
Study on the Microwave-Assisted Batch and Continuous Flow Synthesis of N-Alkyl-Isoindolin-1-One-3-Phosphonates by a Special Kabachnik–Fields Condensation

 ,
, 
Abstract

1. Introduction
2. Results and Discussion
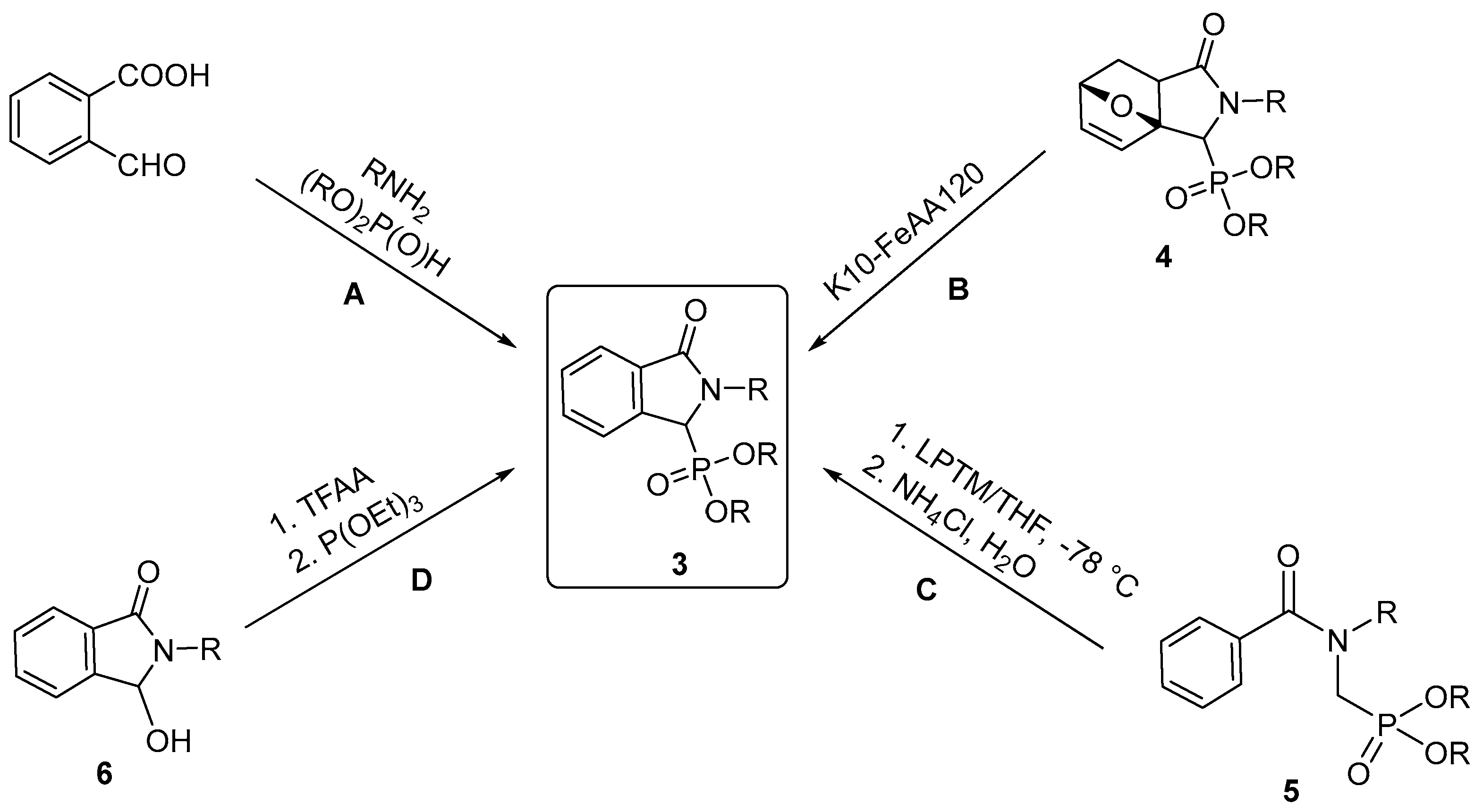
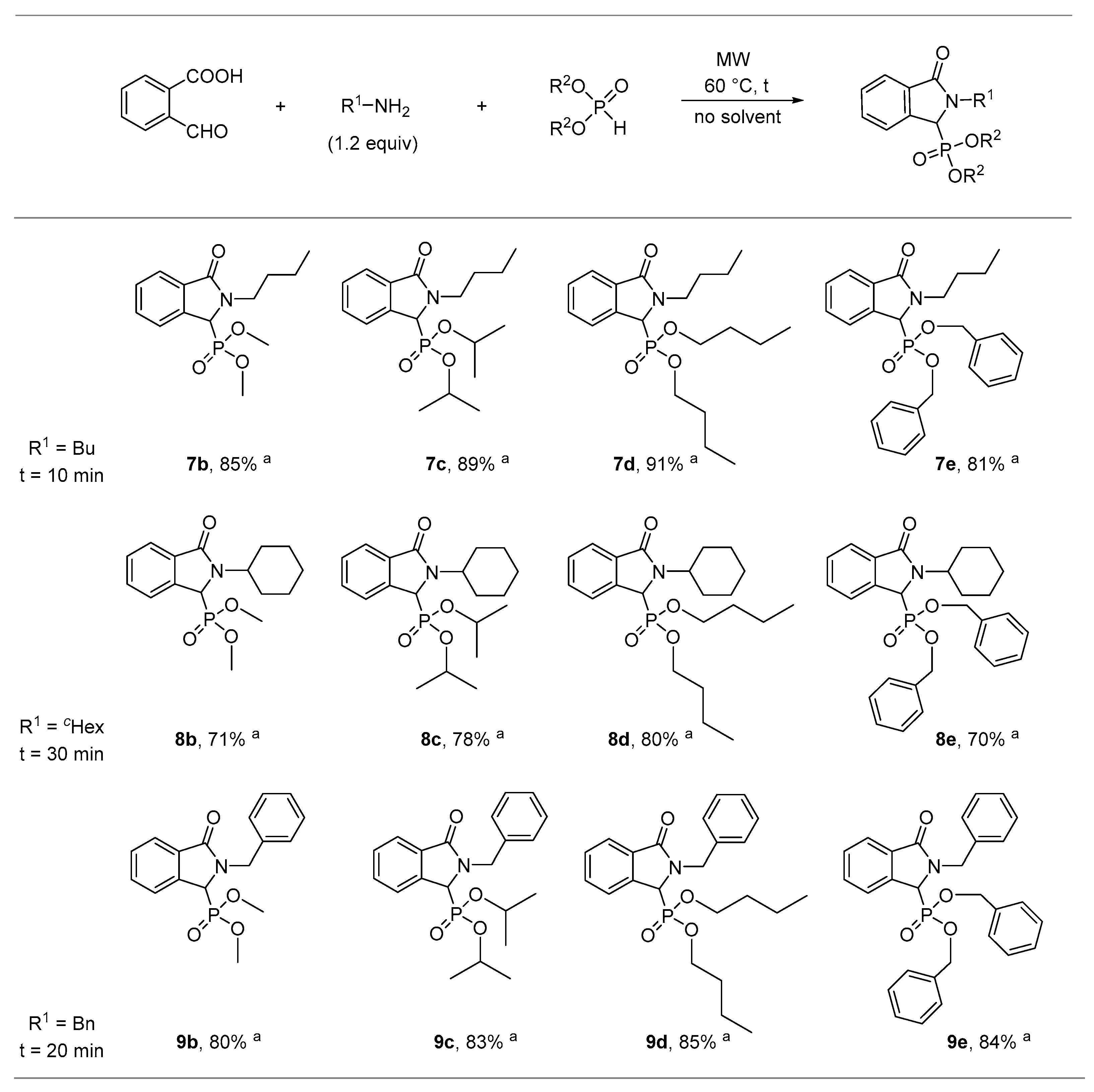
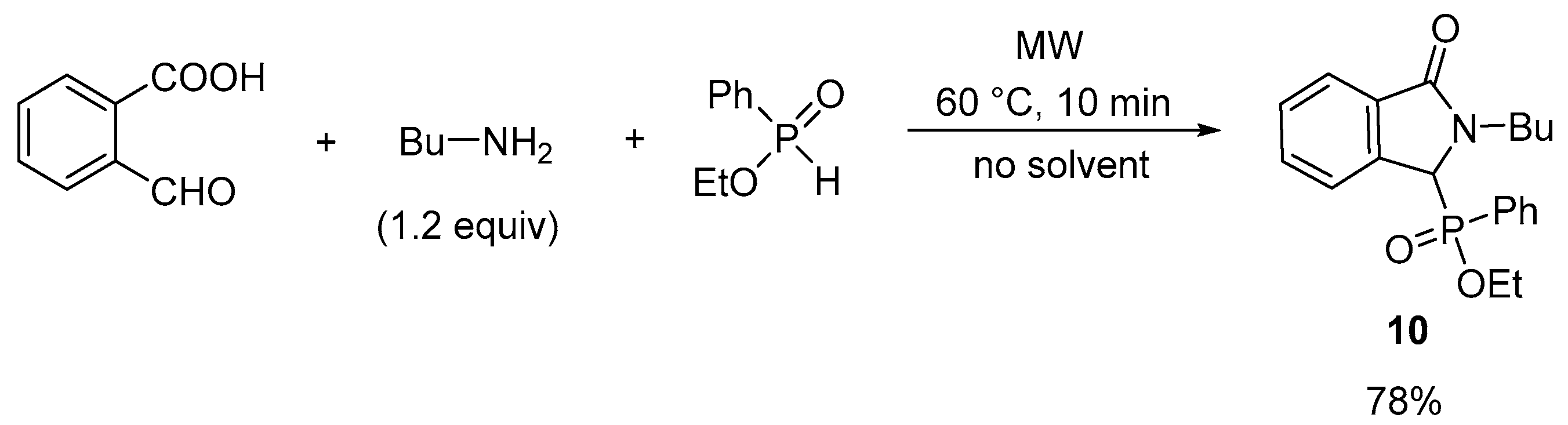
2.1. Synthesis of Isoindolin-1-one-3-phosphonates
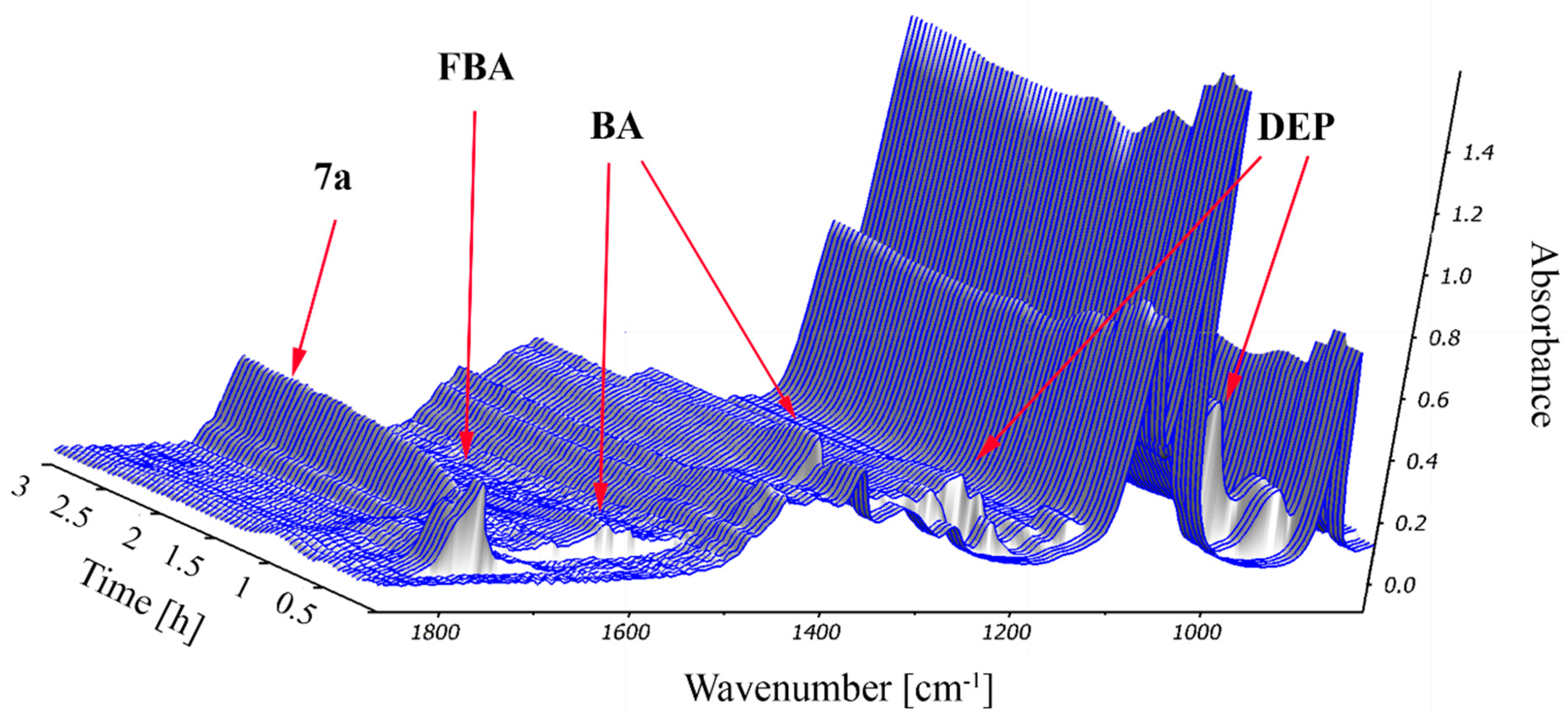
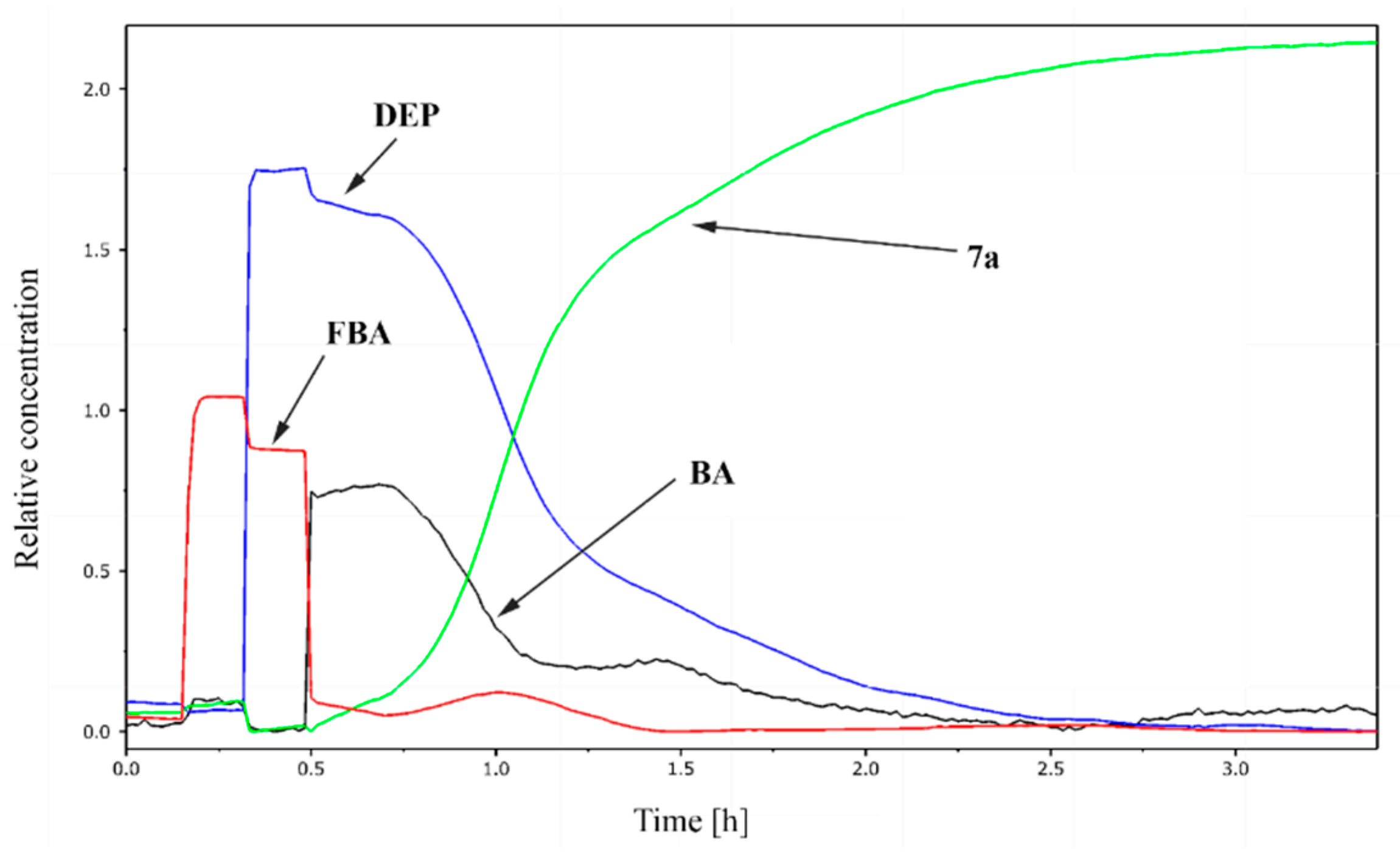
2.2. Study on the Condensation of 2-Formylbenzoic Acid, Butylamine and Diethyl Phosphite by In Situ FT-IR Spectroscopy
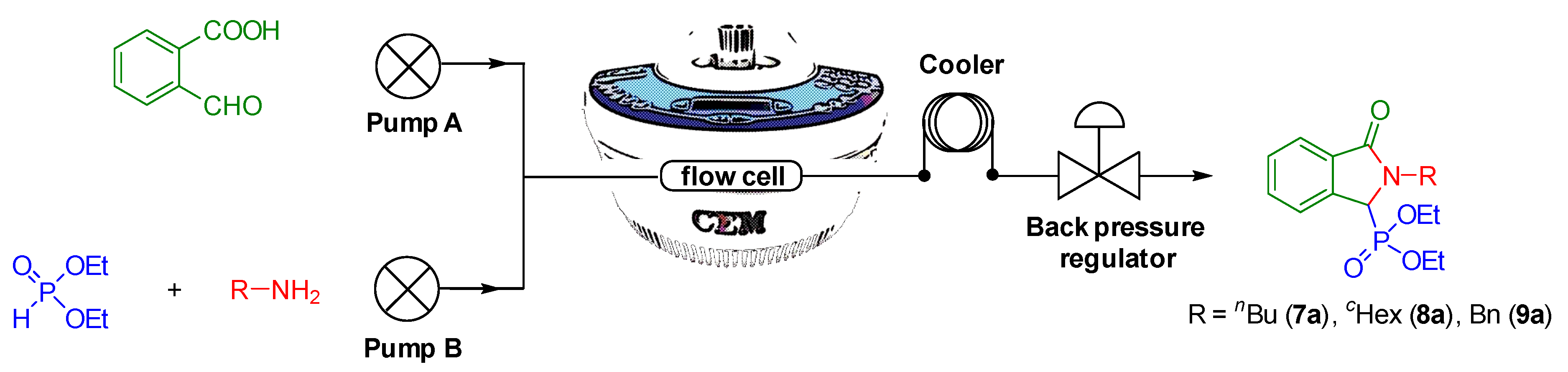
2.3. Condensation of 2-Formylbenzoic Acid, Aliphatic Primary Amines and Diethyl Phosphite in a Continuous Flow Microwave Reactor
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Isoindolin-1-One-3-Phosphonates (7–9) and Isoindolin-1-One-3-Phosphinate (10)
3.2.1. Diethyl (2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (7a)
3.2.2. Dimethyl (2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (7b)
3.2.3. Diisopropyl (2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (7c)
3.2.4. Dibutyl (2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (7d)
3.2.5. Dibenzyl (2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (7e)
3.2.6. Diethyl (2-Cyclohexyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (8a)
3.2.7. Dimethyl (2-Cyclohexyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (8b)
3.2.8. Diisopropyl (2-Cyclohexyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (8c)
3.2.9. Dibutyl (2-Cyclohexyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (8d)
3.2.10. Dibenzyl (2-Cyclohexyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (8e)
3.2.11. Diethyl (2-Benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (9a)
3.2.12. Dimethyl (2-Benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (9b)
3.2.13. Diisopropyl (2-Benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (9c)
3.2.14. Dibutyl (2-Benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (9d)
3.2.15. Dibenzyl (2-Benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (9e)
3.2.16. Ethyl (2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)(phenyl)phosphinate (10)
3.3. Procedure for the Real Time Monitoring of the Condensation of 2-Formylbenzoic Acid, Butylamine and Diethyl Phosphite by In Situ FT-IR Spectroscopy
3.4. Procedure for the Continuous Flow Synthesis of Diethyl (2-butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonate (7a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bhatia, R.K. Isoindole derivatives: Propitious anticancer structural motifs. Curr. Top. Med. Chem. 2017, 17, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Palillero-Cisneros, A.; Bedolla-Medrano, M.; Ordóñez, M. Efficient PhB(OH)2-catalyzed one-pot synthesis of 3-substituted isoindolin-1-ones and isobenzofuran-1(3H)-ones under solvent free conditions. Tetrahedron 2018, 74, 4174–4181. [Google Scholar] [CrossRef]
- Ordóñez, M.; Palillero-Cisneros, A.; Labastida-Galván, V.; Terán-Vázquez, J.L. Practical synthesis of 3-(2-arylethylidene)isoindolin-1-ones (analogues of AKS-182) and 3-(2-arylethylidene)isobenzofuran-1(3H)-ones. Tetrahedron 2020, 76, 130838. [Google Scholar] [CrossRef]
- Wang, J.; Teng, Q.; Lin, C.; Gao, F.; Liu, X.; Shen, L. Co(II)-Catalyzed desilylative annulation of benzamides and acrylamides with alkynylsilanes: Access to 3-methyleneisoindolin-1-one and 5-methylene-1H-pyrrol-2(5H)-one derivatives. Synthesis 2020, 52, 1969–1980. [Google Scholar] [CrossRef]
- Gordon, C.P.; Byrne, N.; McCluskey, A. A facile, protic ionic liquid route to N-substituted 5-hydroxy-4-methyl-3-oxoisoindoline-1-carboxamides and N-substituted 3-oxoisoindoline-4-carboxylic acids. Green Chem. 2010, 12, 1000–1006. [Google Scholar] [CrossRef]
- Couture, A.; Moreau, A.; Deniau, E.; Grandclaudon, P. A convenient synthesis of 4-alkoxylated isoindolin-1-ones. Synthesis 2004, 10, 1664–1670. [Google Scholar] [CrossRef]
- Bunce, R.A.; Muddala, N.P.; Nammalwar, B. Expeditous synthesis of (±)-diethyl 2-alkyl- and 2-aryl-(3-oxoisoindolin-1-yl)phosphonates catalysed by OSU-6. RSC Adv. 2015, 5, 28389–28393. [Google Scholar] [CrossRef]
- Macsari, I.; Besidski, Y.; Csjernyik, G.; Nilsson, L.I.; Sandberg, L.; Yngve, U.; Ahlin, K.; Bueters, T.; Eriksson, A.B.; Lund, P.-E.; et al. 3 Oxoisoindoline-1-carboxamides: Potent, state-dependent blockers of voltage-gated sodium channel Nav1.7 with efficacy in rat pain models. J. Med. Chem. 2012, 55, 6866–6880. [Google Scholar] [CrossRef]
- Bjore, A.; Bostrom, J.; Davidsson, O.; Emtenas, H.; Gran, U.; Iliefski, T.; Kajanus, J.; Olsson, R.; Strandlund, G.; Sundell, J.; et al. New Compounds I/418. U.S. Patent 001,523,7A1, January 2008. [Google Scholar]
- Kukhar, V.P.; Hudson, H.R. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; Wiley: Chichester, UK, 2000; Volume 14, ISBN 978-0-471-89149-9. [Google Scholar]
- Phillips, W.G. Phosphinylphthalimidines and their use as plant growth regulants. U.S. Patent 4,164,406, August 1979. [Google Scholar]
- Ordóñez, M.; Tibhe, G.D.; Zamudio-Medina, A.; Viveros-Ceballos, J.V. An easy approach for the synthesis of N-substituted isoindolin-1-ones. Synthesis 2012, 44, 569–574. [Google Scholar] [CrossRef]
- Ordóñez, M.; Reyes-González, M.A.; Zamudio-Medina, A. Practical and high stereoselective synthesis of 3-(arylmethylene)isoindolin-1-ones from 2-formylbenzoic acid. Tetrahedron Lett. 2012, 53, 5756–5758. [Google Scholar] [CrossRef]
- Reyes-González, M.A.; Zamudio-Medina, A.; Ramírez-Marroquin, O.A.; Ordóñez, M. Synthesis of 3-alkylideneisoindolinones and isoindolones by a Horner–Wadsworth–Emmons reaction. Monatsch. Chem. 2014, 145, 1001–1007. [Google Scholar] [CrossRef]
- Ordóñez, M.; Viveros-Ceballos, J.L.; Cativiela, C. One-pot three-component highly diastereoselective synthesis of isoindolin-1-one-3-phosphonates under solvent and catalyst free-conditions. Tetrahedron: Asymmetry 2011, 22, 1479–1484. [Google Scholar] [CrossRef]
- Failla, S.; Finocchiaro, P. Unexpected course of dimethyl phosphite addition to the condensation products obtained from o-carboxybenzaldehyde and aliphatic amines. Phosphorus Sulfur Silicon Relat. Elem. 1995, 105, 195–203. [Google Scholar] [CrossRef]
- Milen, M.; Dancsó, A.; Földesi, T.; Slégel, P.; Volk, B. Propylphosphonic anhydride (T3P) mediated one-pot three-component synthesis of racemic dialkyl (2-substituted3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonates. Tetrahedron 2016, 72, 5091–5099. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields reaction: Mechanism and synthetic use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef]
- Claeys, D.D.; Stevens, C.V.; Roman, B.I.; Van De Caveye, P.; Waroquier, M.; Van Speybroeck, V. Experimental and computational study of the ring opening of tricyclic oxanorbornenes to polyhydro isoindole phosphonates. Org. Biomol. Chem. 2010, 8, 3644–3654. [Google Scholar] [CrossRef]
- Couture, A.; Deniau, E.; Woisel, P.; Grandclaudon, P. A short and efficient synthesis of 3-diphenylphosphoryl- and 3-diethoxyphosphoryl(aza)isoindolinones: Extension to the sulfonylated analogs. Synthesis 1997, 12, 1439–1445. [Google Scholar] [CrossRef]
- Kachkovskyi, G.O.; Kolodiazhnyi, O.I. Synthesis of phosphonic acids possessing isoindolin-1-one moiety: Unexpected acid-catalyzed C-P-bond cleavage. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 890–907. [Google Scholar] [CrossRef]
- Kachkovskyi, G.O.; Kolodiazhnyi, O.I. Synthesis of the phosphonoanalogue of benzo[c]pyroglutamic acid. Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 2441–2448. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Bálint, E.; Keglevich, G. Microwave-assisted syntheses in organic chemistry. In Milestones in Microwave Chemistry; Keglevich, G., Ed.; Springer: Basel, Switzerland, 2016; pp. 11–45. ISBN 978-3-319-30632-2. [Google Scholar]
- Kappe, C.O.; Stadler, A.; Dallinger, D. Microwaves in organic and medicinal chemistry, 2nd ed.; Wiley: Weinheim, Germany, 2012; Volume 52, ISBN 978-3-527-33185-7. [Google Scholar]
- Jiang, B.; Shi, F.; Tu, S.J. Microwave-assisted multicomponent reactions in the heterocyclic chemistry. Curr. Org. Chem. 2010, 14, 357–378. [Google Scholar] [CrossRef]
- Bálint, E.; Keglevich, G. The Spread of the Application of the Microwave Technique in Organic Synthesis. In Milestones in Microwave Chemistry; Keglevich, G., Ed.; Springer: Basel, Switzerland, 2016; pp. 1–10. ISBN 978-3-319-30632-2. [Google Scholar]
- de la Hoz, A.; Loupy, A. Microwaves in Organic Synthesis, 3rd ed.; Wiley: Weinheim, Germany, 2012; ISBN 978-3-527-65131-3. [Google Scholar]
- Estela, L.; Pouxb, M.; Benamaraa, N.; Polaerta, I. Continuous flow-microwave reactor: Where are we? Chem. Eng. Process. 2016, 113, 56–64. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Ladányi-Pára, K.; Tóth, N.; Mátravölgyi, B.; Keglevich, G. Continuous flow synthesis of α-aryl-α-aminophosphonates. Pure Appl. Chem. 2019, 91, 67–76. [Google Scholar] [CrossRef]
- Sterk, H. Untersuchungen zur Temperaturabhängigkeit charakteristischer IR-Absorptionen. Monatsch. Chem. 1968, 99, 1764–1769. [Google Scholar] [CrossRef]
- Bowden, K.; Taylor, G.R. Ring-chain tautomerism. Part I. 2-Acyl- and 2-aroyl-benzoic acids. J. Chem. Soc. B 1971, 1390–1394. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Ádám, A.; Csontos, I.; Karaghiosoff, K.; Czugler, M.; Ábrányi-Balogh, P.; Keglevich, G. The synthesis of α-aryl-α-aminophosphonates and α-aryl-α-aminophosphine oxides by the microwave-assisted Pudovik reaction. Beilstein J. Org. Chem. 2017, 13, 76–86. [Google Scholar] [CrossRef]
- Tajti, Á.; Tóth, N.; Bálint, E.; Keglevich, G. Esterification of benzoic acid in a continuous flow microwave reactor. J. Flow Chem. 2018, 8, 11–19. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Tóth, N.; Keglevich, G. Continuous flow alcoholysis of dialkyl H-phosphonates with aliphatic alcohols. Molecules 2018, 23, 1618. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 7–10 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Mode of Heating | R | Amine (equiv) | DEP (equiv) | T (°C) | t (min) | Conversion a (%) | Yield b (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | - | Bu | 1 | 1 | 25 | 180c | 77 | - |
| 2 | MW | Bu | 1 | 1 | 40 | 10 | 61 | - |
| 3 | MW | Bu | 1 | 1 | 60 | 10 | 85 | - |
| 4 | MW | Bu | 1 | 1 | 80 | 10 | 87 | - |
| 5 | MW | Bu | 1 | 1 | 60 | 20 | 90 | - |
| 6 | MW | Bu | 1 | 1 | 60 | 30 | 91 | - |
| 7 | MW | Bu | 1 | 1.1 | 60 | 10 | 87 | - |
| 8 | MW | Bu | 1 | 1.2 | 60 | 10 | 89 | - |
| 9 | MW | Bu | 1.1 | 1 | 60 | 10 | 92 | - |
| 10 | MW | Bu | 1.2 | 1 | 60 | 10 | 100 | 94 (7a) |
| 11 | - | Bu | 1.2 | 1 | 60 | 10 | 90 | - |
| 12 | MW | cHex | 1.2 | 1 | 60 | 10 | 82 | - |
| 13 | MW | cHex | 1.2 | 1 | 60 | 20 | 93 | - |
| 14 | MW | cHex | 1.2 | 1 | 60 | 30 | 100 | 84 (8a) |
| 15 | - | cHex | 1.2 | 1 | 60 | 30 | 84 | - |
| 16 | MW | Bn | 1.2 | 1 | 60 | 10 | 89 | - |
| 17 | MW | Bn | 1.2 | 1 | 60 | 20 | 100 | 90 (9a) |
| 18 | - | Bn | 1.2 | 1 | 60 | 20 | 88 | - |

| Entry | R | Amine (equiv) | DEP (equiv) | Flow Rate (mL/min) | τ (min) | Conversion a (%) | Yield b (%) |
|---|---|---|---|---|---|---|---|
| 1 | Bu | 1.2 | 1 | 0.70 | 10 | 43 | - |
| 2 | Bu | 1.2 | 1 | 0.35 | 20 | 52 | - |
| 3 | Bu | 1.2 | 1 | 0.25 | 30 | 56 | - |
| 4 | Bu | 1.5 | 1 | 0.35 | 20 | 70 | - |
| 5 | Bu | 2.0 | 1 | 0.35 | 20 | 73 | - |
| 6 | Bu | 1.5 | 1.2 | 0.35 | 20 | 81 | - |
| 7 | Bu | 1.5 | 1.5 | 0.35 | 20 | 95 | - |
| 8 | Bu | 1.5 | 1.5 | 0.25 | 30 | 100 | 95% (7a) |
| 9 | cHex | 1.5 | 1.5 | 0.15 | 45 | 100 | 86% (8a) |
| 10 | Bn | 1.5 | 1.5 | 0.18 | 40 | 100 | 91% (9a) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajti, Á.; Tóth, N.; Rávai, B.; Csontos, I.; Szabó, P.T.; Bálint, E. Study on the Microwave-Assisted Batch and Continuous Flow Synthesis of N-Alkyl-Isoindolin-1-One-3-Phosphonates by a Special Kabachnik–Fields Condensation. Molecules 2020, 25, 3307. https://doi.org/10.3390/molecules25143307
Tajti Á, Tóth N, Rávai B, Csontos I, Szabó PT, Bálint E. Study on the Microwave-Assisted Batch and Continuous Flow Synthesis of N-Alkyl-Isoindolin-1-One-3-Phosphonates by a Special Kabachnik–Fields Condensation. Molecules. 2020; 25(14):3307. https://doi.org/10.3390/molecules25143307
Chicago/Turabian StyleTajti, Ádám, Nóra Tóth, Bettina Rávai, István Csontos, Pál Tamás Szabó, and Erika Bálint. 2020. "Study on the Microwave-Assisted Batch and Continuous Flow Synthesis of N-Alkyl-Isoindolin-1-One-3-Phosphonates by a Special Kabachnik–Fields Condensation" Molecules 25, no. 14: 3307. https://doi.org/10.3390/molecules25143307
APA StyleTajti, Á., Tóth, N., Rávai, B., Csontos, I., Szabó, P. T., & Bálint, E. (2020). Study on the Microwave-Assisted Batch and Continuous Flow Synthesis of N-Alkyl-Isoindolin-1-One-3-Phosphonates by a Special Kabachnik–Fields Condensation. Molecules, 25(14), 3307. https://doi.org/10.3390/molecules25143307