Study on a Three-Step Rapid Assembly of Zolpidem and Its Fluorinated Analogues Employing Microwave-Assisted Chemistry


Abstract
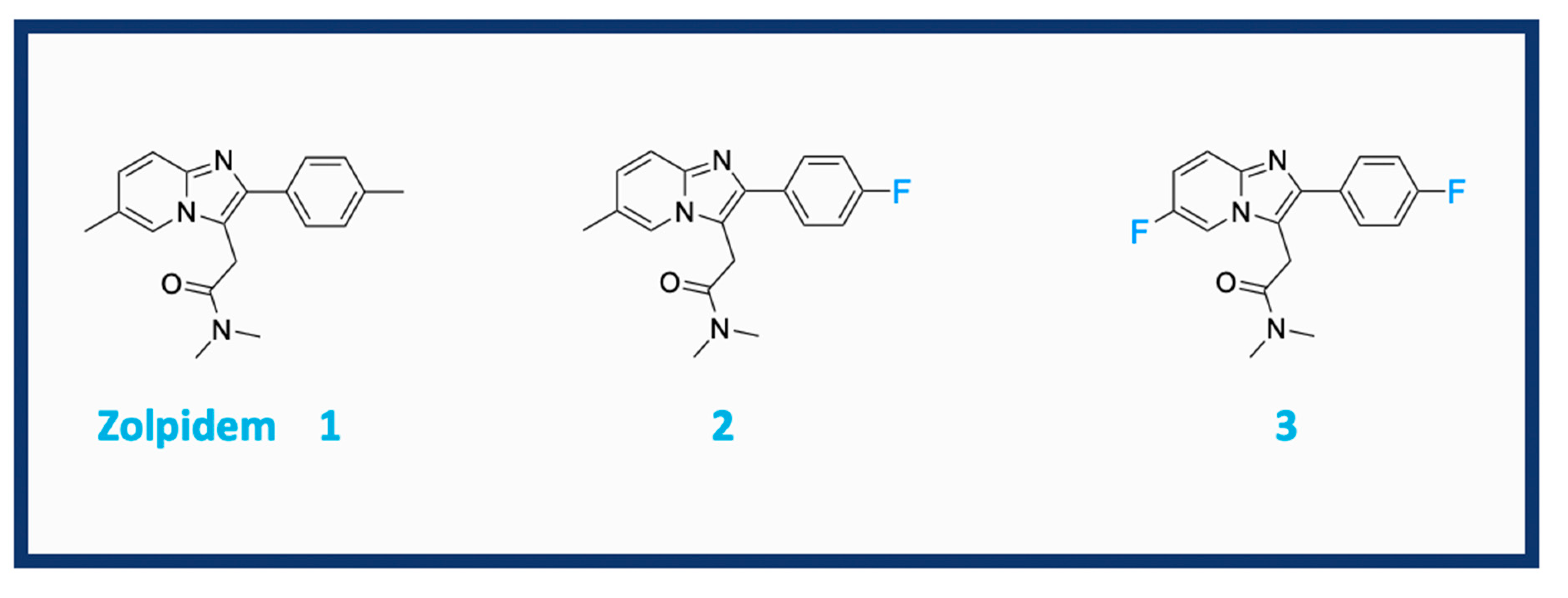
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Preparation of Water-Soluble Salts
2.3. Evaluation of the Particles’ Size
3. Materials and Methods
3.1. General Methods
3.2. Compound Characterization
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Harward, J.L.; Clinard, V.B.; Jiroutek, M.R.; Lingerfeldt, B.H.; Muzyk, A.J. Impact of a US Food and Drug Administration Drug Safety Communication on Zolpidem Dosing: An Observational Retrospective Cohort. Prim Care Companion CNS Disord. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.P.; George, P. Imidazo[1, 2-A] Pyridine Derivatives and Their Application as Pharmaceuticals. U.S. Patent 4,382,938A, 10 May 1983. [Google Scholar]
- George, P.; Allen, J. Imidazopyridines, leur Preparation et leur Application en Therapeutique. FR 2,606,409B1, 5 June 1989. [Google Scholar]
- Kaufmann, C.N.; Spira, A.P.; Alexander, G.C.; Rutkow, L. Trends in Prescribing of Sedative-Hypnotic Medications in the USA: 1993–2010. Pharmacoepidemiol. Drug Saf. 2016, 25, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Scatton, B.; Claustre, Y.; Dennis, T.; Nishikawa, T. Zolpidem, a novel nonbenzodiazepine hypnotic II. Effects on cerebellar cyclic GMP levels and cerebral monoamines. J. Pharmacol. Exp. Ther. 1986, 237, 659–665. [Google Scholar] [PubMed]
- Sutton, J.A.; Clauss, R.P. A review of the evidence of zolpidem efficacy in neurological disability after brain damage due to stroke, trauma and hypoxia: A justification of further clinical trials. Brain Inj. 2017, 31, 1019–1027. [Google Scholar] [CrossRef]
- Daniele, A.; Panza, F.; Greco, A.; Logroscino, G.; Seripa, D. Can a Positive Allosteric Modulation of GABAergic Receptors Improve Motor Symptoms in Patients with Parkinson’s Disease? The Potential Role of Zolpidem in the Treatment of Parkinson’s Disease. Park Dis. 2016, 2016, 2531812. [Google Scholar] [CrossRef]
- Puthiyathu, M.; Greenspan, H.; Levitt, W.; Riccardi, G.; Montemurro, N.; Nandu, B.; Waseem, S. The Analgesic Properties of Zolpidem. Psychiatr Ann. 2013, 43, 6–7. [Google Scholar] [CrossRef]
- Mierzejewski, P.; Kolaczkowski, M.; Marcinkowska, M.; Wesolowska, A.; Samochowiec, J.; Pawlowski, M.; Bienkowski, P. Antipsychotic-like effects of zolpidem in Wistar rats. Eur. J. Pharmacol. 2016, 773, 51–58. [Google Scholar] [CrossRef]
- Marcinkowska, M.; Kołaczkowski, M.; Kamiński, K.; Bucki, A.; Pawłowski, M.; Siwek, A.; Karcz, T.; Mordyl, B.; Starowicz, G.; Kubowicz, P.; et al. Design, synthesis, and biological evaluation of fluorinated imidazo[1,2-a]pyridine derivatives with potential antipsychotic activity. Eur. J. Med. Chem. 2016, 124, 456–467. [Google Scholar] [CrossRef]
- Marcinkowska, M.; Kołaczkowski, M.; Kamiński, K.; Bucki, A.; Pawłowski, M.; Siwek, A.; Tarcz, T.; Starowicz, G.; Bienkowski, P.; Mierzejewski, P.; et al. 3-Aminomethyl Derivatives of 2-Phenylimidazo[1,2-a]-pyridine as Positive Allosteric Modulators of GABAA Receptor with Potential Antipsychotic Activity. ACS Chem. Neurosci. 2017, 8, 1291–1298. [Google Scholar] [CrossRef]
- Nair, D.K.; Mobin, S.M.; Namboothiri, I.N.N. Synthesis of Imidazopyridines from the Morita–Baylis–Hillman Acetates of Nitroalkenes and Convenient Access to Alpidem and Zolpidem. Org. Lett. J. Am. Chem. Soc. 2012, 14, 4580–4583. [Google Scholar]
- Zhang, B.; Shan, G.; Ma, Q.; Xu, Q.; Lei, X. An improved and scalable synthesis of zolpidem via a CuI/BINOL-mediated tandem reaction of imine and alkyne. Heterocycl. Commun. 2017, 23, 445–448. [Google Scholar] [CrossRef]
- Sumatatha, Y.; Reddy, T.R.; Pratap, P. A simple and efficient synthesis of hypnotic agent, zolpidem and its related substances. Arkivoc 2009, 2, 315–320. [Google Scholar]
- Kappe, C.O.; Pieber, B.; Dallinger, D. Microwave Effects in Organic Synthesis: Myth or Reality? Angew. Chem. Int. Ed. 2013, 52, 1088–1094. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. Engl. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, S.; Chudoba, T.; Wojnarowicz, J.; Łojkowski, W. Current Trends in the Development of Microwave Reactors for the Synthesis of Nanomaterials in Laboratories and Industries: A Review. Crystals 2018, 8, 379. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Sui, Y.; She, Z. Effect of particle size on solubility, dissolution rate, and oral bioavailability: Evaluation using coenzyme Q10 as naked nanocrystals. Int. J. Nanomed. 2012, 7, 5733–5744. [Google Scholar]
- Hoz A de la Díaz-Ortiz, Á.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, S.; Osawa, A.; Abe, M.; Serpone, N. On the generation of hot-spots by microwave electric and magnetic fields and their impact on a microwave-assisted heterogeneous reaction in the presence of metallic Pd nanoparticles on an activated carbon support. J. Phys. Chem. 2011, 115, 23030–23035. [Google Scholar] [CrossRef]
- Schloemer, G.C. Process for the Preparation Imidazo[1,2-A]pyridine-3-acetamides. U.S. Patent 6,861,525, 1 March 2015. [Google Scholar]
- Eades, K. A Process for the Preparation of 2-Phenyl-Imidazo[1,2-A] Pyridine-3-Acetamides. U.S. Patent 6,384,226, 7 May 2002. [Google Scholar]
- Rossey, G.; Long, D. Process for the Preparation of Imidazopyridines. U.S. Patent 4,794,185A, 27 December 1988. [Google Scholar]
- Mousavipour, S.H.; Saheb, V.; Pirhadi, F.; Dehbozorgi, M.R. Experimental and theoretical study on the kinetics and mechanism of thermal decomposition of 1,2-dichloroethane. J. Iran. Chem. Soc. 2007, 4, 279–298. [Google Scholar] [CrossRef]
- Prinsloo, J.J.; Van Berge, P.C.; Zlotnick, J. The catalytic decomposition of 1,2-dichloroethane with activated carbon catalysts. J. Catal. 1974, 32, 466–469. [Google Scholar] [CrossRef]
- Alvarez-Manzaneda, E.J.; Chahboun, R.; Cabrera Torres, E.; Alvarez, E.; Alvarez-Manzaneda, R.; Haidour, A.; Lopez-Ramos, J.M. Reaction of allylic and benzylic alcohols and esters with PPh3/I2: One-pot synthesis of β,γ-unsaturated compounds. Tetrahedron Lett. 2005, 46, 3755–3759. [Google Scholar] [CrossRef]
- Fitzgerald, S.P.; McConnell, R.I. Immunoassay with Extended Detection Window. U.S. Patent 8,673,652, 18 March 2014. [Google Scholar]
- Ettema, G.; Lemmens, J.; Peters, T.; Pìcha, F. Zolpidem Salts. NL Patent 1,014,634C1, 8 March 2000. [Google Scholar]
- Ettema, G.J.B.; Lemmens, J.M.; Peters, T.H.A.; Picha, F. Zolpidem Salts. EP1,163,241B1, 15 June 2005. [Google Scholar]
- Foda, N.H.; Ali, S.M. Chapter 11—Zolpidem Tartrate. In Profiles Drug Subst Excip Relat Methodol; Brittain, H.G., Ed.; Academic Press: Amsterdam, The Netherlands, 2012; pp. 413–438. [Google Scholar]
- Krupa, A.; Descamps, M.; Willart, J.-F.; Strach, B.; Wyska, E.; Jachowicz, R.; Danède, F. High-Energy Ball Milling as Green Process to Vitrify Tadalafil and Improve Bioavailability. Mol. Pharm. 2016, 13, 3891–3902. [Google Scholar] [CrossRef]
- Ettema, G.J.B.; Lemmens, J.M.; Peters, T.H.A.; Picha, F. Zolpidem Salt Forms. U.S. Patent 6,242,460B1, 5 June 2001. [Google Scholar]
- Marcinkowska, M.; Kotańska, M.; Zagórska, A.; Śniecikowska, J.; Kubacka, M.; Siwek, A.; Bucki, A.; Pawlowski, M.; Bednarski, M.; Sapa, J.; et al. Synthesis and biological evaluation of N-arylpiperazine derivatives of 4,4-dimethylisoquinoline-1,3(2H,4H)-dione as potential antiplatelet agents. J. Enzyme Inhib. Med. Chem. 2018, 33, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Jagodzinska, M.; Huguenot, F.; Candiani, G.; Zanda, M. Assessing the bioisosterism of the trifluoromethyl group with a protease probe. Chem. Med. Chem. 2009, 4, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, M.J.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Hoffmanne, V.; László, P.; Reiter, J.; Simig, G.; Koncz, L.; Rivo, E.; Donath, G.; Nagy, K. Process for the Preparation of 6-Methyl-2-(4-methyl-phenyl)-imidazo 1,2-a]pyridine-3-(n,n-dimethyl-acetamide) and Intermediates. WO 2001/038327, 22 November 2001. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Time (minutes) | Yield (%) a |
| 1 | ethanol | 78 | 30 | 82.9 |
| 2 | methanol | 64 | 30 | 0 |
| 3 | acetone | 56 | 30 | 84 |
| 4 | dioxane | 101 | 30 | 88.5 |
| 5 | acetonitrile | 82 | 30 | 46.3 |
| 6 | THF | 66 | 30 | 90.7 |
| 7 | chloroform | 61 | 30 | 77.3 |
| 8 | toluene | 110 | 30 | 95.4 |
 | ||||||
| Compound | Conventional Method a | Microwave Assisted Method b | Yield (%) c | |||
|---|---|---|---|---|---|---|
| R1, R2 | Time (h) | Yield (%) c | Time | Solvent Amount (mL) | ||
| R1 = CH3 R2 = CH3 | 4 | 12 | 70 | 40 min | 5 | 71 |
| 3 | 90 | |||||
| R1 = CH3 R2 = F | 5 | 12 | 78 | 40 min | 5 | 75 |
| 3 | 82 | |||||
| R1 = F R2 = F | 6 | 12 | 85 | 40 min | 5 | 86 |
| 3 | 89 | |||||
 | |||||
| Compound | Conventional Method a | Microwave Assisted Method b | |||
|---|---|---|---|---|---|
| R1, R2 | Time (h) | Yield (%) c | Time (h) | Yield (%) c | |
| R1 = CH3 R2 = CH3 | 7 | 12 | 65 | 1 | 81 |
| R1 = CH3 R2 = F | 8 | 12 | 62 | 1 | 86 |
| R1 = F R2 = F | 9 | 12 | 57 | 1 | 88 |
 | ||||||
| Compound | Conventional Method a | Microwave Assisted Method b | ||||
|---|---|---|---|---|---|---|
| R1, R2 | Time | Yield (%) c | Time | Solvent | Yield (%) c | |
| R1 = CH3 R2 = CH3 | 1 | 6 | 70 | 45 min | THF | 71 |
| dioxane | 58 | |||||
| R1 = CH3 R2 = F | 2 | 12 | 70 | 45 min | THF | 82 |
| dioxane | 75 | |||||
| R1 = F R2 = F | 3 | 12 | 43 | 45 min | THF | 76 |
| dioxane | 58 | |||||
 | |||
| Standard Crystalization (Method A) | Direct Evaporation (Method B) | Crystalization + Et2O (Method C) | |
|---|---|---|---|
| Purity % | 99% | 88% | 99% |
| Yield % | 45% | 100% | 73% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajkis, N.; Marcinkowska, M.; Gryzło, B.; Krupa, A.; Kolaczkowski, M. Study on a Three-Step Rapid Assembly of Zolpidem and Its Fluorinated Analogues Employing Microwave-Assisted Chemistry. Molecules 2020, 25, 3161. https://doi.org/10.3390/molecules25143161
Fajkis N, Marcinkowska M, Gryzło B, Krupa A, Kolaczkowski M. Study on a Three-Step Rapid Assembly of Zolpidem and Its Fluorinated Analogues Employing Microwave-Assisted Chemistry. Molecules. 2020; 25(14):3161. https://doi.org/10.3390/molecules25143161
Chicago/Turabian StyleFajkis, Nikola, Monika Marcinkowska, Beata Gryzło, Anna Krupa, and Marcin Kolaczkowski. 2020. "Study on a Three-Step Rapid Assembly of Zolpidem and Its Fluorinated Analogues Employing Microwave-Assisted Chemistry" Molecules 25, no. 14: 3161. https://doi.org/10.3390/molecules25143161
APA StyleFajkis, N., Marcinkowska, M., Gryzło, B., Krupa, A., & Kolaczkowski, M. (2020). Study on a Three-Step Rapid Assembly of Zolpidem and Its Fluorinated Analogues Employing Microwave-Assisted Chemistry. Molecules, 25(14), 3161. https://doi.org/10.3390/molecules25143161