Alkyl and Aryl Derivatives Based on p-Coumaric Acid Modification and Inhibitory Action against Leishmania braziliensis and Plasmodium falciparum

 , and
, and 
Abstract
1. Introduction
2. Results and Discussion
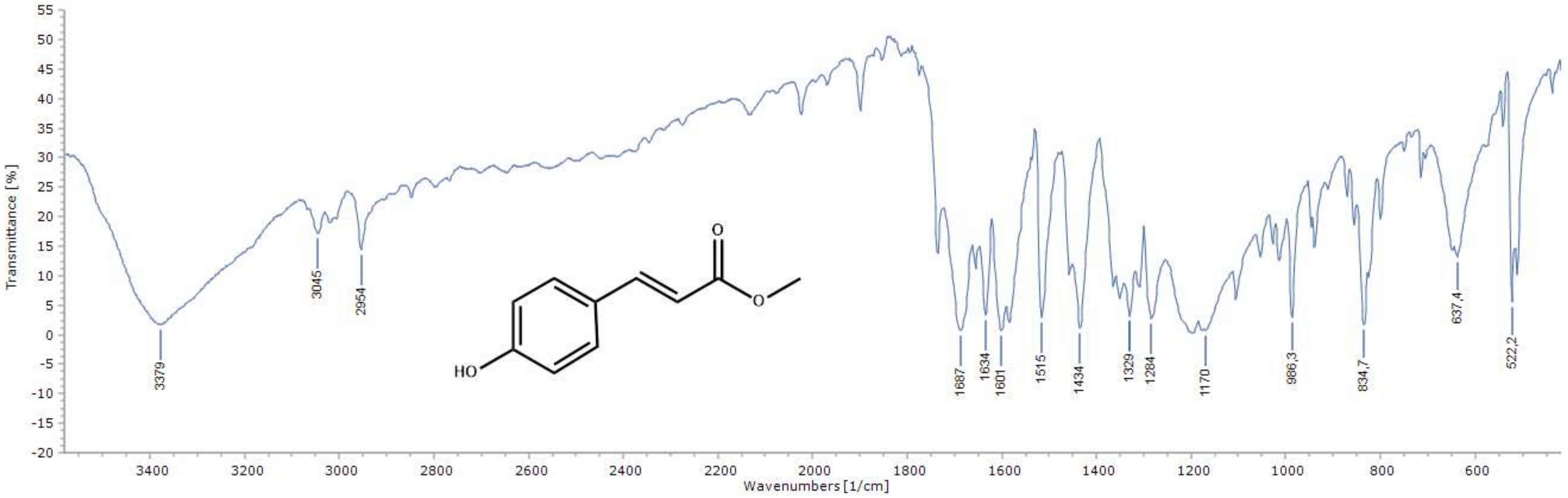
2.1. Chemistry of the Compounds 1–12
2.2. Antileishmanial and Antiplasmodial Activity of Compounds 1–12
2.3. Antileishmanial Activity
2.4. Antiplasmodial Activity
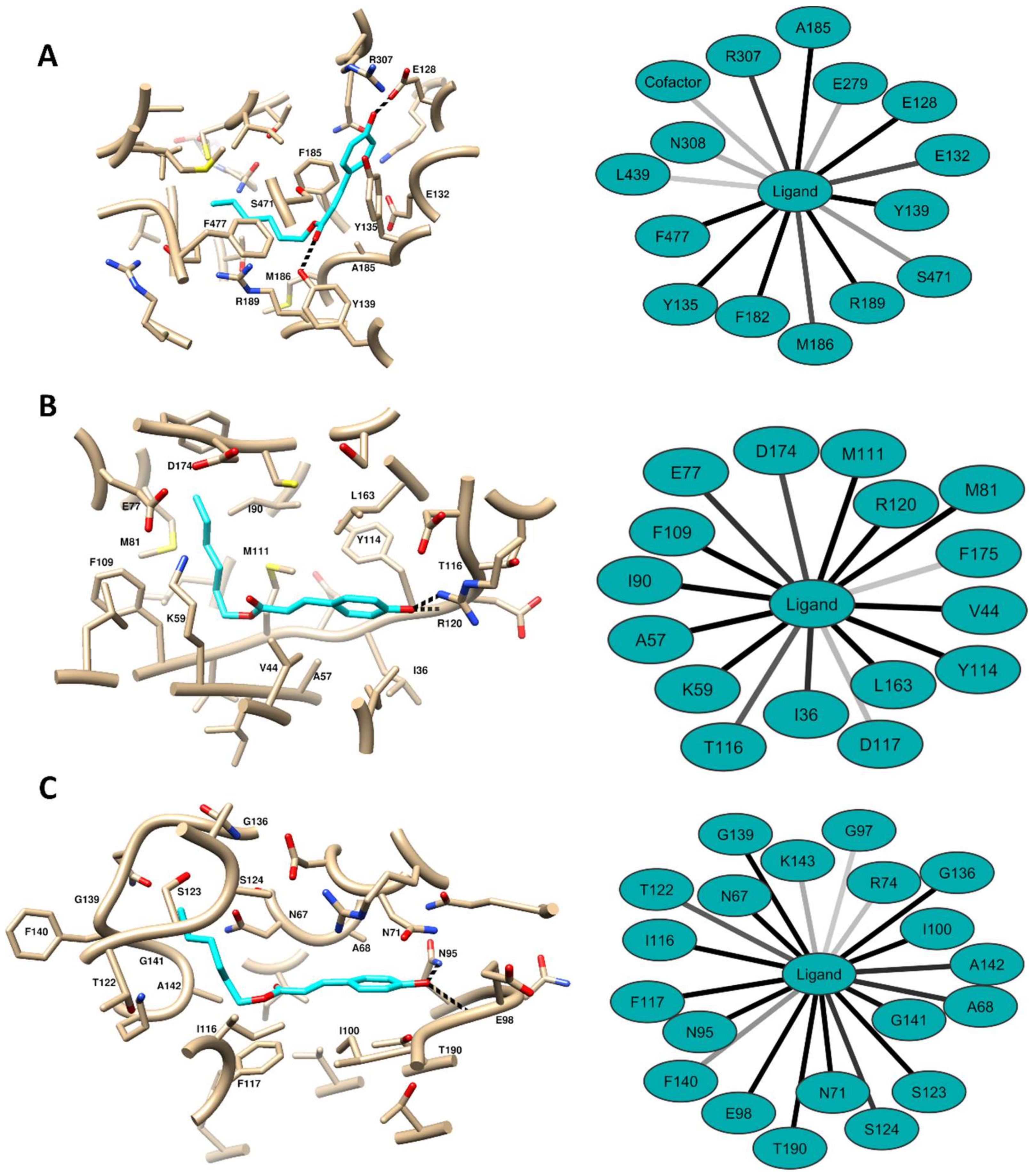
2.5. Computational Methods
3. Conclusions
4. Materials and Methods
4.1. Reagents and Chemical Characterization
4.2. General Procedure for Preparation of Compounds 1–8
4.3. Preparation of Compounds 9–12 by Mitsunobu Reaction
4.4. Cell Culture
4.5. In Vitro Cytotoxicity
4.6. In Vitro Antileishmanial Activity
4.7. In Vitro Antiplasmodial Activity
4.8. Statistical Analysis
4.9. Computational Methods
4.9.1. Target Selection
4.9.2. Molecular Docking
4.9.3. Molecular Dynamics Simulations and MM-PBSA Calculations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bezerra, J.W.A.; Coronel, C.; Gomez, M.C.V.; Rolón, M.; Nunez, C.V.; da Silva, D.R.; da Silva, L.A.; Rodrigues, F.C.; Boligon, A.A.; de Souza, M.A.; et al. Evaluation of antiparasitary, cytotoxic and antioxidant activity and chemical analysis of Tarenaya spinosa (Jacq.) Raf.(Cleomaceae). S. Afr. J. Bot. 2019, 124, 546–555. [Google Scholar] [CrossRef]
- Santos-Valle, A.B.C.; Souza, G.R.R.; Paes, C.Q.; Miyazaki, T.; Silva, A.H.; Altube, M.J.; Pittella, F. Nanomedicine strategies for addressing major needs in neglected tropical diseases. Annu. Rev. Control. 2019, 48, 423–441. [Google Scholar] [CrossRef]
- Santos, S.S.; De Araújo, R.V.; Giarolla, J.; Seoud, O.E.; Ferreira, E.I. Searching drugs for Chagas disease, leishmaniasis and schistosomiasis: A brief review. Int. J. Antimicrob. Agents 2020, 24, 105906. [Google Scholar] [CrossRef]
- Chami, G.F.; Bundy, D.A.P. More medicines alone cannot ensure the treatment of neglected tropical diseases. Lancet Infect. Dis. 2019, 199, 330–336. [Google Scholar] [CrossRef]
- Rampazzo, R.C.P.; Graziani, A.C.; Leite, K.K.; Surdi, J.A.; Biondo, C.A.; Costa, M.L.N.; Jacomasso, T.; Cereda, M.; De Fazio, M.; Bianchessi, M.A.; et al. Proof of Concept for a Portable Platform for Molecular Diagnosis of Tropical Diseases: On-Chip Ready-to-Use Real-Time Quantitative PCR for Detection of Trypanosoma cruzi or Plasmodium spp. J. Mol. Diagn. 2019, 5, 839–851. [Google Scholar] [CrossRef]
- Vargas, E.; Echeverri, F.; Upegui, Y.A.; Robledo, S.M.; Quiñones, W. Hydrazone Derivatives Enhance Antileishmanial Activity of Thiochroman-4-ones. Molecules 2018, 23, 70. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Brogi, S.; Grillo, A.; Campiani, G.; Gemma, S.; Vieira, P.C.; Maquiaveli, C.D.C. Cinnamic acids derived compounds with antileishmanial activity target Leishmania amazonensis arginase. Chem. Biol. Drug. Des. 2019, 93, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.; Rocha, H.; Carvalho, C.; Bandeira, V.; Fonseca, C.; Rosalino, L.M.; Cunha, M.V. Molecular detection and characterization of Leishmania infantum in free-ranging Egyptian mongoose (Herpestes ichneumon). Int. J. Parasitol. Parasites Wildl. 2020, 11, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Diotallevi, A.; Buffi, G.; Ceccarelli, M.; Neitzke-Abreu, H.C.; Gnutzmann, L.V.; da Costa Lima, M.S., Jr.; Di Domenico, A.; De Santi, M.; Magnani, M.; Galluzzi, L. Real-time PCR to differentiate among Leishmania (Viannia) subgenus, Leishmania (Leishmania) infantum and Leishmania (Leishmania) amazonensis: Application on Brazilian clinical samples. Acta Trop. 2020, 201. [Google Scholar] [CrossRef] [PubMed]
- Bott, E.; López, M.G.; Lammel, E.M.; Carfagna, I.E.; Durante de Isola, E.L.; Ruybal, P.; Taboga, O.; Gimenez, G.; Belaunzarán, M.L. Cellular localization, cloning and expression of Leishmania braziliensis Phospholipase A1. Microb. Pathog. 2020, 28. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.A.; Vasconcelos dos Santos, T.; do Rêgo Lima, L.V.; Ramos, P.K.S.; Campos, M.B.; Silveira, F.T. First report on feline leishmaniasis caused by Leishmania (Leishmania) amazonensis in Amazonian Brazil. Vet. Parasitol. Reg. Stud. Rep. 2020, 19. [Google Scholar] [CrossRef]
- Dolat, E.; Salarabadi, S.S.; Layegh, P.; Jaafari, M.R.; Sazgarnia, S.; Sazgarnia, A. The effect of UV radiation in the presence of TiO2-NPs on Leishmania major promastigotes. Biochim. Biophys. Acta Gen. Subj. 2020, 1864. [Google Scholar] [CrossRef] [PubMed]
- Akkawi, M.; Abu-Lafi, S.; Attieh, H.; Abu-Remeleh, Q.; Makhamra, S.; Qutob, M. Preparative HPLC fractionation of Cinnamomum cassia Water Extract and their in-vitro Antimalarial Activities. J. Appl. Pharm. Sci. 2017, 7, 129–134. [Google Scholar] [CrossRef][Green Version]
- Mukherjee, D.; Chora, Â.F.; Mota, M.M. Microbiota, a Third Player in the Host–Plasmodium Affair. Trends Parasitol. 2019, 36, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Deepa, P.; Thirumeignanam, D. Understanding the potency of malarial ligand (D44) in Plasmodium FKBP35 and modelled halogen atom (Br, Cl, F) functional groups. J. Mol. Graph. Model. 2020, 97. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Galluzzi, L.; Diotallevi, A.; Andreoni, F.; Fowler, H.; Petersen, C.; Magnani, M. The use of kDNA minicircle subclass relative abundance to differentiate between Leishmania (L.) infantum and Leishmania (L.) amazonensis. Parasit Vectors 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Demari-Silva, B.; Laporta, G.Z.; Oliveira, T.; Sallum, M. Plasmodium infection in Kerteszia cruzii (Diptera: Culicidae) in the Atlantic tropical rain forest, southeastern Brazil. Infect. Genet. Evol. 2019, 78, 78. [Google Scholar] [CrossRef]
- Wahab, A.; Shaukat, A.; Ali, Q.; Hussain, M.; Khan, T.A.; Khan, M.A.U.; Rashid, I.; Saleem, M.A.; Evans, M.; Sargison, N.D.; et al. A novel metabarcoded 18S ribosomal DNA sequencing tool for the detection of Plasmodium species in malaria positive patients. Infect. Genet. Evol. 2020, 82. [Google Scholar] [CrossRef]
- Qidwai, T. Exploration of copy number variation in genes related to anti-malarial drug resistance in Plasmodium falciparum. Gene 2020, 736. [Google Scholar] [CrossRef]
- Nqoro, X.; Tobeka, N.; Aderibigbe, B.A. Quinoline-Based Hybrid Compounds with Antimalarial Activity. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Gayam, V.; Ravi, S. Cinnamoylated chloroquine analogues: A new structural class of antimalarial agents. Eur. J. Med. Chem. 2017, 135, 382–391. [Google Scholar] [CrossRef]
- Tajuddeen, N.; Van Heerden, F.R. Antiplasmodial natural products: An update. Malar. J. 2019, 18, 404. [Google Scholar] [CrossRef]
- Gao, C.; Chang, L.; Xu, Z.; Yan, X.F.; Ding, C.; Zhao, F.; Wu, X.; Feng, L.S. Recent advances of tetrazole derivatives as potential anti-tubercular and anti-malarial agents. Eur. J. Med. Chem. 2019, 163, 404–412. [Google Scholar] [CrossRef]
- Silva, E.C.O.; Santos, F.M.; Ribeiro, A.; Souza, S.T.; Barreto, E.; Fonseca, E.J.S. Drug-induced anti-inflammatory response in A549 cells, as detected by Raman spectroscopy: A comparative analysis of the actions of dexamethasone and p-coumaric acid. Analyst 2019, 144, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M.A.; Kotyńska, J.; Kusaczuk, M.; Gál, M.; Naumowicz, M. The Modulating Effect of p-Coumaric Acid on The Surface Charge Density of Human Glioblastoma Cell Membranes. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Eroğlu, C.; Avcı, E.; Vural, H.; Kurar, E. Anticancer mechanism of Sinapic acid in PC-3 and LNCaP human prostate cancer cell lines. Gene 2018, 671, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.; Matsumoto, H. Urinary excretion rate and bioavailability of chlorogenic acid, caffeic acid, p-coumaric acid, and ferulic acid in non-fasted rats maintained under physiological conditions. Heliyon 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jia, Z.; Wan, G.; Wang, S.; Min, D. Enhancing isolation of p-coumaric and ferulic acids from sugarcane bagasse by sequential hydrolysis. Chem. Pap. 2019, 74, 499–507. [Google Scholar] [CrossRef]
- Kumar, D.; Shahid, M. (Eds.) Natural Materials and Products from Insects: Chemistry and Applications; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Ferreira, P.S.; Victorelli, F.D.; Fonseca-Santos, B.; Chorilli, M. A Review of Analytical Methods for p-Coumaric Acid in Plant-Based Products, Beverages, and Biological Matrices. Crit. Rev. Anal. Chem. 2019, 49, 21–31. [Google Scholar] [CrossRef]
- Rodriguez, A.; Chen, Y.; Khoomrung, S.; Özdemira, E.; Borodinaa, I.; Nielsen, J. Comparison of the metabolic response to over-production of p-coumaric acid in two yeast strains. Metab. Eng. 2017, 44, 265–272. [Google Scholar] [CrossRef]
- Zabad, O.M.; Samra, Y.A.; Eissa, L.A. p-Coumaric acid alleviates experimental diabetic nephropathy through modulation of toll like receptor-4 in rats. Life Sci. 2019, 238, 116965. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Magotra, A.; Rath, S.K.; Wazir, P.; Nandi, U.; Koul, S.; Sangwan, P.L.; Gupta, A.P.; Singh, G. In-vitro and in-vivo pharmacokinetics of IS01957, p-coumaric acid derivative using a validated LC–ESI–MS/MS method in mice plasma. J. Pharm. Investig. 2018, 48, 565–574. [Google Scholar] [CrossRef]
- Boo, Y.C. p-Coumaric Acid as An Active Ingredient in Cosmetics: A Review Focusing on its Antimelanogenic Effects. Antioxidants 2019, 8. [Google Scholar] [CrossRef]
- Contardi, M.; Heredia-Guerrero, J.A.; Guzman-Puyol, S.; Summa, M.; Benítez, J.J.; Goldoni, L.; Caputo, G.; Cusimano, G.; Picone, P.; Di Carlo, M.; et al. Combining dietary phenolic antioxidants with polyvinylpyrrolidone: Transparent biopolymer films based on p-coumaric acid for controlled release. J. Mater. Chem. B 2019, 7, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Contardi, M.; Alfaro-Pulido, A.; Picone, P.; Guzman-Puyol, S.; Goldoni, L.; Benítez, J.J.; Heredia, A.; Barthel, M.J.; Ceseracciu, L.; Cusimano, G.; et al. Low molecular weight ε-caprolactone-p-coumaric acid copolymers as potential biomaterials for skin regeneration applications. PLoS ONE 2019, 14, e0214956. [Google Scholar] [CrossRef]
- Yue, Y.; Shen, P.; Xu, Y.; Park, Y. p-Coumaric acid improves oxidative and osmosis stress responses in Caenorhabditis elegans. J. Sci. Food Agric. 2019, 99, 1190–1197. [Google Scholar]
- Long, R.; Li, T.; Tong, C.; Wu, L.; Shi, S. Molecularly imprinted polymers coated CdTe quantum dots with controllable particle size for fluorescent determination of p-coumaric acid. Talanta 2019, 196, 579–584. [Google Scholar] [CrossRef]
- Shen, Y.; Song, X.; Li, L.; Sun, J.; Jaiswal, Y.; Huang, J.; Liu, C.; Yang, W.; Williams, L.; Zhang, H.; et al. Protective effects of p-coumaric acid against oxidant and hyperlipidemia-an in vitro and in vivo evaluation. Biomed. Pharmacother. 2019, 111, 579–587. [Google Scholar] [CrossRef]
- Silva, A.T.; Bento, C.M.; Pena, A.C.; Figueiredo, L.M.; Prudêncio, C.; Aguiar, L.; Silva, T.; Ferraz, R.; Gomes, M.S.; Teixeira, C.; et al. Cinnamic Acid Conjugates in the Rescuing and Repurposing of Classical Antimalarial Drugs. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Monzote, L.; Córdova, W.H.P.; García, M.; Piñón, A.; Setzer, W.N. In-vitro and In-vivo Activities of Phenolic Compounds Against Cutaneous Leishmaniasis. Rec. Nat. Prod. 2016, 10, 269–276. [Google Scholar]
- Sudi, S.; Hassan, W.R.M.; Ali, A.H.; Basir, R.; Embi, N.; Sidek, H.M. A derivative of cinnamic acid, methyl-4-hydroxycinnamate modulates inflammatory cytokine levels in malaria-infected mice through inhibition of gsk3β. Malays Appl. Biol. 2018, 47, 153–157. [Google Scholar]
- Narayanaswamy, R.; Kok, L.; Ismail, I.S. Natural Compounds as Inhibitors of Plasmodium Falciparum Enoyl-acyl Carrier Protein Reductase (PfENR): An In silico Study. J. Chosun. Nat. Sci. 2017, 10, 1–6. [Google Scholar] [CrossRef][Green Version]
- Lopes, S.P.; Castillo, Y.P.; Monteiro, M.L.; Menezes, R.R.P.P.B.; Almeida, R.N.; Martins, A.M.C.; Sousa, D.P. Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Nóbrega, F.R.; Silva, L.V.; Filho, C.S.M.B.; Lima, T.C.; Castillo, Y.P.; Bezerra, D.P.; Lima, T.K.S.; De Sousa, D.P. Design, Antileishmanial Activity, and QSAR Studies of a Series of Piplartine Analogues. J. Chem. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Lima, T.C.; Souza, R.J.; Santos, A.D.; Moraes, M.H.; Biondo, N.E.; Barison, A.; Steindel, M.; Biavatti, M.W. Evaluation of leishmanicidal and trypanocidal activities of phenolic compounds from Calea uniflora Less. Nat. Prod. Res. 2016, 30, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Lira, A.B.; Montenegro, C.A.; Oliveira, K.M.; Filho, A.A.O.; Paz, A.R.; Araújo, M.O.; Sousa, D.M.; Almeida, C.L.F.; Silva, T.G.; Lima, C.M.B.L. Isopropyl Caffeate: A Caffeic Acid Derivative-Antioxidant Potential and Toxicity. Oxid. Med. Cell Longev. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.A.; Schmid, T.J. A Comprehensive QSAR Study on Antileishmanial and Antitrypanosomal Cinnamate Ester Analogues. Molecules 2019, 24(23), 4358. [Google Scholar] [CrossRef]
- Bernal, F.A.; Kaiser, M.; Wünsch, B.; Schmidt, T.J. Structure—Activity Relationships of Cinnamate Ester Analogs as Potent Antiprotozoal Agents. ChemMedChem 2019, 15, 68–78. [Google Scholar] [CrossRef]
- Otero, E.; García, E.; Palacios, G.; Yepes, L.M.; Carda, M.; Agut, R.; Vélez, I.D.; Cardona, W.I.; Robledo, S.M. Triclosan-caffeic acid hybrids: Synthesis, leishmanicidal, trypanocidal and cytotoxic activities. Eur. J. Med. Chem. 2017, 141, 73–83. [Google Scholar] [CrossRef]
- Steverding, D.; Da Nóbrega, F.R.; Rushworth, S.A.; De Sousa, D.P. Trypanocidal and cysteine protease inhibitory activity of isopentyl caffeate is not linked in Trypanosoma brucei. Parasitol. Res. 2016, 115, 4397–4403. [Google Scholar] [CrossRef]
- Otero, E.; Robledo, S.; Diaz, S.; Carda, M.; Muñoz, D.; Paños, J.; Vélez, I.; Cardona, W. Synthesis and Leishmanicidal Activity of Cinnamic Acid Esters: Structure-Activity Relationship. Med. Chem. Res. 2014, 23, 1378–1386. [Google Scholar] [CrossRef]
- Jorda, R.; Sacerdoti-Sierra, N.; Voller, J.; Havlíček, L.; Kráčalíková, K.; Nowicki, M.W.; Nasereddin, A.; Kryštof, V.; Strnad, M.; Walkinshaw, M.D.; et al. Anti-leishmanial activity of disubstituted purines and related pyrazolo[4,3-d]pyrimidines. Bioorg. Med. Chem. Lett. 2011, 21, 4233–4237. [Google Scholar] [CrossRef] [PubMed]
- Sghaier, R.M.; Aissa, I.; Attia, H.; Bali, A.; Leon Martinez, P.A.; Mkannez, G.; Guerfali, F.Z.; Gargouri, Y.; Laouini, D. Treatment with synthetic lipophilic tyrosyl ester controls Leishmania major infection by reducing parasite load in BALB/c mice. Parasitology 2016, 143, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Júnior, D.; Froes, T.; Silva, P.; Egito, M.; Moreira, P.; Varotti, F.; Castilho, M.; Neto, R.; Albuquerque, J.; et al. Antileishmanial activity evaluation of thiazolidine-2,4-dione against Leishmania infantum and Leishmania braziliensis. Parasitol Res. 2020, 10. [Google Scholar] [CrossRef]
- Debnath, M.; Abbasi, M.; Sasmal, S.; Datta, R.; Haldar, D. m-Nitrocinnamic Acid Containing Lipophilic Peptide Exhibits Selective Growth Inhibition Activity against Leishmania major. Chem. Select. 2019, 4, 116–122. [Google Scholar] [CrossRef]
- Alson, S.G.; Jansen, O.; Cieckiewicz, E.; Rakotoarimanana, H.; Rafatro, H.; Degotte, G.; Francotte, P.; Frederich, M. In-vitro and in-vivo antimalarial activity of caffeic acid and some of its derivatives. J. Pharm. Pharmacol. 2018, 70, 1349–1356. [Google Scholar] [CrossRef]
- Arsianti, A.; Astuti, H.; Fadilah, F.; Simadibrata, D.; Adyasa, Z.; Amartya, D.; Bahtiar, A.; Tanimoto, H.; Kakiuchi, K. Synthesis and in Vitro Antimalarial Activity of Alkyl Esters Gallate as a Growth Inhibitors of Plasmodium Falciparum. Orient. J. Chem. 2018, 34, 655–662. [Google Scholar] [CrossRef]
- Arsianti, A.; Astuty, H.; Fadilah, T.; Bahtiar, A.; Tanimoto, H.; Kakiuchi, K. Design and screening of gallic acid derivatives as inhibitors of malarial dihydrofolate reductase by in silico docking. Asian J. Pharm. Clin. Res. 2017, 10, 330–334. [Google Scholar] [CrossRef][Green Version]
- Cheng, C.H.; Cheng, Y.P.; Chang, I.L.; Chen, H.Y.; Wu, C.C.; Hsieh, C.P. Dodecyl gallate induces apoptosis by upregulating the caspase-dependent apoptotic pathway and inhibiting the expression of anti-apoptotic Bcl-2 family proteins in human osteosarcoma cells. Mol. Med. Rep. 2016, 13, 1495–1500. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef]
- Iturrate, P.M.; Sebastián-Pérez, V.; Nácher-Vázquez, M.; Tremper, C.S.; Smirlis, D.; Martín, J.; Martínez, A.; Campillo, N.E.; Rivas, L.; Gila, C. Towards discovery of new leishmanicidal scaffolds able to inhibit Leishmania GSK-3. J. Enzyme Inhib. Med. Chem. 2020, 35, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, T.; Mukherjee, M.; Das, A.; Mandal, C.; Das, R.; Mukherjee, T.; Majumder, H.K. Characterization of the ATPase activity of topoisomerase II from Leishmania donovani and identification of residues conferring resistance to etoposide. Biochem. J. 2005, 390, 419–426. [Google Scholar] [CrossRef]
- Keighobadi, M.; Fakhar, M.; Emami, S. Hypothesis: The potential application of doxorubicin against cutaneous leishmaniasis. Trop. Parasitol. 2015, 5, 69–70. [Google Scholar] [PubMed]
- Turkez, H.; Nóbrega, F.R.; da Ozdemir, O.; da Silva, C.B.F.; Almeida, R.N.; Tejera, E.; Castillo, Y.P.; Sousa, D.P. NFBTA: A Potent Cytotoxic Agent against Glioblastoma. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, R.D.M.; Duarte, M.C.; Mattos, E.C.; Martins, V.T.; Lage, P.S.; Chávez-Fumagalli, M.A.; Daniela, P.; Lage, D.P.; Menezes-Souza, D.; Régis, W.C.B.; et al. Identification of Differentially Expressed Proteins from Leishmania amazonensis Associated with the Loss of Virulence of the Parasites. PLoS Negl. Trop. Dis. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Melzer, I.M.; Kruse, M.; Sander-Juelch, C.; Wiese, M. LmxMPK4, a mitogen-activated protein (MAP) kinase homologue essential for promastigotes and amastigotes of Leishmania mexicana. Kinetoplastid. Biol. Dis. 2005, 4, 4. [Google Scholar] [CrossRef]
- Dacher, M.; Morales, M.A.; Pescher, P.; Leclercq, O.; Rachidi, N.; Prina, E.; Cayla, M.; Descoteaux, A.; Späth, G.F. Probing druggability and biological function of essential proteins in Leishmania combining facilitated null mutant and plasmid shuffle analyses. Mol. Microbiol. 2014, 93, 146–166. [Google Scholar] [CrossRef]
- Jones, N.G.; Catta-Preta, C.M.C.; Lima, A.P.C.A.; Mottram, J.C. Genetically Validated Drug Targets in Leishmania: Current Knowledge and Future Prospects. ACS Infect. Dis. 2018, 4, 467–477. [Google Scholar] [CrossRef]
- Balaña-Fouce, R.; Álvarez-Velilla, R.; Fernández-Prada, C.; García-Estrada, C.; Reguera, R.M. Trypanosomatids topoisomerase re-visited. New structural findings and role in drug discovery. Int. J. Parasitol. Drugs Drug. Resist. 2014, 4, 326–337. [Google Scholar] [CrossRef]
- Khatkar, A.; Nanda, A.; Kumar, P.; Narasimhan, B. Synthesis, antimicrobial evaluation and QSAR studies of p-coumaric acid derivaties. Arab. J. Chem. 2017, 10, 3804–3815. [Google Scholar] [CrossRef]
- Nishimura, K.; Takenaka, Y.; Kishi, M.; Tanahashi, T.; Yoshida, H.; Okuda, C.; Mizushin, Y. Synthesis and DNA Polymerase α and β Inhibitory Activity of Alkyl p-Coumarates and Related Compounds. Chem. Pharm. Bull. 2009, 57, 476–480. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, V.M.; Cedeño, D.L.; Muñoz, D.L.; Jones, M.A.; Lash, T.D.; Young, A.M.; Constantino, M.H.; Esposito, N.; Velez, I.D.; Robledo, S.M. In Vitro and in Vivo studies of the utility of dimethyl and diethyl carbaporphyrin ketals in treatment of cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2011, 55, 4755–4764. [Google Scholar] [CrossRef] [PubMed]
- Pulido, S.A.; Muñoz, D.L.; Restrepo, A.M.; Mesa, C.V.; Alzate, J.F.; Velez, I.D.; Robledo, S.M. Improvement of the green fluorescent protein reporter system in Leishmania spp. for the in vitro and in vivo screening of antileishmanial drugs. Acta Trop. 2012, 122, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Insuasty, D.; Robledo, S.M.; Vélez, I.D.; Cuervo, P.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J.; Abonia, R. A Schmidt rearrangement-mediated synthesis of novel tetrahydro-benzo[1,4]diazepin-5-ones as potential anticancer and antiprotozoal agents. Eur. J. Med. Chem. 2017, 141, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Finney, D.J. Statistical Method in Biological Assay (Mathematics in Medicine Series); Hodder Arnold: London, UK, 1978. [Google Scholar]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA. Santa Fe, NM: OpenEye Scientific Software. 2020. Available online: http://www.eyesopen.com (accessed on 8 July 2020).
- OpenEye Scientific Software. QUACPAC. Santa Fe, NM: OpenEye Scientific Software. 2020. Available online: http://www.eyesopen.com (accessed on 8 July 2020).
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1-12 are available from authors (or from MDPI). |




{kind=link}
{kind=link}
{kind=link}
| p-Coumaric Acid Esters | ||||||
|---|---|---|---|---|---|---|
| Leishmania braziliensis Amastigotes | Plasmodium falciparum | U-937 Macrophages | Molar Mass (g/mol) | |||
| EC50 (μg/mL)a | SI | EC50 (μg/mL)b | SI | LC50 (μg/mL) | ||
| 1 | 8.28 ± 0.14 | 0.76 | 64.59 ± 2.89 | 0.1 | 6.26 ± 0.03 | 178.18 |
| 2 | 4.91 ± 0.64 | 0.21 | 110.31 ± 30.84 | 0.01 | 1.06 ± 1.08 | 192.21 |
| 3 | 6.3 ± 1.14 | 1.89 | 139.04 ± 4.07 | 0.09 | 11.89 ± 0.8 | 206.23 |
| 4 | 28.33 ± 0.27 | 3.4 | 106.08 ± 9.97 | 0.91 | 96.23 ± 18.89 | 206.23 |
| 5 | 86.98 ± 18.13 | 1.46 | 98.66 ± 17.7 | 1.28 | 126.71 ± 16.5 | 222.23 |
| 6 | 323.29 ± 50.04 | 0.1 | 198.84 ± 7.9 | 0.16 | 30.9 ± 6.32 | 220.26 |
| 7 | 32.03 ± 5.05 | 0.5 | 95.48 ± 1.84 | 0.17 | 16.09 ± 3.81 | 234.29 |
| 8 | 6.02 ± 0.22 | 3.25 | 156.37 ± 8.2 | 0.13 | 19.58 ± 1.76 | 234.29 |
| 9 | 4.14 ± 0.55 | 2.72 | 206.35 ± 46.4 | 0.05 | 11.24 ± 0.86 | 248.31 |
| 10 | 16.34 ± 0.49 | 0.94 | 81.19 ± 8.54 | 0.19 | 15.43 ± 1.84 | 332.47 |
| 11 | 5.69 ± 0.3 | 3.66 | 83.98 ± 15.34 | 0.25 | 20.81 ± 3.47 | 268.30 |
| 12 | 11.21 ± 0.2 | 3.14 | 93.66 ± 14.17 | 0.38 | 35.2 ± 13.94 | 296.36 |
| AMBc | 0.3 ± 0.07 | 128 | 38.4 ± 6.3 | |||
| CQd | 3.35 ± 0.39 | 14.1 | 47.26 ± 4.9 | |||
| L. braziliensis Target (a) | Description | ID |
|---|---|---|
| A4HED7_LEIBR | Serine/threonine-protein kinase | PKC |
| A4HE56_LEIBR | Aldehyde dehydrogenase, mitochondrial | ALDH2 |
| A4HP40_LEIBR | Aldehyde dehydrogenase | ALDH |
| A4HG21_LEIBR | Aldo-keto reductase protein | AKR |
| A4HGT2_LEIBR | DNA topoisomerase 2 | TOP2 |
| A4H9D1_LEIBR | Putative glycogen synthase kinase | GSK3 |
| A4HBR4_LEIBR | Histone deacetylase | HDA1 |
| A4HDK5_LEIBR | Histone deacetylase | HDA2 |
| A4HA94_LEIBR | Mitogen-activated protein kinase | MPK4 |
| A4H9X2_LEIBR | Mitogen-activated protein kinase | CBPKC |
| A4HJJ7_LEIBR | Prostaglandin-F synthase protein | PGFS |
| A4HK26_LEIBR | Prostaglandin f synthase | PGFS2 |
| A4H9L8_LEIBR | Protein kinase A catalytic subunit | PKAC3 |
| A4HN71_LEIBR | Protein kinase A catalytic subunit isoform 1 | PKAC2a |
| A4HEL2_LEIBR | Protein kinase domain-containing protein | CRK7 |
| A4H8N9_LEIBR | Tyrosyl or methionyl-tRNA synthetase-like protein | YARS1 |
| Target | Conformer | CHEMPLP | GoldScore | ChemScore | ASP | Consensus Z-Score | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | Z-Score | Score | Z-Score | Score | Z-Score | Score | Z-Score | |||
| PKC | 1 | 64.76 | 2.69 | 29.03 | 1.06 | 24.23 | 1.32 | 24.27 | 1.68 | 1.69 |
| 2 | 60.60 | 1.31 | 29.05 | 1.07 | 23.04 | 0.71 | 23.60 | 1.35 | 1.11 | |
| ALDH2 | 1 | 63.77 | 2.22 | 34.27 | 1.46 | 21.22 | 0.76 | 25.99 | 0.47 | 1.23 |
| ALDH | 1 | 64.64 | 1.97 | 66.89 | 1.54 | 20.48 | 1.65 | 27.38 | 0.79 | 1.49 |
| 2 | 59.38 | 0.56 | 69.68 | 2.24 | 20.25 | 1.54 | 27.18 | 0.71 | 1.26 | |
| 3 | 62.51 | 1.40 | 66.04 | 1.32 | 17.22 | 0.09 | 29.32 | 1.63 | 1.11 | |
| 4 | 62.25 | 1.33 | 65.00 | 1.06 | 17.98 | 0.45 | 29.19 | 1.57 | 1.10 | |
| AKR | 1 | 60.32 | 2.04 | 10.04 | 1.10 | 25.09 | 2.50 | 37.69 | 2.35 | 2.00 |
| 2 | 61.32 | 2.17 | −8.41 | 0.13 | 21.15 | 1.84 | 26.77 | 1.04 | 1.30 | |
| 3 | 51.69 | 0.90 | −2.36 | 0.45 | 18.34 | 1.37 | 29.66 | 1.39 | 1.03 | |
| TOP2 | 1 | 57.19 | 1.26 | 33.54 | 1.29 | 19.27 | 1.92 | 23.73 | 1.36 | 1.46 |
| 2 | 57.11 | 1.24 | 32.47 | 1.01 | 16.63 | 0.85 | 22.31 | 0.92 | 1.00 | |
| GSK3 (a) | 1 | 70.94 | 2.37 | 34.80 | 1.27 | 24.32 | 1.97 | 32.04 | 2.70 | 2.08 |
| 2 | 67.43 | 1.62 | 29.45 | 0.22 | 23.34 | 1.40 | 29.85 | 1.66 | 1.22 | |
| GSK3 SBS (a) | 1 | 50.52 | 1.64 | 25.70 | 0.50 | 13.30 | 0.87 | 18.51 | 1.87 | 1.22 |
| 2 | 48.54 | 1.17 | 27.61 | 0.72 | 14.49 | 1.32 | 17.12 | 1.34 | 1.14 | |
| HDA1 | 1 | 65.54 | 1.28 | 29.53 | 0.57 | 24.30 | 0.79 | 31.58 | 1.40 | 1.01 |
| HDA2 | 1 | 67.10 | 0.94 | 40.20 | 1.55 | 27.36 | 1.03 | 35.93 | 1.33 | 1.21 |
| MPK4 | 1 | 58.40 | 2.11 | 32.17 | 1.73 | 20.59 | 0.66 | 20.43 | 0.70 | 1.30 |
| 2 | 55.89 | 1.23 | 26.35 | 0.12 | 24.14 | 1.92 | 21.72 | 1.27 | 1.14 | |
| 3 | 57.14 | 1.67 | 33.77 | 2.17 | 20.50 | 0.63 | 18.31 | −0.25 | 1.05 | |
| CBPKC | 1 | 54.89 | 2.04 | 24.42 | 1.07 | 17.09 | 0.06 | 19.03 | 0.89 | 1.01 |
| PGFS | 1 | 50.11 | 1.89 | 24.68 | 1.70 | 13.91 | 1.14 | 30.81 | 1.82 | 1.64 |
| PGFS2 | 1 | 32.76 | 1.06 | 13.87 | 1.33 | 7.74 | 1.27 | 20.71 | 1.51 | 1.29 |
| PKAC3 | 1 | 58.29 | 1.47 | 36.02 | 1.16 | 23.65 | 1.83 | 24.29 | 1.08 | 1.39 |
| 2 | 57.86 | 1.33 | 31.27 | 0.57 | 21.71 | 1.01 | 26.05 | 1.82 | 1.18 | |
| 3 | 56.43 | 0.84 | 36.64 | 1.24 | 21.06 | 0.73 | 24.93 | 1.35 | 1.04 | |
| PKAC2a | 1 | 61.50 | 2.15 | 32.33 | 1.09 | 24.27 | 1.72 | 20.13 | −0.17 | 1.20 |
| CRK7 | 1 | 66.15 | 2.13 | 36.34 | 2.04 | 24.01 | 1.60 | 25.98 | 1.43 | 1.80 |
| 2 | 65.15 | 1.86 | 34.07 | 1.38 | 23.39 | 1.36 | 24.01 | 0.65 | 1.31 | |
| 3 | 63.92 | 1.53 | 33.69 | 1.27 | 21.55 | 0.62 | 25.55 | 1.26 | 1.17 | |
| YARS1 | 1 | 61.62 | 2.08 | 21.92 | 1.15 | 21.95 | 1.56 | 19.98 | 0.23 | 1.26 |
| 2 | 57.26 | 0.97 | 16.22 | 0.80 | 21.32 | 1.24 | 23.33 | 1.52 | 1.13 | |
| Target | Conformer | MM-PBSA Components | ΔG TOTAL(h) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| VDWAALS(a) | EEL(b) | EPB(c) | ENPOLAR(d) | EDISPER(e) | ΔG Gas(f) | ΔG Solv(g) | |||
| PKC | 1 | −31.35 | −27.22 | 37.23 | −27.27 | 43.17 | −58.57 | 53.12 | −5.45 |
| 2 | −33.93 | −15.64 | 29.63 | −28.73 | 46.01 | −49.57 | 46.91 | −2.66 | |
| ALDH2 | 1 | −29.85 | −26.61 | 36.34 | −25.56 | 43.08 | −56.46 | 53.86 | −2.60 |
| ALDH | 1 | −38.10 | −15.73 | 36.01 | −29.50 | 48.51 | −53.83 | 55.02 | 1.19 |
| 2 | −37.59 | −22.81 | 36.17 | −30.23 | 49.25 | −60.40 | 55.19 | −5.21 | |
| 3 | −34.69 | −23.13 | 37.63 | −28.16 | 46.95 | −57.82 | 56.43 | −1.39 | |
| 4 | −38.78 | −30.30 | 40.58 | −30.43 | 50.68 | −69.08 | 60.83 | −8.25 | |
| AKR | 1 | −30.88 | −13.99 | 28.85 | −25.79 | 40.67 | −44.87 | 43.74 | −1.13 |
| 2 | −28.48 | −7.63 | 22.63 | −22.28 | 36.82 | −36.11 | 37.17 | 1.06 | |
| 3 | −29.47 | −13.77 | 32.42 | −23.82 | 39.14 | −43.24 | 47.74 | 4.50 | |
| TOP2 | 1 | −42.66 | −24.71 | 42.55 | −31.82 | 51.87 | −67.37 | 62.60 | −4.76 |
| 2 | −46.13 | −16.83 | 35.84 | −31.37 | 52.34 | −62.96 | 56.80 | −6.15 | |
| GSK3 | 1 | −28.68 | −11.34 | 31.25 | −21.69 | 37.74 | −40.02 | 47.30 | 7.27 |
| 2 | −34.81 | −17.30 | 30.70 | −28.87 | 46.12 | −52.11 | 47.95 | −4.16 | |
| GSK3 (SBS) | 1 | −18.60 | −14.21 | 23.44 | −16.13 | 26.89 | −32.81 | 34.20 | 1.39 |
| 2 | −27.34 | −9.22 | 26.18 | −20.94 | 35.99 | −36.56 | 41.23 | 4.67 | |
| HDA1 | 1 | −28.68 | −16.20 | 31.32 | −22.81 | 37.85 | −44.88 | 46.36 | 1.47 |
| HDA2 | 1 | −25.51 | −24.37 | 35.37 | −22.39 | 35.83 | −49.88 | 48.80 | −1.08 |
| MPK4 | 1 | −30.88 | −24.72 | 35.63 | −26.12 | 42.49 | −55.60 | 51.99 | −3.61 |
| 2 | −36.54 | −13.69 | 31.88 | −29.36 | 46.60 | −50.23 | 49.11 | −1.11 | |
| 3 | −39.58 | −17.44 | 32.41 | −30.01 | 47.48 | −57.02 | 49.88 | −7.14 | |
| CBPKC | 1 | −32.78 | −22.89 | 35.50 | −27.42 | 44.34 | −55.67 | 52.42 | −3.25 |
| PGFS | 1 | −26.09 | −9.33 | 19.97 | −20.90 | 34.35 | −35.42 | 33.41 | −2.01 |
| PGFS2 | 1 | −22.24 | −11.63 | 20.66 | −18.94 | 30.60 | −33.87 | 32.32 | −1.55 |
| PKAC3 | 1 | −36.72 | −34.98 | 57.32 | −29.95 | 47.33 | −71.70 | 74.69 | 2.99 |
| 2 | −38.00 | −12.24 | 33.67 | −28.84 | 47.80 | −50.25 | 52.63 | 2.38 | |
| 3 | −37.74 | −34.00 | 57.82 | −30.26 | 47.61 | −71.74 | 75.18 | 3.44 | |
| PKAC2a | 1 | −33.06 | −7.86 | 27.04 | −25.35 | 43.07 | −40.92 | 44.76 | 3.84 |
| CRK7 | 1 | −40.13 | −18.73 | 38.07 | −31.07 | 48.14 | −58.86 | 55.14 | −3.72 |
| 2 | −34.61 | −12.95 | 30.38 | −27.47 | 45.17 | −47.56 | 48.08 | 0.52 | |
| 3 | −38.51 | −15.27 | 35.40 | −30.09 | 48.31 | −53.78 | 53.62 | −0.16 | |
| YARS1 | 1 | −25.99 | −11.84 | 23.28 | −21.71 | 35.63 | −37.83 | 37.19 | −0.64 |
| 2 | −24.81 | −17.62 | 28.89 | −22.35 | 34.35 | −42.43 | 40.89 | −1.54 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, S.P.; Yepes, L.M.; Pérez-Castillo, Y.; Robledo, S.M.; de Sousa, D.P. Alkyl and Aryl Derivatives Based on p-Coumaric Acid Modification and Inhibitory Action against Leishmania braziliensis and Plasmodium falciparum. Molecules 2020, 25, 3178. https://doi.org/10.3390/molecules25143178
Lopes SP, Yepes LM, Pérez-Castillo Y, Robledo SM, de Sousa DP. Alkyl and Aryl Derivatives Based on p-Coumaric Acid Modification and Inhibitory Action against Leishmania braziliensis and Plasmodium falciparum. Molecules. 2020; 25(14):3178. https://doi.org/10.3390/molecules25143178
Chicago/Turabian StyleLopes, Susiany P., Lina M. Yepes, Yunierkis Pérez-Castillo, Sara M. Robledo, and Damião P. de Sousa. 2020. "Alkyl and Aryl Derivatives Based on p-Coumaric Acid Modification and Inhibitory Action against Leishmania braziliensis and Plasmodium falciparum" Molecules 25, no. 14: 3178. https://doi.org/10.3390/molecules25143178
APA StyleLopes, S. P., Yepes, L. M., Pérez-Castillo, Y., Robledo, S. M., & de Sousa, D. P. (2020). Alkyl and Aryl Derivatives Based on p-Coumaric Acid Modification and Inhibitory Action against Leishmania braziliensis and Plasmodium falciparum. Molecules, 25(14), 3178. https://doi.org/10.3390/molecules25143178