
N-Benzyl Residues as the P1′ Substituents in Phosphorus-Containing Extended Transition State Analog Inhibitors of Metalloaminopeptidases

 , ,
, ,  and
and
Abstract

1. Introduction
2. Results and Discussion
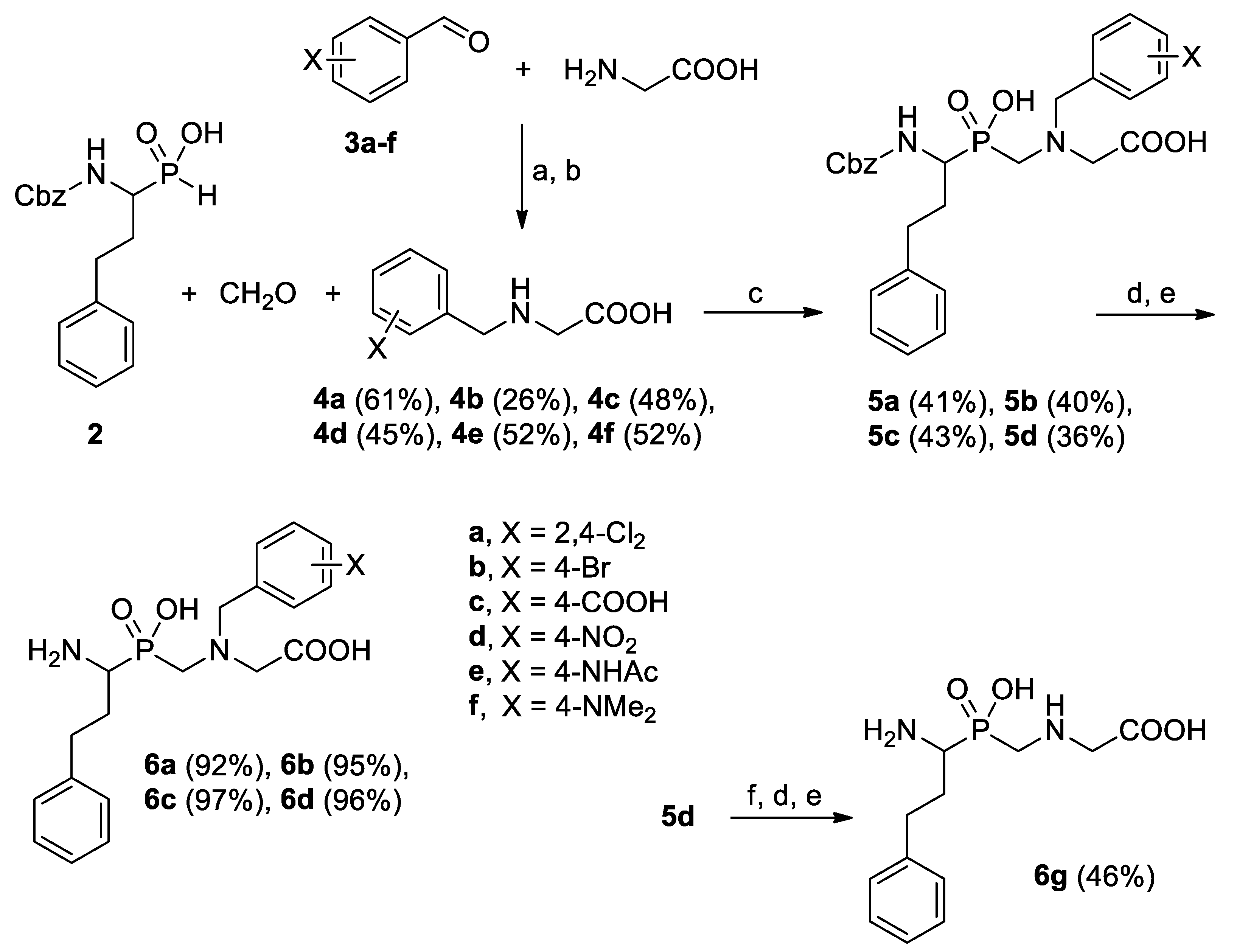
2.1. Chemistry
2.2. Enzyme Inhibition
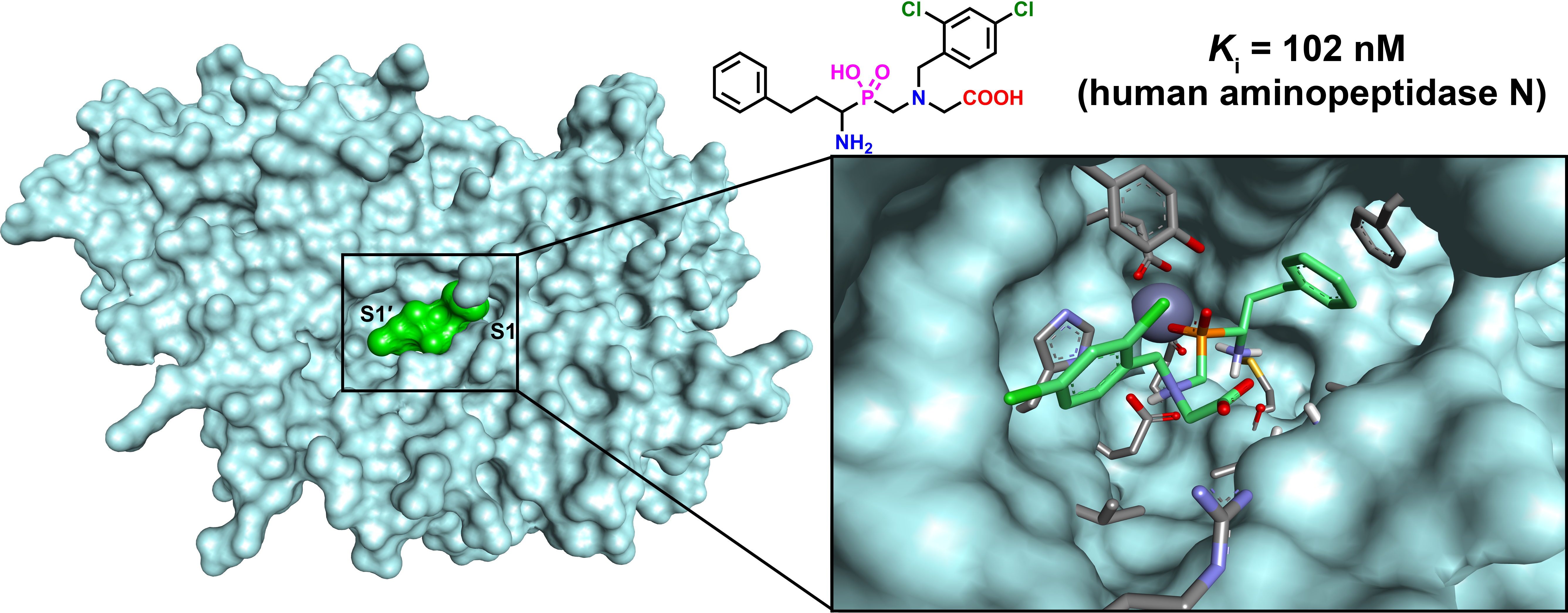
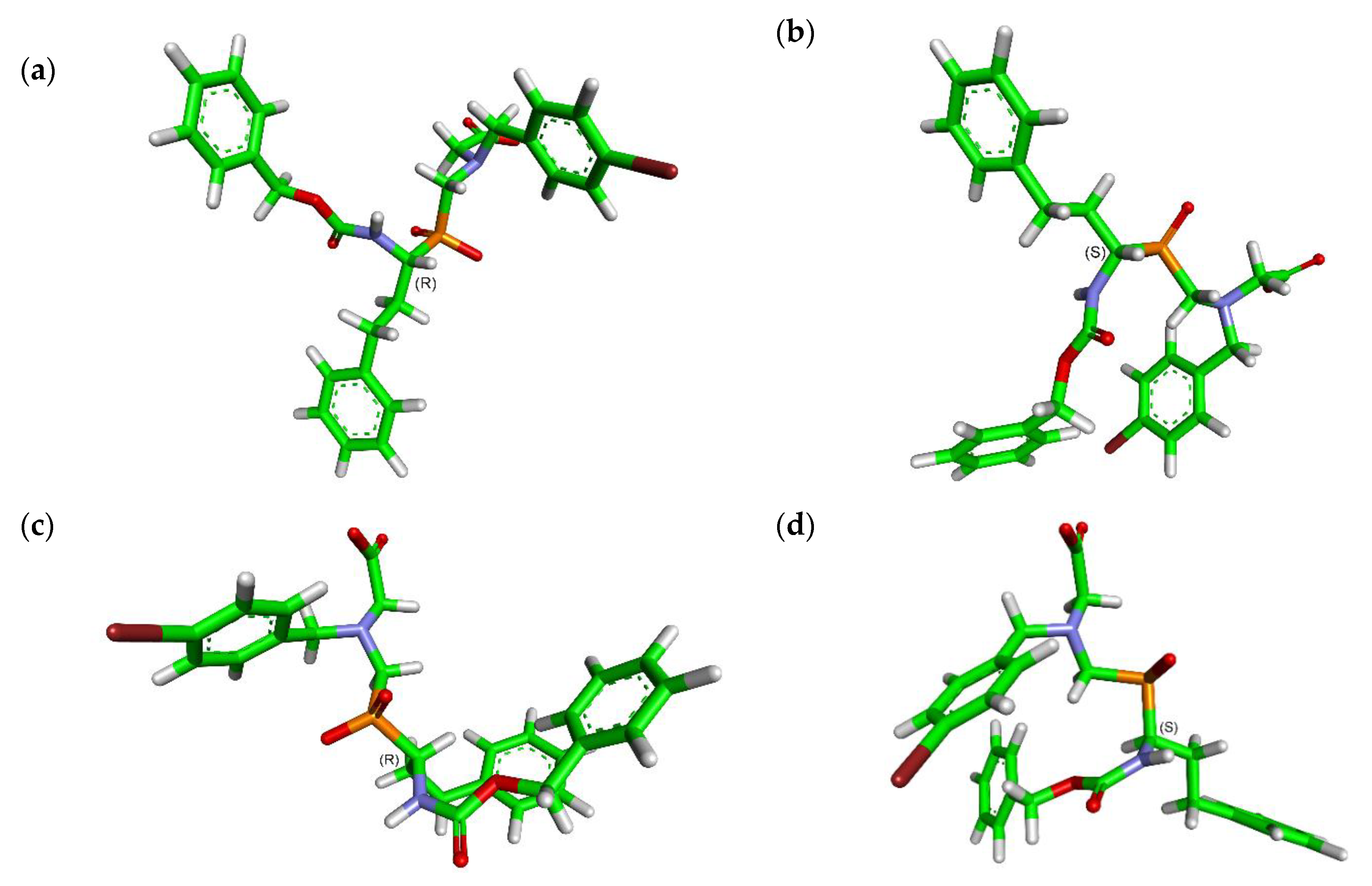
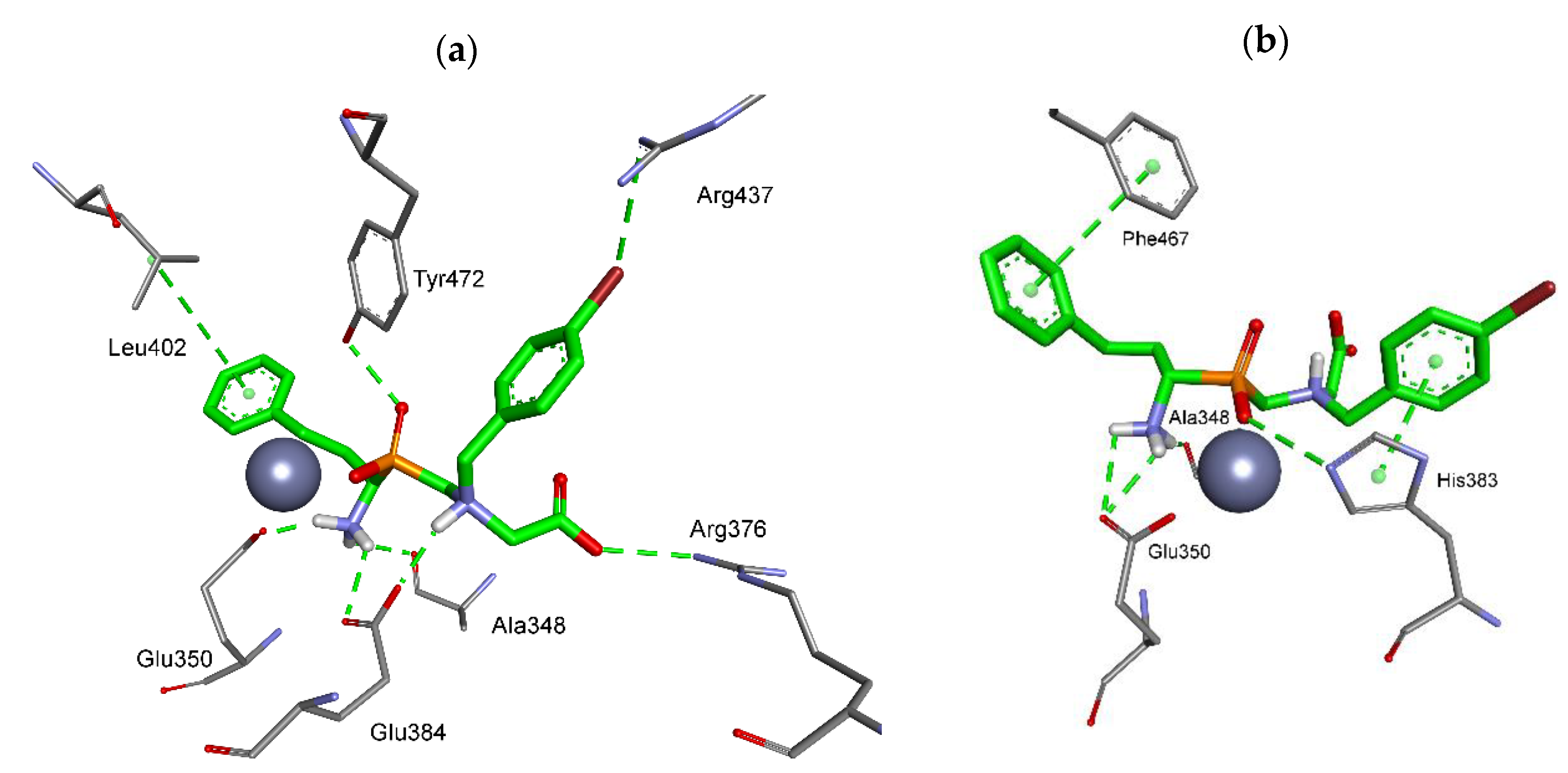
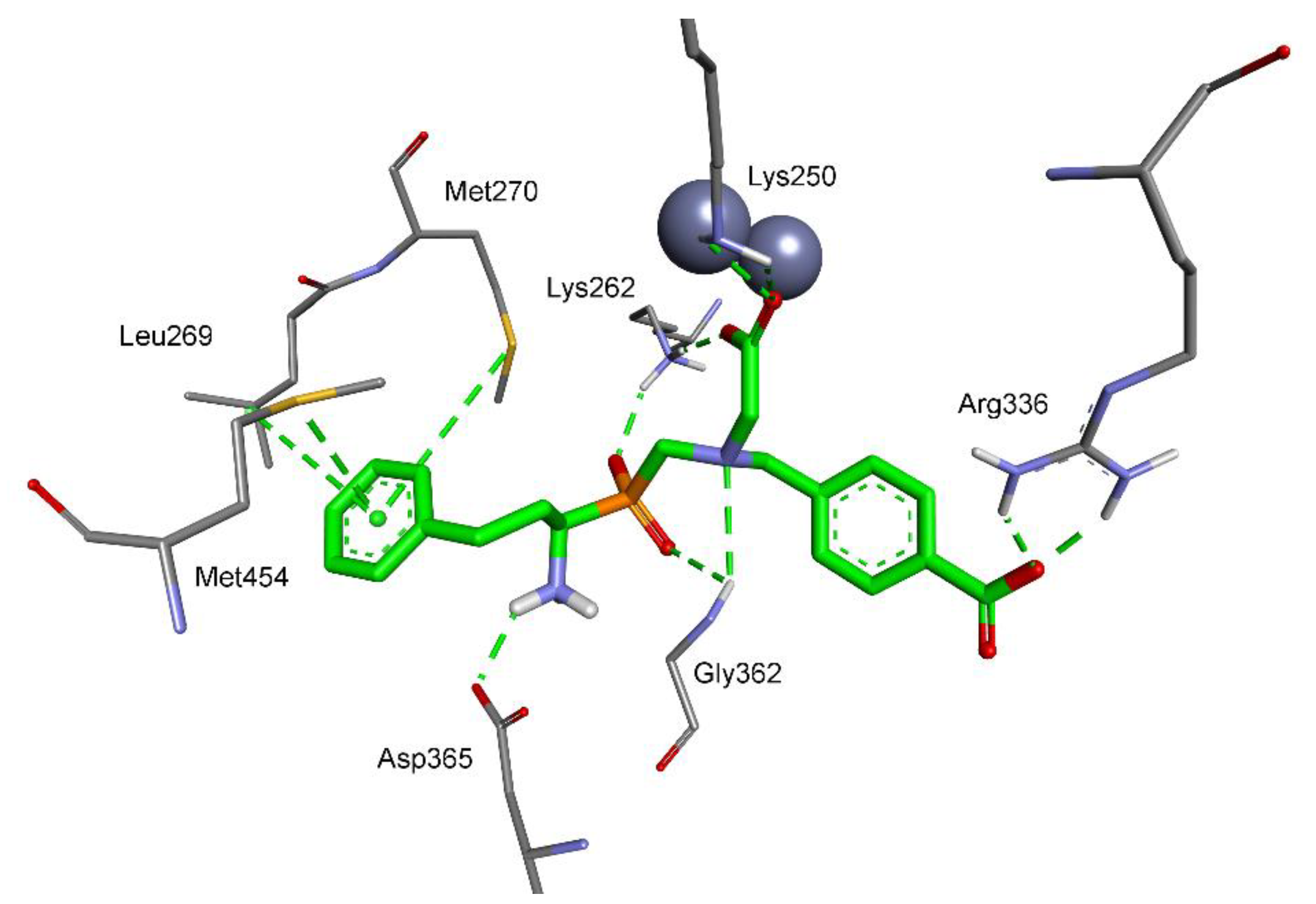
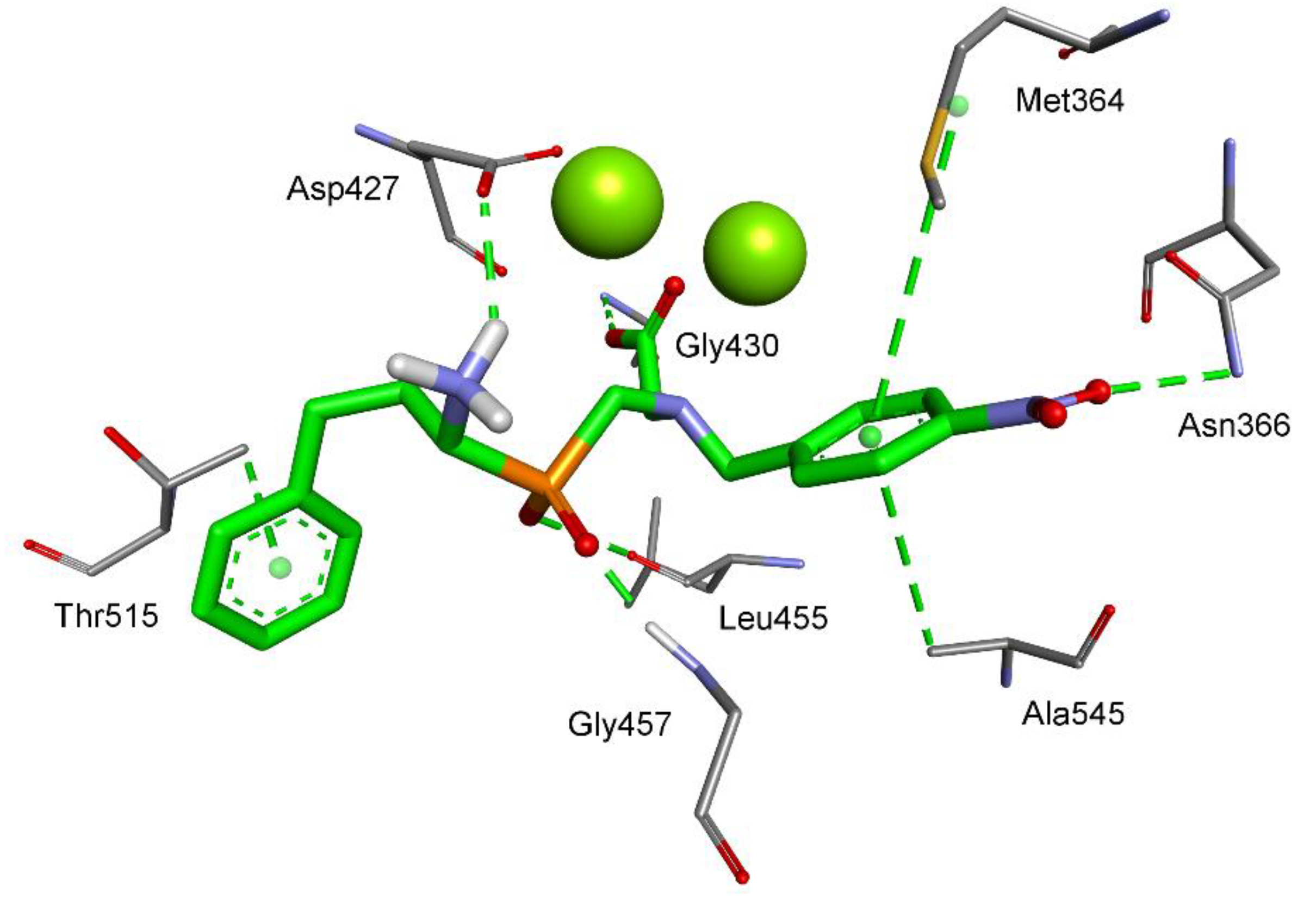
2.3. Molecular Modeling
2.4. Summary
3. Materials and Methods
3.1. General
3.2. Synthetic Procedure
3.3. Inhibition Studies
3.4. Modeling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kafarski, L.; Lejczak, B. The biological activity of phosphono- and phosphinopeptides. In Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons: Chichester, UK, 2000; pp. 407–442. [Google Scholar]
- Bartlett, P.A.; Marlowe, C.K. Possible role for water dissociation in the slow binding of phosphorus-containing transition-state-analogue inhibitors of thermolysin. Biochemistry 1987, 26, 8553–8561. [Google Scholar] [CrossRef] [PubMed]
- Holden, H.M.; Tronrud, D.E.; Monzingo, A.F.; Weaver, L.H.; Matthews, B.W. Slow- and fast-binding inhibitors of thermolysin display different modes of binding: Crystallographic analysis of extended phosphonamidate transition-state analogs. Biochemistry 1987, 26, 8542–8553. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 1988, 21, 333–340. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef]
- Mucha, A.; Grembecka, J.; Cierpicki, T.; Kafarski, P. Hydrolysis of the phosphonamidate bond in phosphono dipeptide analogues—The influence of the nature of the N-terminal functional group. Eur. J. Org. Chem. 2003, 4797–4803. [Google Scholar] [CrossRef]
- Dziełak, A.; Pawełczak, M.; Mucha, A. A three-component Mannich-type condensation leading to phosphinic dipeptides–extended transition state analogue inhibitors of aminopeptidases. Tetrahedron Lett. 2011, 52, 3141–3145. [Google Scholar] [CrossRef]
- Macegoniuk, K.; Dziełak, A.; Mucha, A.; Berlicki, Ł. Bis(aminomethyl)phosphinic acid, a highly promising scaffold for the development of bacterial urease inhibitors. ACS Med. Chem. Lett. 2015, 6, 146–150. [Google Scholar] [CrossRef]
- Moedritzer, K.; Irani, R.R. The direct synthesis of α-aminomethylphosphonic acids. Mannich-type reactions with orthophosphorous acid. J. Org. Chem. 1966, 31, 1603–1607. [Google Scholar] [CrossRef]
- Maier, L.; Smith, M.J. Organic phosphorus compounds. Part 72. Preparation and properties of bis(N-hydroxycarbonylmethylaminomethyl)phosphinic acid, (HO2CCH2NHCH2)2P(O)OH, and its derivatives. Phosphorus Sulfur Silicon Relat. Elem. 1980, 8, 67–71. [Google Scholar] [CrossRef]
- Dhansay, M.A.; Linder, P.W.; Torrington, R.G.; Modro, T.A. Organophosphorus herbicides and plant growth regulators. Part 1. Synthesis and protonation behavior of glyphosate and related compounds. J. Phys. Org. Chem. 1990, 3, 248–254. [Google Scholar] [CrossRef]
- Kiss, T.; Farkas, E.; Jezowska-Bojczuk, M.; Kozlowski, H.; Kowalik, E. Copper(II) complexes of ligands containing both aminocarboxylate and aminophosphinate moieties. J. Chem. Soc. Dalton Trans. 1990, 1, 377–379. [Google Scholar] [CrossRef]
- Baylis, E.K.; Campbell, C.D.; Dingwall, J.G. 1-Aminoalkylphosphonous acids. Part 1. Isosteres of the protein amino acids. J. Chem. Soc. Perkin Trans. I 1984, 0, 2845–2853. [Google Scholar] [CrossRef]
- Wilson, J.G. Phenolic analogs of amino carboxylic acid ligands for 99mTc. 4. N-(2-Hydroxybenzyl)glycines (hbg). Aust. J. Chem. 1990, 43, 1283–1289. [Google Scholar] [CrossRef]
- Talma, M.; Mucha, A. P1′ residue-oriented virtual screening for potent and selective phosphinic (dehydro)dipeptide inhibitors of metallo-aminopeptidases. Biomolecules 2020, 10, 659. [Google Scholar] [CrossRef]
- Hirschmann, R.; Yager, K.M.; Taylor, C.M.; Witherington, J.; Sprengeler, P.A.; Phillips, B.W.; Moore, W.; Smith, A.B., III. Phosphonate diester and phosphonamide synthesis. Reaction coordinate analysis by 31P NMR spectroscopy: Identification of pyrophosphonate anhydrides and highly reactive phosphonylammonium salts. J. Am. Chem. Soc. 1997, 119, 8177–8190. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Talma, M.; Maślanka, M.; Mucha, A. Recent developments in the synthesis and applications of phosphinic peptide analogs. Bioorg. Med. Chem. Lett. 2019, 29, 1031–1042. [Google Scholar] [CrossRef]
- Hooper, N.M.; Lendeckel, U. Aminopeptidases in Biology and Disease; Springer: New York, NY, USA, 2004. [Google Scholar]
- Matsui, M.; Fowler, J.H.; Walling, L.L. Leucine aminopeptidases: Diversity in structure and function. Biol. Chem. 2006, 387, 1535–1544. [Google Scholar] [CrossRef]
- Drinkwater, N.; Lee, J.; Yang, W.; Malcolm, T.R.; McGowan, S. M1 aminopeptidases as drug targets: Broad applications or therapeutic niche? FEBS J. 2017, 284, 1473–1488. [Google Scholar] [CrossRef]
- Oszywa, B.; Makowski, M.; Pawełczak, M. Purification and partial characterization of aminopeptidase from barley (Hordeum vulgare L.) seeds. Plant Physiol. Biochem. 2013, 65, 75–80. [Google Scholar] [CrossRef]
- Chen, L.; Lin, Y.-L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.M.; Zhou, D.; Rini, J.M. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J. Biol. Chem. 2012, 287, 36804–36813. [Google Scholar] [CrossRef] [PubMed]
- Straeter, N.; Lipscomb, W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: Active site solvent structure and binding mode of l-leucinal, a gem-diolate transition state analog, by X-ray crystallography. Biochemistry 1995, 34, 14792–14800. [Google Scholar] [CrossRef] [PubMed]
- Duprez, K.; Scranton, M.A.; Walling, L.L.; Fan, L. Structure of tomato wound-induced leucine aminopeptidase sheds light on substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Lejczak, B.; Kafarski, P.; Zygmunt, J. Inhibition of aminopeptidases by aminophosphonates. Biochemistry 1989, 28, 3549–3555. [Google Scholar] [CrossRef]
- Oszywa, B.; Pawełczak, M.; Kafarski, P. The influence of alpha-aminophosphonic acids on the activity of aminopeptidase from barley seeds—An approach to determine the enzyme specificity. Acta Physiol. Plant. 2015, 37, 44. [Google Scholar] [CrossRef][Green Version]
- Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Phosphonic acid analogues of phenylglycine as inhibitors of aminopeptidases: Comparison of porcine aminopeptidase N, bovine leucine aminopeptidase, tomato acidic leucine aminopeptidase and aminopeptidase from barley seeds. Pharmaceuticals 2019, 12, 139. [Google Scholar] [CrossRef]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef]
- Pícha, J.; Liboska, R.; Buděšínský, M.; Jiráček, J.; Pawełczak, M.; Mucha, A. Unusual activity pattern of leucine aminopeptidase inhibitors based on phosphorus containing derivatives of methionine and norleucine. J. Enz. Inhib. Med. Chem. 2011, 26, 155–161. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-4: Protein Preparation Wizard; Epik; Schrödinger, LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-4: Induced Fit Docking Protocol; Glide; Schrödinger, LLC: New York, NY, USA, 2018.
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry |  | Ki [μM] | |||
| SsAPN | HsAPN | SsLAP | HwLAP | ||
| 1 |  | 3.0 [7] | NT | 52 [7] | NT |
| 6a |  | 7.7 | 0.10 | 107 | 37 |
| 6b |  | 5.8 | 0.13 | 168 | 94 |
| 6c |  | 34 | 1.4 | 27 a | 36 |
| 6d |  | 17 | 0.24 | 53 a | 13 |
| 6g | H | 63 | NT | 29 | 173 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janiszewska, K.; Talma, M.; Oszywa, B.; Pawełczak, M.; Kafarski, P.; Mucha, A. N-Benzyl Residues as the P1′ Substituents in Phosphorus-Containing Extended Transition State Analog Inhibitors of Metalloaminopeptidases. Molecules 2020, 25, 4334. https://doi.org/10.3390/molecules25184334
Janiszewska K, Talma M, Oszywa B, Pawełczak M, Kafarski P, Mucha A. N-Benzyl Residues as the P1′ Substituents in Phosphorus-Containing Extended Transition State Analog Inhibitors of Metalloaminopeptidases. Molecules. 2020; 25(18):4334. https://doi.org/10.3390/molecules25184334
Chicago/Turabian StyleJaniszewska, Kamila, Michał Talma, Bartosz Oszywa, Małgorzata Pawełczak, Paweł Kafarski, and Artur Mucha. 2020. "N-Benzyl Residues as the P1′ Substituents in Phosphorus-Containing Extended Transition State Analog Inhibitors of Metalloaminopeptidases" Molecules 25, no. 18: 4334. https://doi.org/10.3390/molecules25184334
APA StyleJaniszewska, K., Talma, M., Oszywa, B., Pawełczak, M., Kafarski, P., & Mucha, A. (2020). N-Benzyl Residues as the P1′ Substituents in Phosphorus-Containing Extended Transition State Analog Inhibitors of Metalloaminopeptidases. Molecules, 25(18), 4334. https://doi.org/10.3390/molecules25184334