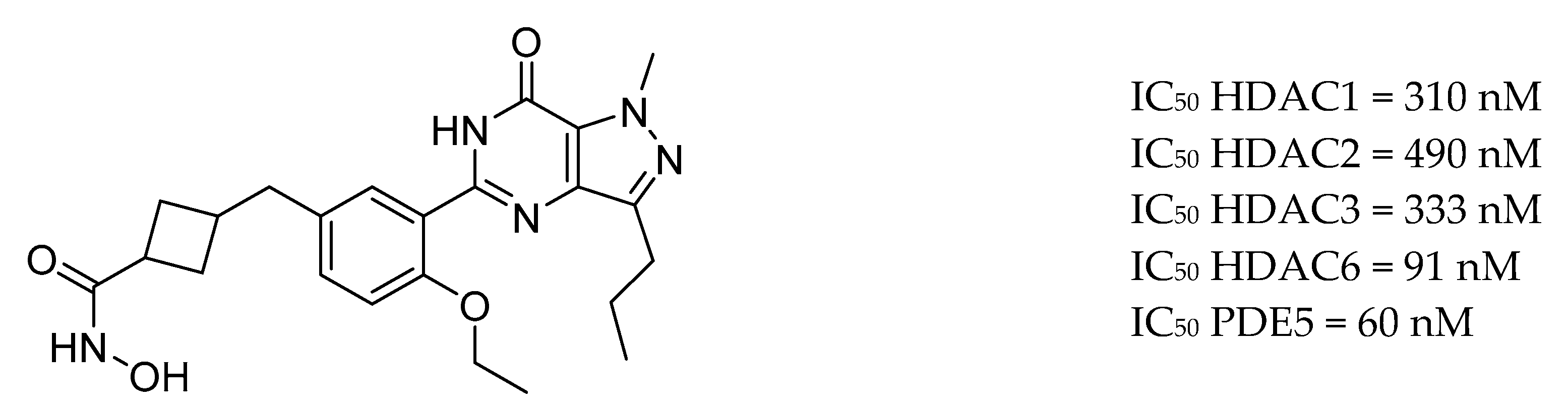
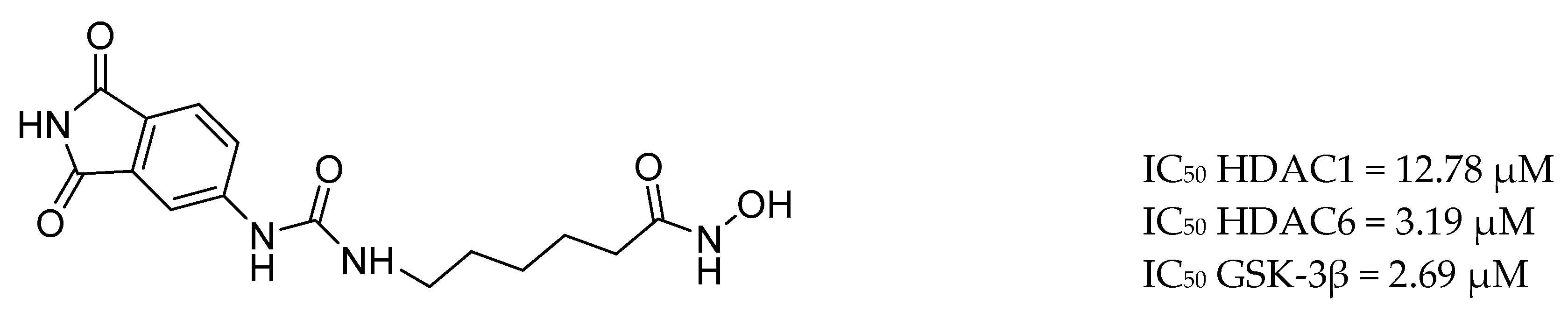
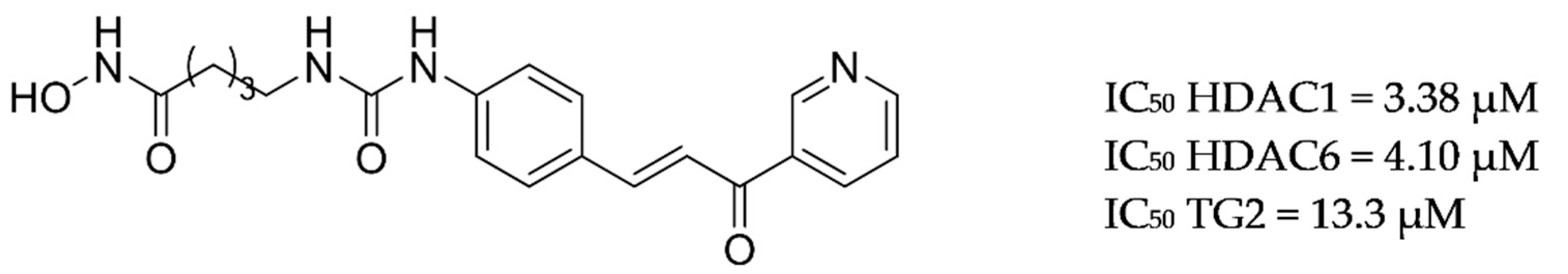
3.9. Other Targets
4. Physicochemical Properties of MTDLs for AD
5. Perspectives for MTDLs in the Treatment of AD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and cognitive impairment in alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S. Lowering of amyloid-beta by β-secretase inhibitors—Some informative failures. N. Engl. J. Med. 2019, 380, 1476–1478. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; St. George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-Mac Lachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele ϵ4 with late-onset familial and sporadic alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Sawmiller, D.; Habib, A.; Hou, H.; Mori, T.; Fan, A.; Tian, J.; Zeng, J.; Giunta, B.; Sanberg, P.R.; Mattson, M.P.; et al. A Novel Apolipoprotein E Antagonist Functionally Blocks Apolipoprotein E Interaction With N-terminal Amyloid Precursor Protein, Reduces β-Amyloid-Associated Pathology, and Improves Cognition. Biol. Psychiatry 2019, 86, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Hauptmann, S.; Keil, U.; Scherping, I.; Bonert, A.; Eckert, A.; Müller, W.E. Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol. 2006, 41, 668–673. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef] [PubMed]
- KIM, H.-S. Carboxyl-terminal fragment of Alzheimer’s APP destabilizes calcium homeostasis and renders neuronal cells vulnerable to excitotoxicity. FASEB J. 2000, 14, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Rogers, J. Anti–inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 1992, 42, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2019, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Lyubartseva, G.; Lovell, M.A. A potential role for zinc alterations in the pathogenesis of Alzheimer’s disease. Biofactors 2012, 38, 98–106. [Google Scholar] [CrossRef]
- Abelein, A.; Gräslund, A.; Danielsson, J. Zinc as chaperone-mimicking agent for retardation of amyloid β peptide fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, 5407–5412. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Papadopoulos, Z.; Herz, J.; Kipnis, J. Meningeal Lymphatics: From Anatomy to Central Nervous System Immune Surveillance. J. Immunol. 2020, 204, 286–293. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; et al. Microbes and Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 51, 979–984. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J. 2017, 31, 3216–3226. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-G.; Wang, J.-L.; Li, L.; Wang, P.-C. MicroRNA-384 regulates both amyloid precursor protein and β-secretase expression and is a potential biomarker for Alzheimer’s disease. Int. J. Mol. Med. 2014, 34, 160–166. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-339-5p down-regulates protein expression of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J. Biol. Chem. 2014, 289, 5184–5198. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Woldemichael, B.T.; Mansuy, I.M. Micro-RNAs in cognition and cognitive disorders: Potential for novel biomarkers and therapeutics. Biochem. Pharmacol. 2016, 104, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.D.; Zhang, S.; Hao, J.J.; Xie, T.R.; Kang, J.S. Cellular model of neuronal atrophy induced by DYNC1I1 deficiency reveals protective roles of RAS-RAF-MEK signaling. Protein Cell 2016, 7, 638–650. [Google Scholar] [CrossRef]
- La Joie, R.; Perrotin, A.; Barré, L.; Hommet, C.; Mézenge, F.; Ibazizene, M.; Camus, V.; Abbas, A.; Landeau, B.; Guilloteau, D.; et al. Region-specific hierarchy between atrophy, hypometabolism, and 2-amyloid (Aβ) load in Alzheimer’s disease dementia. J. Neurosci. 2012, 32, 16265–16273. [Google Scholar] [CrossRef]
- Dyrba, M.; Mohammadi, R.; Grothe, M.J.; Kirste, T.; Teipel, S.J. Gaussian Graphical Models Reveal Inter-Modal and Inter-Regional Conditional Dependencies of Brain Alterations in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Mensah-Nyagan, A.G.; Eckert, A. Alzheimer, mitochondria and gender. Neurosci. Biobehav. Rev. 2016, 67, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Seiça, R.M.; Moreira, P.I. Mitochondria as a target for neuroprotection: Implications for Alzheimer’s disease. Expert Rev. Neurother. 2017, 17, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Lejri, I.; Hallé, F.; Schmitt, M.; Götz, J.; Bihel, F.; Eckert, A. Mitochondria modulatory effects of new TSPO ligands in a cellular model of tauopathies. J. Neuroendocrinol. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Glasgow, N.G.; Povysheva, N.V. Recent insights into the mode of action of memantine and ketamine. Curr. Opin. Pharmacol. 2015, 20, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.Y.; Fu, W.M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24. [Google Scholar] [CrossRef]
- Melchiorre, C.; Andrisano, V.; Bolognesi, M.L.; Budriesi, R.; Cavalli, A.; Cavrini, V.; Rosini, M.; Tumiatti, V.; Recanatini, M. Acetylcholinesterase noncovalent inhibitors based on a polyamine backbone for potential use against Alzheimer’s disease. J. Med. Chem. 1998, 41, 4186–4189. [Google Scholar] [CrossRef]
- Marco-Contelles, J. Facts, Results, and Perspectives of the Current Alzheimer’s Disease Research. ACS Chem. Neurosci. 2019, 10, 1127–1128. [Google Scholar] [CrossRef]
- Kim, J.; Onstead, L.; Randle, S.; Price, R.; Smithson, L.; Zwizinski, C.; Dickson, D.W.; Golde, T.; McGowan, E. Aβ40 inhibits amyloid deposition in vivo. J. Neurosci. 2007, 27, 627–633. [Google Scholar] [CrossRef]
- Ghezzi, L.; Scarpini, E.; Galimberti, D. Disease-modifying drugs in Alzheimer’s disease. Drug Des. Devel. Ther. 2013, 7, 1471–1478. [Google Scholar] [CrossRef]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Price, D.L.; Tanzi, R.E.; Borchelt, D.R.; Sisodia, S.S. ALZHEIMER’S DISEASE: Genetic Studies and Transgenic Models. Annu. Rev. Genet. 1998, 32, 461–493. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Zecca, C.; Barull, M.R.; Bellomo, A.; Pilotto, A.; et al. Tau-Centric Targets and Drugs in Clinical Development for the Treatment of Alzheimer’s Disease. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- Avila, J.; Wandosell, F.; Hernández, F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 2010, 10, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem. 2012, 120, 9–21. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of Glycogen Synthase Kinase 3 in Alzheimer’s Disease. Curr. Alzheimer Res. 2012, 9, 864–879. [Google Scholar] [CrossRef]
- Rossner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef]
- Sun, X.; Bromley-Brits, K.; Song, W. Regulation of β-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s disease. J. Neurochem. 2012, 120, 62–70. [Google Scholar] [CrossRef]
- Ly, P.T.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Godridge, H.; Reynolds, G.P.; Czudek, C.; Calcutt, N.A.; Benton, M. Alzheimer-like neurotransmitter deficits in adult Down’s syndrome brain tissue. J. Neurol. Neurosurg. Psychiatry 1987, 50, 775–778. [Google Scholar] [CrossRef] [PubMed]
- H. Ferreira-Vieira, T.; M. Guimaraes, I.; R. Silva, F.; M. Ribeiro, F. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.J.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor—Mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Dunger, N.; Schölmerich, J.; Falk, W.; Obermeier, F. Glycogen synthase kinase 3-β: A master regulator of toll-like receptor-mediated chronic intestinal inflammation. Inflamm. Bowel Dis. 2010, 16, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, E.; Tang, Y.P.; Rampon, C.; Tsien, J.Z. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science 2000, 290, 1170–1174. [Google Scholar] [CrossRef]
- Geerts, H.; Grossberg, G.T. Pharmacology of acetylcholinesterase inhibitors and N-methyl-D-aspartate receptors for combination therapy in the treatment of Alzheimer’s disease. J. Clin. Pharmacol. 2006, 46. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Inaba-Hasegawa, K. Type A and B monoamine oxidase in age-related neurodegenerative disorders: Their distinct roles in neuronal death and survival. Curr. Top. Med. Chem. 2012, 12, 2177–2188. [Google Scholar] [CrossRef]
- Naoi, M.; Riederer, P.; Maruyama, W. Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: Genetic and environmental factors involved in type A MAO expression. J. Neural Transm. 2016, 123, 91–106. [Google Scholar] [CrossRef]
- Hirvonen, J.; Kailajärvi, M.; Haltia, T.; Koskimies, S.; Någren, K.; Virsu, P.; Oikonen, V.; Sipilä, H.; Ruokoniemi, P.; Virtanen, K.; et al. Assessment of MAO-B Occupancy in the Brain With PET and [11C]-L-Deprenyl-D2: A Dose-Finding Study With a Novel MAO-B Inhibitor, EVT 301. Clin. Pharmacol. Ther. 2009, 85, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H.; Fridkin, M.; Zheng, H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech. Ageing Dev. 2005, 126, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147. [Google Scholar] [CrossRef]
- Riederer, P.; Danielczyk, W.; Grünblatt, E. Monoamine Oxidase-B Inhibition in Alzheimer’s Disease. Neurotoxicology 2004, 25, 271–277. [Google Scholar] [CrossRef]
- Gökhan-Kelekçi, N.; Yabanoǧlu, S.; Küpeli, E.; Salgin, U.; Özgen, Ö.; Uçar, G.; Yeşilada, E.; Kendi, E.; Yeşilada, A.; Bilgin, A.A. A new therapeutic approach in Alzheimer disease: Some novel pyrazole derivatives as dual MAO-B inhibitors and antiinflammatory analgesics. Bioorg. Med. Chem. 2007, 15, 5775–5786. [Google Scholar] [CrossRef]
- Crouch, P.J.; White, A.R.; Bush, A.I. The modulation of metal bio-availability as a therapeutic strategy for the treatment of Alzheimer’s disease. FEBS J. 2007, 274, 3775–3783. [Google Scholar] [CrossRef]
- Kepp, K.P. Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Lu, Z.; Zheng, X.; Ni, W.; Zhu, J.; Fu, Y.; Lian, F.; Zhang, N.; Li, J.; et al. Development of Multifunctional Pyrimidinylthiourea Derivatives as Potential Anti-Alzheimer Agents. J. Med. Chem. 2016, 59, 8326–8344. [Google Scholar] [CrossRef]
- Basha, S.J.; Mohan, P.; Yeggoni, D.P.; Babu, Z.R.; Kumar, P.B.; Rao, A.D.; Subramanyam, R.; Damu, A.G. New Flavone-Cyanoacetamide Hybrids with a Combination of Cholinergic, Antioxidant, Modulation of β-Amyloid Aggregation, and Neuroprotection Properties as Innovative Multifunctional Therapeutic Candidates for Alzheimer’s Disease and Unraveling Their Mechan. Mol. Pharm. 2018, 15, 2206–2223. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Marjo, C.; Rutlidge, H.; Rich, A.; Jayasena, T.; Inestrosa, N.C.; Sachdev, P. Metal and complementary molecular bioimaging in alzheimer’s disease. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Matsumoto, M.; Togashi, H.; Mori, K.; Ueno, K.; Ohashi, S.; Kojima, T.; Yoshioka, M. Evidence for Involvement of Central 5-HT4 Receptors in Cholinergic Function Associated with Cognitive Processes: Behavioral, Electrophysiological, and Neurochemical Studies. J. Pharmacol. Exp. Ther. 2001, 296, 676–682. [Google Scholar] [PubMed]
- Upton, N.; Chuang, T.T.; Hunter, A.J.; Virley, D.J. 5-HT6 Receptor Antagonists as Novel Cognitive Enhancing Agents for Alzheimer’s Disease. Neurotherapeutics 2008, 5, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Lezoualc’h, F. 5-HT4 receptor and Alzheimer’s disease: The amyloid connection. Exp. Neurol. 2007, 205, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Fréon, A.M.; et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor (5-HT4R) Agonist Activities As Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef]
- Kubo, M.; Kishi, T.; Matsunaga, S.; Iwata, N. Histamine H3 Receptor Antagonists for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. J. Alzheimer’s Dis. 2015, 48, 667–671. [Google Scholar] [CrossRef]
- Prickaerts, J.; Heckman, P.R.A.; Blokland, A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 1033–1048. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Z.; Huang, Y.-Y.; Wu, D.; Luo, H.-B. Novel Phosphodiesterase Inhibitors for Cognitive Improvement in Alzheimer’s Disease. J. Med. Chem. 2018, 61, 5467–5483. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Talarek, S.; Listos, J.; Nabavi, S.F.; Devi, K.P.; Roberto de Oliveira, M.; Tewari, D.; Argüelles, S.; Mehrzadi, S.; Hosseinzadeh, A.; et al. Phosphodiesterase inhibitors say NO to Alzheimer’s disease. Food Chem. Toxicol. 2019, 134. [Google Scholar] [CrossRef]
- Arai, H.; Takahashi, T. A combination therapy of Donepezil and Cilostazol for patients with moderate Alzheimer disease: Pilot follow-up study. Am. J. Geriatr. Psychiatry 2009, 17, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Nishino, M.; Taguchi, A.; Yamamoto, Y.; Hattori, Y.; Saito, S.; Takahashi, Y.; Tsuji, M.; Kasahara, Y.; Takata, Y.; et al. Cilostazol add-on therapy in patients with mild dementia receiving donepezil: A retrospective study. PLoS ONE 2014, 9, e89516. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Maurya, P.K.; Yadav, B.S.; Singh, S.; Mani, A. Current Therapeutic Targets for Alzheimer’s Disease. J. Biomed. 2018, 3, 74–84. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Melchiorre, C. Memoquin: A Multi-Target-Directed Ligand as an Innovative Therapeutic Opportunity for Alzheimer’s Disease. Neurotherapeutics 2009, 6, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Capurro, V.; Busquet, P.; Lopes, J.P.; Bertorelli, R.; Tarozzo, G.; Bolognesi, M.L.; Piomelli, D.; Reggiani, A.; Cavalli, A. Pharmacological Characterization of Memoquin, a Multi-Target Compound for the Treatment of Alzheimer’s Disease. PLoS ONE 2013, 8, e56870. [Google Scholar] [CrossRef]
- Pan, W.; Hu, K.; Bai, P.; Yu, L.; Ma, Q.; Li, T.; Zhang, X.; Chen, C.; Peng, K.; Liu, W.; et al. Design, synthesis and evaluation of novel ferulic acid-memoquin hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2016, 26, 2539–2543. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Maspero, M.; Volpato, D.; Cirillo, D.; Yuan Chen, N.; Messerer, R.; Sotriffer, C.; De Amici, M.; Holzgrabe, U.; Dallanoce, C. Tacrine-xanomeline and tacrine-iperoxo hybrid ligands: Synthesis and biological evaluation at acetylcholinesterase and M1 muscarinic acetylcholine receptors. Bioorg. Chem. 2020, 96, 103633. [Google Scholar] [CrossRef]
- Oukoloff, K.; Coquelle, N.; Bartolini, M.; Naldi, M.; Le Guevel, R.; Bach, S.; Josselin, B.; Ruchaud, S.; Catto, M.; Pisani, L.; et al. Design, biological evaluation and X-ray crystallography of nanomolar multifunctional ligands targeting simultaneously acetylcholinesterase and glycogen synthase kinase-3. Eur. J. Med. Chem. 2019, 168, 58–77. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Hong, K.H.; Ning, Y.; Yu, P.; Ren, J.; Ji, M.; Cai, J. Design, synthesis and evaluation of a novel metal chelator as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 87, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Q.; Zhu, K.K.; Zhang, J.; Song, J.L.; Muehlmann, L.A.; Jiang, C.S.; Liu, C.L.; Zhang, H. Molecular-docking-guided design and synthesis of new IAA-tacrine hybrids as multifunctional AChE/BChE inhibitors. Bioorg. Chem. 2019, 83, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Q.; Song, J.L.; Zhu, K.; Zhang, J.; Jiang, C.S.; Zhang, H. Total synthesis of pulmonarin B and design of brominated phenylacetic acid/tacrine hybrids: Marine pharmacophore inspired discovery of new CHE and Aβ aggregation inhibitors. Mar. Drugs 2018, 16, 293. [Google Scholar] [CrossRef]
- Lopes, J.P.B.; Silva, L.; da Costa Franarin, G.; Antonio Ceschi, M.; Seibert Lüdtke, D.; Ferreira Dantas, R.; de Salles, C.M.C.; Paes Silva-Jr, F.; Roberto Senger, M.; Alvim Guedes, I.; et al. Design, synthesis, cholinesterase inhibition and molecular modelling study of novel tacrine hybrids with carbohydrate derivatives. Bioorg. Med. Chem. 2018, 26, 5566–5577. [Google Scholar] [CrossRef] [PubMed]
- Chufarova, N.; Czarnecka, K.; Skibiński, R.; Cuchra, M.; Majsterek, I.; Szymański, P. New tacrine-acridine hybrids as promising multifunctional drugs for potential treatment of Alzheimer’s disease. Arch. Pharm. (Weinheim.) 2018, 351, 1800050. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Radchenko, E.V.; et al. Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer’s disease treatment: Synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg. Chem. 2020, 94, 103387. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, H.; Chen, Y.; Lin, H.; Li, Q.; Mo, J.; Bian, Y.; Pei, Y.; Sun, H. Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2018, 33, 496–506. [Google Scholar] [CrossRef]
- Habibpour, R.; Eslami, M.; Amani, P.; Bagheri Novir, S. Tacrine-Flavonoid Quercetin Hybride as a MTDL ligand against Alzheimer’s disease with metal chelating and AChE, BChE, AChE-induced Aβ aggregation inhibition properties: A computational study. Phys. Chem. Res. 2019, 7, 561–579. [Google Scholar] [CrossRef]
- Hepnarova, V.; Korabecny, J.; Matouskova, L.; Jost, P.; Muckova, L.; Hrabinova, M.; Vykoukalova, N.; Kerhartova, M.; Kucera, T.; Dolezal, R.; et al. The concept of hybrid molecules of tacrine and benzyl quinolone carboxylic acid (BQCA) as multifunctional agents for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 292–306. [Google Scholar] [CrossRef]
- Costa, M.; Josselin, R.; Silva, D.F.; Cardoso, S.M.; May, N.V.; Chaves, S.; Santos, M.A. Donepezil-based hybrids as multifunctional anti-Alzheimer’s disease chelating agents: Effect of positional isomerization. J. Inorg. Biochem. 2020, 206, 111039. [Google Scholar] [CrossRef]
- Chaves, S.; Resta, S.; Rinaldo, F.; Costa, M.; Josselin, R.; Gwizdala, K.; Piemontese, L.; Capriati, V.; Pereira-Santos, A.R.; Cardoso, S.M.; et al. Design, synthesis, and in vitro evaluation of hydroxybenzimidazole-donepezil analogues as multitarget-directed ligands for the treatment of Alzheimer’s disease. Molecules 2020, 25, 985. [Google Scholar] [CrossRef]
- Cai, P.; Fang, S.Q.; Yang, H.L.; Yang, X.L.; Liu, Q.H.; Kong, L.Y.; Wang, X.B. Donepezil-butylated hydroxytoluene (BHT) hybrids as Anti-Alzheimer’s disease agents with cholinergic, antioxidant, and neuroprotective properties. Eur. J. Med. Chem. 2018, 157, 161–176. [Google Scholar] [CrossRef]
- Cai, P.; Fang, S.Q.; Yang, X.L.; Wu, J.J.; Liu, Q.H.; Hong, H.; Wang, X.B.; Kong, L.Y. Rational Design and Multibiological Profiling of Novel Donepezil-Trolox Hybrids against Alzheimer’s Disease, with Cholinergic, Antioxidant, Neuroprotective, and Cognition Enhancing Properties. ACS Chem. Neurosci. 2017, 8, 2496–2511. [Google Scholar] [CrossRef]
- Wang, X.B.; Yin, F.C.; Huang, M.; Jiang, N.; Lan, J.S.; Kong, L.Y. Chromone and donepezil hybrids as new multipotent cholinesterase and monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. RSC Med. Chem. 2020, 11, 225–233. [Google Scholar] [CrossRef]
- Malek, R.; Refouvelet, B.; Benchekroun, M.; Iriepa, I.; Moraleda, I.; Andrys, R.; Musilek, K.; Marco-Contelles, J.; Ismaili, L. Synthesis and Biological Evaluation of Novel Chromone+Donepezil Hybrids for Alzheimer’s Disease Therapy. Curr. Alzheimer Res. 2019, 16, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Thamban Chandrika, N.; Fosso, M.Y.; Tsodikov, O.V.; LeVine, H.; Garneau-Tsodikova, S. Combining Chalcones with Donepezil to Inhibit Both Cholinesterases and Aβ Fibril Assembly. Molecules 2019, 25, 77. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, K.; Shi, J.; Zhang, P.; Yang, D.; Fan, X.; Zhang, Z.; Liu, W.; Sang, Z. The development of 2-acetylphenol-donepezil hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2019, 29, 126625. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Liu, X.; Xie, J.; Ma, F. Novel Deoxyvasicinone-Donepezil Hybrids as Potential Multitarget Drug Candidates for Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; Abdel-Raziq, M.S. Design and synthesis of donepezil analogues as dual AChE and BACE-1 inhibitors. Bioorg. Chem. 2018, 80, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Tomás, D.; Hiremathad, A.; Capriati, V.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enzyme Inhib. Med. Chem. 2018, 33, 1212–1224. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.M.; Li, X.M.; Li, F.; Wu, J.J.; Kong, L.Y.; Wang, X.B. Synthesis and evaluation of multi-target-directed ligands for the treatment of Alzheimer’s disease based on the fusion of donepezil and melatonin. Bioorg. Med. Chem. 2016, 24, 4324–4338. [Google Scholar] [CrossRef] [PubMed]
- Girek, M.; Szymański, P. Tacrine hybrids as multi-target-directed ligands in Alzheimer’s disease: Influence of chemical structures on biological activities. Chem. Pap. 2019, 73, 269–289. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Nepovimova, E.; Dolezal, R.; Mezeiova, E.; Soukup, O.; Kuca, K. Multitarget Tacrine Hybrids with Neuroprotective Properties to Confront Alzheimer’s Disease; Bentham Science Publishers: Sharjah, UAE, 2017; Volume 17. [Google Scholar]
- Mahdavi, M.; Hariri, R.; Mirfazli, S.S.; Lotfian, H.; Rastergari, A.; Firuzi, O.; Edraki, N.; Larijani, B.; Akbarzadeh, T.; Saeedi, M. Synthesis and Biological Activity of Some Benzochromenoquinolinones: Tacrine Analogs as Potent Anti-Alzheimer’s Agents. Chem. Biodivers. 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Aso, E.; Frattini, D.; López-González, I.; Espargaró, A.; Sabaté, R.; Di Pietro, O.; Luque, F.J.; Clos, M.V.; Ferrer, I.; et al. Novel Levetiracetam Derivatives That Are Effective against the Alzheimer-like Phenotype in Mice: Synthesis, in Vitro, ex Vivo, and in Vivo Efficacy Studies. J. Med. Chem. 2015, 58, 6018–6032. [Google Scholar] [CrossRef]
- Zha, X.; Lamba, D.; Zhang, L.; Lou, Y.; Xu, C.; Kang, D.; Chen, L.; Xu, Y.; Zhang, L.; De Simone, A.; et al. Novel Tacrine-Benzofuran Hybrids as Potent Multitarget-Directed Ligands for the Treatment of Alzheimers Disease: Design, Synthesis, Biological Evaluation, and X-ray Crystallography. J. Med. Chem. 2016, 59, 114–131. [Google Scholar] [CrossRef]
- Liao, S.; Deng, H.; Huang, S.; Yang, J.; Wang, S.; Yin, B.; Zheng, T.; Zhang, D.; Liu, J.; Gao, G.; et al. Design, synthesis and evaluation of novel 5,6,7-trimethoxyflavone-6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015, 25, 1541–1545. [Google Scholar] [CrossRef]
- Green, K.D.; Fosso, M.Y.; Garneau-Tsodikova, S. Multifunctional donepezil analogues as cholinesterase and BACE1 inhibitors. Molecules 2018, 23, 3252. [Google Scholar] [CrossRef]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef]
- Irannejad, H.; Amini, M.; Khodagholi, F.; Ansari, N.; Tusi, S.K.; Sharifzadeh, M.; Shafiee, A. Synthesis and in vitro evaluation of novel 1,2,4-triazine derivatives as neuroprotective agents. Bioorg. Med. Chem. 2010, 18, 4224–4230. [Google Scholar] [CrossRef]
- Sinha, A.; Tamboli, R.S.; Seth, B.; Kanhed, A.M.; Tiwari, S.K.; Agarwal, S.; Nair, S.; Giridhar, R.; Chaturvedi, R.K.; Yadav, M.R. Neuroprotective Role of Novel Triazine Derivatives by Activating Wnt/β Catenin Signaling Pathway in Rodent Models of Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 638–652. [Google Scholar] [CrossRef]
- Shidore, M.; Machhi, J.; Shingala, K.; Murumkar, P.; Sharma, M.K.; Agrawal, N.; Tripathi, A.; Parikh, Z.; Pillai, P.; Yadav, M.R. Benzylpiperidine-Linked Diarylthiazoles as Potential Anti-Alzheimer’s Agents: Synthesis and Biological Evaluation. J. Med. Chem. 2016, 59, 5823–5846. [Google Scholar] [CrossRef] [PubMed]
- Więckowska, A.; Więckowski, K.; Bajda, M.; Brus, B.; Sałat, K.; Czerwińska, P.; Gobec, S.; Filipek, B.; Malawska, B. Synthesis of new N-benzylpiperidine derivatives as cholinesterase inhibitors with β-amyloid anti-aggregation properties and beneficial effects on memory in vivo. Bioorg. Med. Chem. 2015, 23, 2445–2457. [Google Scholar] [CrossRef]
- Panek, D.; Więckowska, A.; Wichur, T.; Bajda, M.; Godyń, J.; Jończyk, J.; Mika, K.; Janockova, J.; Soukup, O.; Knez, D.; et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur. J. Med. Chem. 2017, 125, 676–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.M.; Cai, P.; Liu, Q.H.; Xu, D.Q.; Yang, X.L.; Wu, J.J.; Kong, L.Y.; Wang, X.B. Rational modification of donepezil as multifunctional acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2016, 123, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Kumari, S.; Manral, A.; Prakash, A.; Saini, V.; Lynn, A.M.; Tiwari, M. Design, synthesis, in-silico and biological evaluation of novel donepezil derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 125, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Panek, D.; Wichur, T.; Godyń, J.; Pasieka, A.; Malawska, B. Advances toward multifunctional cholinesterase and β-amyloid aggregation inhibitors. Future Med. Chem. 2017, 9, 1835–1854. [Google Scholar] [CrossRef]
- Fernández-Bachiller, M.I.; Pérez, C.; Monjas, L.; Rademann, J.; Rodríguez-Franco, M.I. New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with cholinergic, antioxidant, and β-amyloid-reducing properties. J. Med. Chem. 2012, 55, 1303–1317. [Google Scholar] [CrossRef]
- Pérez-Areales, F.J.; Betari, N.; Viayna, A.; Pont, C.; Espargaró, A.; Bartolini, M.; De Simone, A.; Rinaldi Alvarenga, J.F.; Pérez, B.; Sabate, R.; et al. Design, synthesis and multitarget biological profiling of second-generation anti-Alzheimer rhein-huprine hybrids. Future Med. Chem. 2017, 9, 965–981. [Google Scholar] [CrossRef]
- Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F.G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F.J.; et al. Synthesis and multitarget biological profiling of a novel family of rhein derivatives as disease-modifying anti-Alzheimer agents. J. Med. Chem. 2014, 57, 2549–2567. [Google Scholar] [CrossRef]
- Sivaprakasam, P.; Han, X.; Civiello, R.L.; Jacutin-Porte, S.; Kish, K.; Pokross, M.; Lewis, H.A.; Ahmed, N.; Szapiel, N.; Newitt, J.A.; et al. Discovery of new acylaminopyridines as GSK-3 inhibitors by a structure guided in-depth exploration of chemical space around a pyrrolopyridinone core. Bioorg. Med. Chem. Lett. 2015, 25, 1856–1863. [Google Scholar] [CrossRef]
- Wu, W.Y.; Dai, Y.C.; Li, N.G.; Dong, Z.X.; Gu, T.; Shi, Z.H.; Xue, X.; Tang, Y.P.; Duan, J.A. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2016, 32, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Bolea, I.; Juárez-Jiménez, J.; De Los Ríos, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(Benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s di. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Esteban, G.; Ojima, M.; Bautista-Aguilera, O.M.; Inokuchi, T.; Moraleda, I.; Iriepa, I.; Samadi, A.; Youdim, M.B.H.; Romero, A.; et al. Donepezil + propargylamine + 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 80, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Esteban, G.; Brogi, S.; Shionoya, M.; Wang, L.; Campiani, G.; Unzeta, M.; Inokuchi, T.; Butini, S.; Marco-Contelles, J. Donepezil-like multifunctional agents: Design, synthesis, molecular modeling and biological evaluation. Eur. J. Med. Chem. 2016, 121, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Estrada, M.; Herrera-Arozamena, C.; Pérez, C.; Viña, D.; Romero, A.; Morales-García, J.A.; Pérez-Castillo, A.; Rodríguez-Franco, M.I. New cinnamic—N-benzylpiperidine and cinnamic—N,N-dibenzyl(N-methyl)amine hybrids as Alzheimer-directed multitarget drugs with antioxidant, cholinergic, neuroprotective and neurogenic properties. Eur. J. Med. Chem. 2016, 121, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhou, Q.; Yan, J.; Du, Z.; Huang, L.; Li, X. A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 62, 745–753. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Guo, Y.; Wang, Z.; Huang, L.; Li, X. Dual functional cholinesterase and MAO inhibitors for the treatment of Alzheimers disease: Synthesis, pharmacological analysis and molecular modeling of homoisoflavonoid derivatives. J. Enzyme Inhib. Med. Chem. 2016, 31, 389–397. [Google Scholar] [CrossRef]
- Benek, O.; Soukup, O.; Pasdiorova, M.; Hroch, L.; Sepsova, V.; Jost, P.; Hrabinova, M.; Jun, D.; Kuca, K.; Zala, D.; et al. Design, Synthesis and in vitro Evaluation of Indolotacrine Analogues as Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease. ChemMedChem 2016, 11, 1264–1269. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, H.; Li, X.; Dong, S.; Liu, W.; Gong, Q.; Wang, T.; Tang, Y.; Zhu, J.; Li, J.; et al. Discovery of novel propargylamine-modified 4-aminoalkyl imidazole substituted pyrimidinylthiourea derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 143, 33–47. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Neuroprotective effects of multifaceted hybrid agents targeting MAO, cholinesterase, iron and β-amyloid in ageing and Alzheimer’s disease. Br. J. Pharmacol. 2016, 173, 2080–2094. [Google Scholar] [CrossRef]
- Safety and Efficacy Study of Ladostigil in Mild to Moderate Probable Alzheimer’s Disease—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01354691 (accessed on 28 May 2020).
- Li, Y.; Qiang, X.; Luo, L.; Yang, X.; Xiao, G.; Zheng, Y.; Cao, Z.; Su, F.; Deng, Y.; Sang, Z. Multitarget drug design strategy against Alzheimer’s disease: Homoisoflavonoid Mannich base derivatives serve as acetylcholinesterase and monoamine oxidase B dual inhibitors with multifunctional properties. Bioorg. Med. Chem. 2017, 25, 714–726. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Minarini, A.; Melchiorre, C. Multi-target Design Strategies in the Context of Alzheimer’s Disease: Acetylcholinesterase Inhibition and NMDA Receptor Antagonism as the Driving Forces. Neurochem. Res. 2014, 39, 1914–1923. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Cavalli, A.; Ceccarini, L.; Pascu, N.; McClymont, D.W.; Tarozzi, A.; Bolognesi, M.L.; Minarini, A.; et al. Inhibition of acetylcholinesterase, β-amyloid aggregation, and NMDA receptors in Alzheimer’s disease: A promising direction for the multi-target-directed ligands gold rush. J. Med. Chem. 2008, 51, 4381–4384. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Soriano, E.; Marco-Contelles, J.; Andrisano, V.; Monti, B.; Windisch, M.; Hutter-Paier, B.; Mcclymont, D.W.; et al. The bivalent ligand approach as a tool for improving the in vitro anti-alzheimer multitarget profile of dimebon. ChemMedChem 2013, 8, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Sokolov, V.B.; Grigoriev, V.V.; Serebryakova, O.G.; Vikhareva, E.A.; Aksinenko, A.Y.; Barreto, G.E.; Aliev, G.; et al. Conjugates of γ 3-Carbolines and Phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer Disease. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Yun, H.-M.; Rhim, H. The Serotonin-6 Receptor as a Novel Therapeutic Target. Exp. Neurobiol. 2011, 20, 159–168. [Google Scholar] [CrossRef]
- Claeysen, S.; Bockaert, J.; Giannoni, P. Serotonin: A new hope in Alzheimer’s disease? ACS Chem. Neurosci. 2015, 6, 940–943. [Google Scholar] [CrossRef]
- Freret, T.; Bouet, V.; Quiedeville, A.; Nee, G.; Dallemagne, P.; Rochais, C.; Boulouard, M. Synergistic effect of acetylcholinesterase inhibition (donepezil) and 5-HT 4 receptor activation (RS67333) on object recognition in mice. Behav. Brain Res. 2012, 230, 304–308. [Google Scholar] [CrossRef]
- Charlier, Y.; Brabant, C.; Serrano, M.E.; Lamberty, Y.; Tirelli, E. The prototypical histamine H3 receptor inverse agonist thioperamide improves multiple aspects of memory processing in an inhibitory avoidance task. Behav. Brain Res. 2013, 253, 121–127. [Google Scholar] [CrossRef]
- Inocente, C.; Arnulf, I.; Bastuji, H.; Thibault-Stoll, A.; Raoux, A.; Reimão, R.; Lin, J.S.; Franco, P. Pitolisant, an inverse agonist of the histamine H3 receptor: An alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin. Neuropharmacol. 2012, 35, 55–60. [Google Scholar] [CrossRef]
- Syed, Y.Y. Pitolisant: First Global Approval. Drugs 2016, 76, 1313–1318. [Google Scholar] [CrossRef]
- Bajda, M.; Łażewska, D.; Godyń, J.; Zaręba, P.; Kuder, K.; Hagenow, S.; Łątka, K.; Stawarska, E.; Stark, H.; Kieć-Kononowicz, K.; et al. Search for new multi-target compounds against Alzheimer’s disease among histamine H3 receptor ligands. Eur. J. Med. Chem. 2020, 185, 111785. [Google Scholar] [CrossRef]
- Huang, W.; Tang, L.; Shi, Y.; Huang, S.; Xu, L.; Sheng, R.; Wu, P.; Li, J.; Zhou, N.; Hu, Y. Searching for the Multi-Target-Directed Ligands against Alzheimer’s disease: Discovery of quinoxaline-based hybrid compounds with AChE, H 3R and BACE 1 inhibitory activities. Bioorg. Med. Chem. 2011, 19, 7158–7167. [Google Scholar] [CrossRef]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimers disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef]
- Li, S.Y.; Wang, X.B.; Xie, S.S.; Jiang, N.; Wang, K.D.G.; Yao, H.Q.; Sun, H.B.; Kong, L.Y. Multifunctional tacrine-flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 69, 632–646. [Google Scholar] [CrossRef]
- Xie, S.S.; Wang, X.B.; Li, J.Y.; Yang, L.; Kong, L.Y. Design, synthesis and evaluation of novel tacrine-coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur. J. Med. Chem. 2013, 64, 540–553. [Google Scholar] [CrossRef]
- Yan, J.; Hu, J.; Liu, A.; He, L.; Li, X.; Wei, H. Design, synthesis, and evaluation of multitarget-directed ligands against Alzheimer’s disease based on the fusion of donepezil and curcumin. Bioorg. Med. Chem. 2017, 25, 2946–2955. [Google Scholar] [CrossRef]
- Wichur, T.; Więckowska, A.; Więckowski, K.; Godyń, J.; Jończyk, J.; Valdivieso, Á.D.R.; Panek, D.; Pasieka, A.; Sabaté, R.; Knez, D.; et al. 1-Benzylpyrrolidine-3-amine-based BuChE inhibitors with anti-aggregating, antioxidant and metal-chelating properties as multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2020, 187, 111916. [Google Scholar] [CrossRef]
- Fuse, S.; Matsumura, K.; Fujita, Y.; Sugimoto, H.; Takahashi, T. Development of dual targeting inhibitors against aggregations of amyloid-β and tau protein. Eur. J. Med. Chem. 2014, 85, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; De Simone, A.; Armirotti, A.; Summa, M.; Pizzirani, D.; Scarpelli, R.; Bertozzi, S.M.; Perez, D.I.; Andrisano, V.; Perez-Castillo, A.; et al. 3,4-Dihydro-1,3,5-triazin-2(1H)-ones as the First Dual BACE-1/GSK-3β Fragment Hits against Alzheimer’s Disease. ACS Chem. Neurosci. 2015, 6, 1665–1682. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Veronesi, M.; Bisignano, P.; Kacker, P.; Favia, A.D.; Cavalli, A. Development and Application of a Virtual Screening Protocol for the Identification of Multitarget Fragments. ChemMedChem 2016, 11, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, R.M.C.; De Simone, A.; Andrisano, V.; Bisignano, P.; Bisi, A.; Gobbi, S.; Rampa, A.; Fato, R.; Bergamini, C.; Perez, D.I.; et al. Versatility of the Curcumin Scaffold: Discovery of Potent and Balanced Dual BACE-1 and GSK-3β Inhibitors. J. Med. Chem. 2016, 59, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Chen, J.; Li, X.; Su, T.; Wang, Y.; Wang, Z.; Huang, L.; Li, X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Wang, B.; Li, W.; Huang, L.; Li, X. Design, synthesis, and evaluation of orally available clioquinol-moracin M hybrids as multitarget-directed ligands for cognitive improvement in a rat model of neurodegeneration in Alzheimer’s disease. J. Med. Chem. 2015, 58, 8616–8637. [Google Scholar] [CrossRef]
- AlFadly, E.D.; Elzahhar, P.A.; Tramarin, A.; Elkazaz, S.; Shaltout, H.; Abu-Serie, M.M.; Janockova, J.; Soukup, O.; Ghareeb, D.A.; El-Yazbi, A.F.; et al. Tackling neuroinflammation and cholinergic deficit in Alzheimer’s disease: Multi-target inhibitors of cholinesterases, cyclooxygenase-2 and 15-lipoxygenase. Eur. J. Med. Chem. 2019, 167, 161–186. [Google Scholar] [CrossRef]
- Castanho, I.; Lunnon, K. Epigenetic Processes in Alzheimer’s Disease. In Chromatin Signaling and Neurological Disorders; Elsevier: Amsterdam, The Netherlands, 2019; pp. 153–180. [Google Scholar]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Maes, T.; Mascaró, C.; Rotllant, D.; Lufino, M.M.P.; Estiarte, A.; Guibourt, N.; Cavalcanti, F.; Griñan-Ferré, C.; Pallàs, M.; Nadal, R.; et al. Modulation of KDM1A with vafidemstat rescues memory deficit and behavioral alterations. PLoS ONE 2020, 15, e0233468. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sánchez-Arias, J.A.; Rabal, O.; Pérez-González, M.; Mederos, S.; Ugarte, A.; Franco, R.; Segura, V.; Perea, G.; et al. A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer’s Disease Mice. Neuropsychopharmacology 2016, 42, 524–539. [Google Scholar] [CrossRef]
- De Simone, A.; La Pietra, V.; Betari, N.; Petragnani, N.; Conte, M.; Daniele, S.; Pietrobono, D.; Martini, C.; Petralla, S.; Casadei, R.; et al. Discovery of the First-in-Class GSK-3β/HDAC Dual Inhibitor as Disease-Modifying Agent to Combat Alzheimer’s Disease. ACS Med. Chem. Lett. 2019, 10, 469–474. [Google Scholar] [CrossRef]
- Selkoe, D.J. Introducing transglutaminase into the study of Alzheimer’s disease. A personal look back. Neurochem Int. 2002, 40, 13–16. [Google Scholar] [CrossRef]
- Basso, M.; Chen, H.H.; Tripathy, D.; Conte, M.; Apperley, K.Y.P.; De Simone, A.; Keillor, J.W.; Ratan, R.; Nebbioso, A.; Sarno, F.; et al. Designing Dual Transglutaminase 2/Histone Deacetylase Inhibitors Effective at Halting Neuronal Death. ChemMedChem 2018, 13, 227–230. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2018, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Kumar, A.; Panda, G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Lan, J.S.; Xie, S.S.; Li, S.Y.; Pan, L.F.; Wang, X.B.; Kong, L.Y. Design, synthesis and evaluation of novel tacrine-(β-carboline) hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2014, 22, 6089–6104. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, H.-T.; Zhong, S.-M.; Wang, Z.-Y.; Chen, Z.-F.; Liang, H. Novel oxoisoaporphine-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation. Bioorg. Med. Chem. Lett. 2012, 22, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards Improvements for Penetrating the Blood–Brain Barrier—Recent Progress from a Material and Pharmaceutical Perspective. Cells 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Ávila, J.; Wang, H.; Cieslikiewicz-bouet, M.; Naldi, M.; Bartolini, M.; Pérez, B.; Servent, D.; et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Eur. J. Med. Chem. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Tavana, J.P.; Rosene, M.; Jensen, N.O.; Ridge, P.G.; Kauwe, J.S.K.; Karch, C.M. RAB10: An alzheimer’s disease resilience locus and potential drug target. Clin. Interv. Aging 2019, 14, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.K.; Lin, S.L.; Schooling, C.M. Re-thinking Alzheimer’s disease therapeutic targets using gene-based tests. EBioMedicine 2018, 37, 461–470. [Google Scholar] [CrossRef]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Rasagiline Rescue in Alzheimer’s Disease Clinical Trial—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02359552 (accessed on 24 June 2020).
- Study of LM11A-31-BHS in Mild-moderate AD Patients—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03069014 (accessed on 24 June 2020).
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Cell. Infect. Microbiol. 2019, 9, 233. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F.; Moore, Z.; Taylor, J.M.; Crack, P.J.; Hemonnot, A.L.; et al. Microglia in Alzheimer disease: Well-known targets and new opportunities. Cells 2019, 9, 3533–3543. [Google Scholar] [CrossRef]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef]
- Moore, Z.; Taylor, J.M.; Crack, P.J. The involvement of microglia in Alzheimer’s disease: A new dog in the fight. Br. J. Pharmacol. 2019, 176, 3533–3543. [Google Scholar] [CrossRef] [PubMed]
- Safety, Tolerability, and Pharmacokinetic Study of Single Ascending Doses of GC021109 in Healthy Subjects—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02254369 (accessed on 20 June 2020).
- Chen, Q.; Du, Y.; Zhang, K.; Liang, Z.; Li, J.; Yu, H.; Ren, R.; Feng, J.; Jin, Z.; Li, F.; et al. Tau-Targeted Multifunctional Nanocomposite for Combinational Therapy of Alzheimer’s Disease. ACS Nano 2018, 12, 1321–1338. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Xu, S.; Yang, P.; Wang, P.; Lu, S.; Sheng, D.; Qian, K.; Cao, J.; Lu, W.; Zhang, Q. A dual-ligand fusion peptide improves the brain-neuron targeting of nanocarriers in Alzheimer’s disease mice. J. Control. Release 2020, 320, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Guo, Z.; Zhang, Y.; Li, C.; Zhang, Y.; Guo, Q.; Chen, Q.; Chen, X.; He, X.; Liu, L.; et al. Microenvironment Remodeling Micelles for Alzheimer’s Disease Therapy by Early Modulation of Activated Microglia. Adv. Sci. 2019, 6. [Google Scholar] [CrossRef]
- Paolini, G.V.; Shapland, R.H.B.; Van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global mapping of pharmacological space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Simoni, E.; Daniele, S.; Bottegoni, G.; Pizzirani, D.; Trincavelli, M.L.; Goldoni, L.; Tarozzo, G.; Reggiani, A.; Martini, C.; Piomelli, D.; et al. Combining galantamine and memantine in multitargeted, new chemical entities potentially useful in Alzheimer’s disease. J. Med. Chem. 2012, 55, 9708–9721. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; De Simone, A.; Bisignano, P.; Armirotti, A.; Summa, M.; Pizzirani, D.; Scarpelli, R.; Perez, D.I.; Andrisano, V.; Perez-Castillo, A.; et al. Multitarget drug discovery for Alzheimer’s disease: Triazinones as BACE-1 and GSK-3β inhibitors. Angew. Chemie—Int. Ed. 2015, 54, 1578–1582. [Google Scholar] [CrossRef]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Oset-Gasque, M.J.; Marco-Contelles, J. Alzheimer’s Disease, the “one-Molecule, One-Target” Paradigm, and the Multitarget Directed Ligand Approach. ACS Chem. Neurosci. 2018, 9, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Cavalli, A.; Bolognesi, M. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef]
- Ertekin-Taner, N. Identifying therapeutic targets for Alzheimer’s disease with big data. Neurodegener. Dis. Manag. 2017, 7, 101–105. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
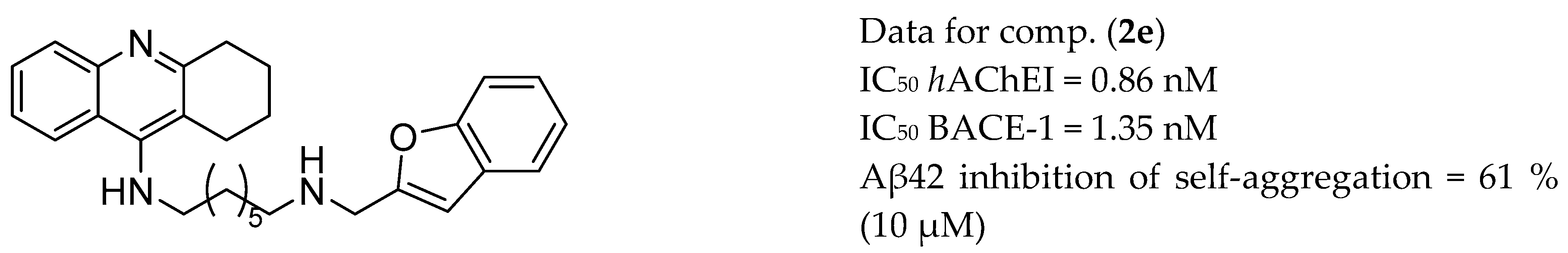{kind=link}
{kind=link}
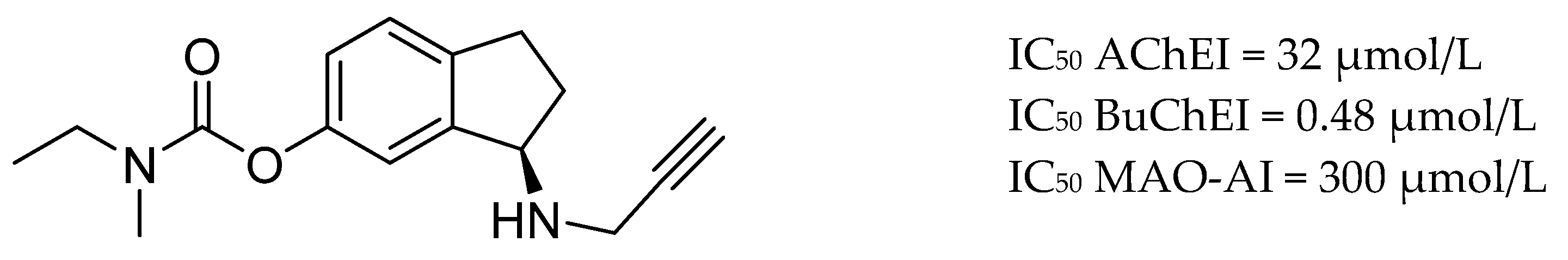{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Structure | Targets | Therapeutic Effects |
|---|---|---|
 | -AChE inhibitor -5-HT2A inducer -ChE inducer -NOS inhibitor/inducer -TNF inhibitor -IL-1β inhibitor/inducer -NMDAR downregulator | -selectively and reversibly inhibits AChE -improves the cognitive and behavioral signs and symptoms of AD -neuroprotective |
 | -AChE inhibitor -ChE inhibitor | -parasympathomimetic and a reversible cholinesterase inhibitor -inhibits both BuChE and AChE -enhances cholinergic function |
 | -AChE competitive and reversible inhibitor -AChR subunit alpha-7 allosteric modulator -N AChR allosteric modulator -ChE inhibitor | -enhances cholinergic function -improve cognitive performance in AD -not considered as a disease-modifying drug |
 | -NMDAR uncompetitive (open-channel) antagonist -Alpha-7 nicotinic cholinergic receptor subunit antagonist | -inhibits calcium influx into cells that is normally caused by chronic NMDAR activation by glutamate -enhances neuronal synaptic plasticity |
| Scaffold | Lead Fragment | Targets | Ref. |
|---|---|---|---|
| Tacrine | xanomeline, iperoxo-fragment | AChE Is and MRs | [99] |
| valmerin | AChE Is and GSK-3α/βIs | [100] | |
| deferasirox | AChE Is and metal chelators | [101] | |
| indole-3acetic acid | AChE/BuChE Is | [102] | |
| pulmonarin B | AChE/BuChE Is | [103] | |
| d-xylose, d-ribose, d-galactose | AChE/BuChE Is | [104] | |
| acridine | AChE/BuChE Is | [105] | |
| 1,2,3-thiadiazole | AChE/BuChE Is | [106] | |
| ferulic acid | AChE/BuChE Is | [107] | |
| flavonoid quercetin | AChE/BuChE Is and metal chelators | [108] | |
| quinolone carboxylic acids | AChE/BuChE Is, M1R Is | [109] | |
| Donepezil | hydroxyphenyl-benzimidazole | AChE Is and metal chelators | [110] |
| hydroxybenzimidazole | AChE Is and metal chelators | [111] | |
| butylated hydroxytoluene hybrids | AChE Is, MAO-B Is, antioxidants | [112] | |
| trolox | AChE Is, MAO-B Is, metal chelators | [113] | |
| chromone | AChE Is, MAO-B Is | [114,115] | |
| chalcones | AChE/BuChE Is | [116] | |
| 2-acetylphenol | AChE/BuChE Is, MAO-A/B Is, metal chelators | [117] | |
| deoxyvasicinone | AChE Is and BACE-1 Is | [118] | |
| introduction of amide bonds | AChE Is and BACE-1 Is | [119] | |
| conjugation of benzylpiperidine moiety benzimidazole or benzofuran | AChE Is, antioxidant, metal chelators | [120] | |
| melatonin | AChE/BuChE Is, antioxydants, metal chelators | [121] |
| Pathway | Targets | Evidence |
|---|---|---|
| Purinergic signalling | -ionotropic P2XRs -metabotropic P2Y -adenosine P1Rs -P2YR-dependent calcium signaling | -P2 × 7R is up-regulated in AD -P2Y2R protective role -P2Y6R—small molecule from GliaCure claims to promote microglial phagocytosis through binding of the microglial purinergic P2Y6 receptor [209] -P2Y12 plays an important role in homeostatic microglia |
| NOD-like receptor family pyrin domain containing 3 (NLRP3) | -triggers TREM2 receptor expressed in myeloid cells 2 | -NLRP3 inflammasome activation is a pathophysiological pathway in AD |
| Toll-like receptors (TLRs) | -TLR2 -TLR4 | -triggers the neuroinflammatory response -downstream TLR signalling through NFκB, activator protein 1 and IFN regulatory factor (IRF) pathways lead to proinflammatory gene transcription |
| microglial fractalkine receptor | -CX3CL1/CX3CR1 | -CX3CL1 exerts an inhibitory signal, maintaining microglia in a resting state |
| Receptor- interacting serine/threonine-protein kinase 1 (RIPK1) | -RIPK1 inhibitor | -enzyme downstream of TNFα signalling, has been shown to mediate microglial responses in AD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagórska, A.; Jaromin, A. Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer′s Disease Therapy. Molecules 2020, 25, 3337. https://doi.org/10.3390/molecules25153337
Zagórska A, Jaromin A. Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer′s Disease Therapy. Molecules. 2020; 25(15):3337. https://doi.org/10.3390/molecules25153337
Chicago/Turabian StyleZagórska, Agnieszka, and Anna Jaromin. 2020. "Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer′s Disease Therapy" Molecules 25, no. 15: 3337. https://doi.org/10.3390/molecules25153337
APA StyleZagórska, A., & Jaromin, A. (2020). Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer′s Disease Therapy. Molecules, 25(15), 3337. https://doi.org/10.3390/molecules25153337