
Covalently Functionalized DNA Duplexes and Quadruplexes as Hybrid Catalysts in an Enantioselective Friedel–Crafts Reaction


Abstract

1. Introduction
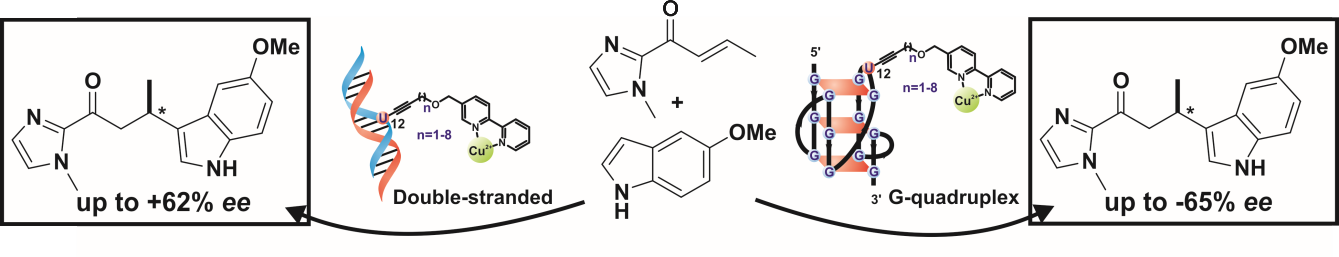
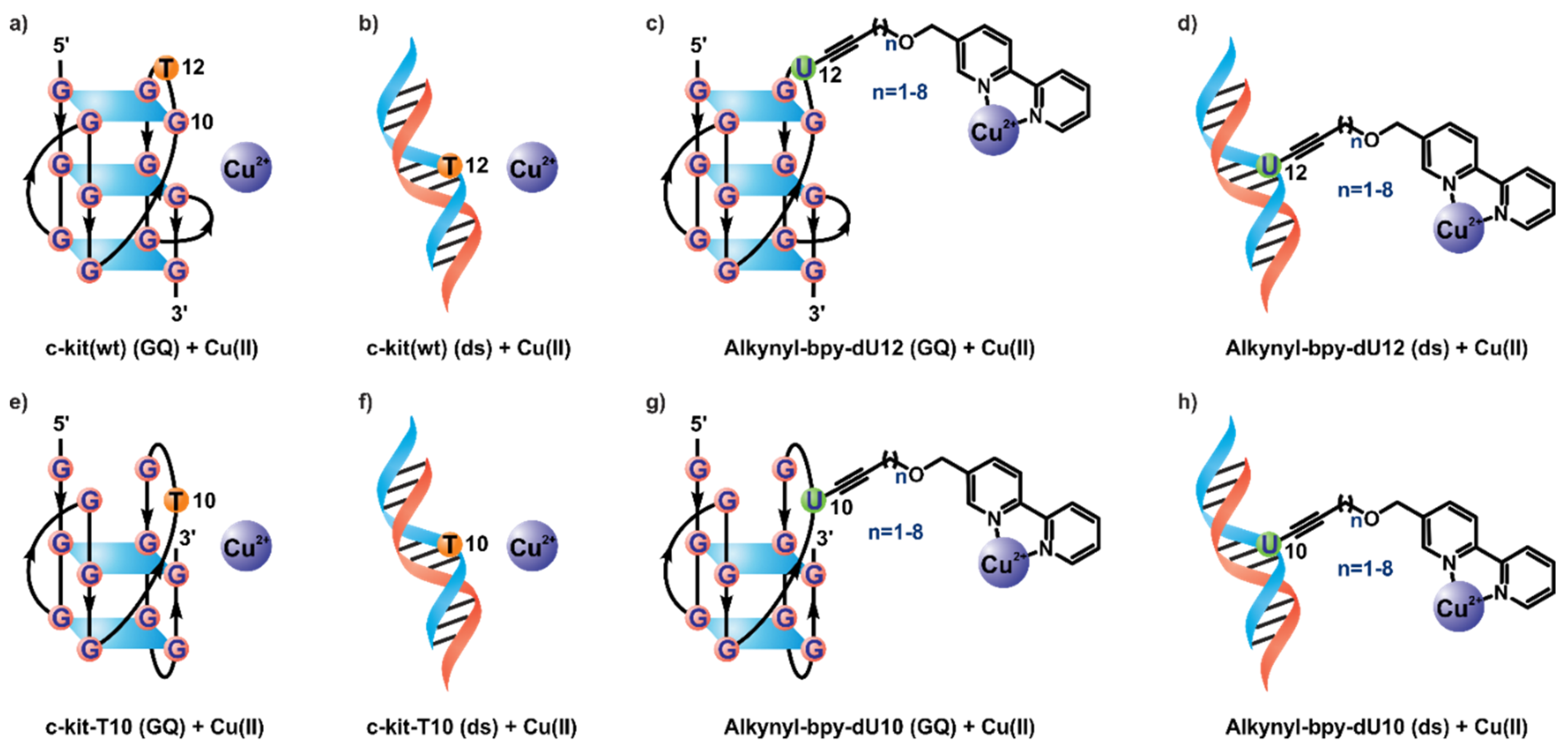
2. Results and Discussion
2.1. Synthesis
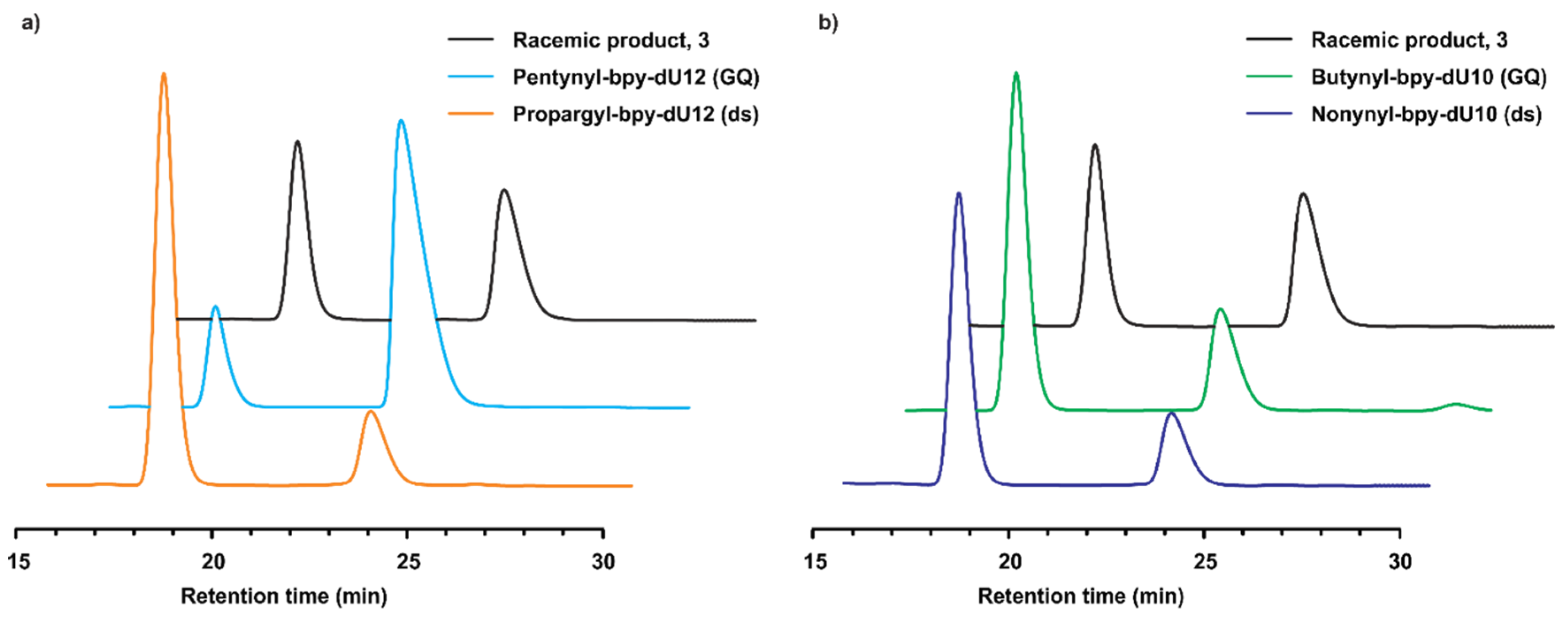
2.2. Catalytic Properties
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedel, C.; Crafts, J.M. C. R. Hebd. Seances Acad. Sci. 1877, 84, 1392. [Google Scholar]
- Friedel, C.; Crafts, J.M. C. R. Hebd. Seances Acad. Sci. 1877, 84, 1450. [Google Scholar]
- Poulsen, T.B.; Jørgensen, K.A. Catalytic Asymmetric Friedel−Crafts Alkylation Reactions—Copper Showed the Way. Chem. Rev. 2008, 108, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Nachtsheim, B.J. A review of new developments in the Friedel–Crafts alkylation – From green chemistry to asymmetric catalysis. Beilstein J. Org. Chem. 2010, 6, 6. [Google Scholar]
- Rueping, M.; Raja, S.; Núñez, A. Asymmetric Brønsted Acid-Catalyzed Friedel–Crafts Reactions of Indoles with Cyclic Imines - Efficient Generation of Nitrogen-Substituted Quaternary Carbon Centers. Adv. Synth. Catal. 2011, 353, 563–568. [Google Scholar] [CrossRef]
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef] [PubMed]
- Rioz-Martínez, A.; Roelfes, G. DNA-based hybrid catalysis. Curr. Opin. Chem. Biol. 2015, 25, 80–87. [Google Scholar] [CrossRef]
- Duchemin, N.; Heath-Apostolopoulos, I.; Smietana, M.; Arseniyadis, S. A decade of DNA-hybrid catalysis: From innovation to comprehension. Org. Biomol. Chem. 2017, 15, 7072–7087. [Google Scholar] [CrossRef]
- Yum, J.H.; Park, S.; Sugiyama, H. G-quadruplexes as versatile scaffolds for catalysis. Org. Biomol. Chem. 2019, 17, 9547–9561. [Google Scholar] [CrossRef]
- Mansot, J.; Vasseur, J.-J.; Arseniyadis, S.; Smietana, M. α,β-Unsaturated 2-Acyl-Imidazoles in Asymmetric Biohybrid Catalysis. ChemCatChem 2019, 11, 5686–5704. [Google Scholar] [CrossRef]
- Steinreiber, J.; Ward, T.R. Artificial metalloenzymes as selective catalysts in aqueous media. Coord. Chem. Rev. 2008, 252, 751–766. [Google Scholar] [CrossRef]
- Boersma, A.J.; Megens, R.P.; Feringa, B.L.; Roelfes, G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Sugiyama, H. DNA-Based Hybrid Catalysts for Asymmetric Organic Synthesis. Angew. Chem., Int. Ed. 2010, 49, 3870–3878. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S.K. DNA as a Versatile Chemical Component for Catalysis, Encoding, and Stereocontrol. Angew. Chem., Int. Ed. 2010, 49, 7180–7201. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.; Roelfes, G. Artificial metalloenzymes for enantioselective catalysis. Curr. Opin. Chem. Biol. 2014, 19, 135–143. [Google Scholar] [CrossRef]
- Jäschke, A. Selective Hybrid Catalysts Based on Nucleic Acids. In Molecular Catalysts; Wiley-VCH Verlag: Weinheim, Germany, 2014; pp. 377–392. [Google Scholar] [CrossRef]
- Roelfes, G.; Feringa, B.L. DNA-based asymmetric catalysis. Angew. Chem., Int. Ed. 2005, 44, 3230–3232. [Google Scholar] [CrossRef]
- Roelfes, G.; Boersma, A.J.; Feringa, B.L. Highly enantioselective DNA-based catalysis. Chem. Commun. 2006, 635–637. [Google Scholar] [CrossRef]
- Boersma, A.J.; Feringa, B.L.; Roelfes, G. alpha,beta-unsaturated 2-acyl imidazoles as a practical class of dienophiles for the DNA-Based catalytic asymmetric diels-alder reaction in water. Org. Lett. 2007, 9, 3647–3650. [Google Scholar] [CrossRef]
- Wang, J.; Benedetti, E.; Bethge, L.; Vonhoff, S.; Klussmann, S.; Vasseur, J.J.; Cossy, J.; Smietana, M.; Arseniyadis, S. DNA vs. Mirror-Image DNA: A Universal Approach to Tune the Absolute Configuration in DNA-Based Asymmetric Catalysis. Angew. Chem. Int. Ed. 2013, 52, 11546–11549. [Google Scholar] [CrossRef]
- Bai, J.K.; Zhou, H.; Sun, X.L.; Chen, D.; Li, C.; Qiao, R.Z. Insight into Stereo-Induction by Minor Modification in the Ligand in DNA-Based Hybrid Catalysis. Catal. Lett. 2018, 148, 3315–3324. [Google Scholar] [CrossRef]
- Mansot, J.; Lauberteaux, J.; Lebrun, A.; Mauduit, M.; Vasseur, J.-J.; Marcia de Figueiredo, R.; Arseniyadis, S.; Campagne, J.-M.; Smietana, M. DNA-Based Asymmetric Inverse Electron-Demand Hetero-Diels–Alder. Chem.—Eur. J. 2020, 26, 3519–3523. [Google Scholar] [CrossRef]
- Boersma, A.J.; Feringa, B.L.; Roelfes, G. Enantioselective Friedel-Crafts Reactions in Water Using a DNA-Based Catalyst. Angew. Chem., Int. Ed. 2009, 48, 3346–3348. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ikehata, K.; Watabe, R.; Hidaka, Y.; Rajendran, A.; Sugiyama, H. Deciphering DNA-based asymmetric catalysis through intramolecular Friedel-Crafts alkylations. Chem. Commun. 2012, 48, 10398–10400. [Google Scholar] [CrossRef] [PubMed]
- Petrova, G.P.; Ke, Z.; Park, S.; Sugiyama, H.; Morokuma, K. The origin of enantioselectivity for intramolecular Friedel–Crafts reaction catalyzed by supramolecular Cu/DNA catalyst complex. Chem. Phys. Lett. 2014, 600, 87–95. [Google Scholar] [CrossRef]
- García-Fernández, A.; Megens, R.P.; Villarino, L.; Roelfes, G. DNA-Accelerated Copper Catalysis of Friedel–Crafts Conjugate Addition/Enantioselective Protonation Reactions in Water. J. Am. Chem. Soc. 2016, 138, 16308–16314. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, D.; Bai, J.K.; Sun, X.L.; Li, C.; Qiao, R.Z. Effect of ligand sequence-specific modification on DNA hybrid catalysis. Org. Biomol. Chem. 2017, 15, 6738–6745. [Google Scholar] [CrossRef]
- Coquiѐre, D.; Feringa, B.L.; Roelfes, G. DNA-Based catalytic enantioselective Michael reactions in water. Angew. Chem., Int. Ed. 2007, 46, 9308–9311. [Google Scholar] [CrossRef]
- Megens, R.P.; Roelfes, G. DNA-based catalytic enantioselective intermolecular oxa-Michael addition reactions. Chem. Commun. 2012, 48, 6366–6368. [Google Scholar] [CrossRef][Green Version]
- Li, Y.H.; Wang, C.H.; Jia, G.Q.; Lu, S.M.; Li, C. Enantioselective Michael addition reactions in water using a DNA-based catalyst. Tetrahedron 2013, 69, 6585–6590. [Google Scholar] [CrossRef]
- Shibata, N.; Yasui, H.; Nakamura, S.; Toru, T. DNA-Mediated enantioselective carbon-fluorine bond formation. Synlett 2007, 1153–1157. [Google Scholar] [CrossRef]
- Boersma, A.J.; Coquiѐre, D.; Geerdink, D.; Rosati, F.; Feringa, B.L.; Roelfes, G. Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst. Nat. Chem. 2010, 2, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Rioz-Martínez, A.; Oelerich, J.; Ségaud, N.; Roelfes, G. DNA-Accelerated Catalysis of Carbene-Transfer Reactions by a DNA/Cationic Iron Porphyrin Hybrid. Angew. Chem. Int. Ed. 2016, 55, 14136–14140. [Google Scholar] [CrossRef] [PubMed]
- Marek, J.J.; Singh, R.P.; Heuer, A.; Hennecke, U. Enantioselective Catalysis by Using Short, Structurally Defined DNA Hairpins as Scaffold for Hybrid Catalysts. Chem.—Eur. J. 2017, 23, 6004–6008. [Google Scholar] [CrossRef] [PubMed]
- Marek, J.J.; Hennecke, U. Why DNA Is a More Effective Scaffold than RNA in Nucleic Acid-Based Asymmetric Catalysis—Supramolecular Control of Cooperative Effects. Chem.—Eur. J. 2017, 23, 6009–6013. [Google Scholar] [CrossRef]
- Bai, J.K.; Chen, D.; Li, C.; Wang, H.S.; Qiao, R.Z. PNA as Hybrid Catalyst Scaffold Catalyzed Asymmetric Friedel–Crafts Alkylation. Catal. Lett. 2020. [Google Scholar] [CrossRef]
- Wang, C.; Hao, M.; Qi, Q.; Dang, J.; Dong, X.; Lv, S.; Xiong, L.; Gao, H.; Jia, G.; Chen, Y.; et al. Highly Efficient Cyclic Dinucleotide Based Artificial Metalloribozymes for Enantioselective Friedel–Crafts Reactions in Water. Angew. Chem. Int. Ed. 2020, 59, 3444–3449. [Google Scholar] [CrossRef]
- Bai, J.; Sun, X.; Wang, H.; Li, C.; Qiao, R. Guanosine-Based Self-Assembly as an Enantioselective Catalyst Scaffold. J. Org. Chem. 2020, 85, 2010–2018. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef]
- Qin, Y.; Hurley, L.H. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie 2008, 90, 1149–1171. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.; Ritson, D.J.; Garner, T.; Searle, M.; Moses, J.E. Tuneable DNA-based asymmetric catalysis using a G-quadruplex supramolecular assembly. Chem. Commun. 2010, 46, 4309–4311. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Li, Y.H.; Jia, G.Q.; Liu, Y.; Lu, S.M.; Li, C. Enantioselective Friedel-Crafts reactions in water catalyzed by a human telomeric G-quadruplex DNA metalloenzyme. Chem. Commun. 2012, 48, 6232–6234. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Jia, G.Q.; Zhou, J.; Li, Y.H.; Liu, Y.; Lu, S.M.; Li, C. Enantioselective Diels-Alder Reactions with G-Quadruplex DNA-Based Catalysts. Angew. Chem., Int. Ed. 2012, 51, 9352–9355. [Google Scholar] [CrossRef]
- Wilking, M.; Hennecke, U. The influence of G-quadruplex structure on DNA-based asymmetric catalysis using the G-quadruplex-bound cationic porphyrin TMPyP4 center dot Cu. Org. Biomol. Chem. 2013, 11, 6940–6945. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, M.; Hao, J.; Wang, C.; Jia, G.; Li, C. Terpyridine-Cu(ii) targeting human telomeric DNA to produce highly stereospecific G-quadruplex DNA metalloenzyme. Chem. Sci. 2015, 6, 5578–5585. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Hao, J.; Cheng, M.; Jia, G.; Li, C. Higher-order human telomeric G-quadruplex DNA metalloenzyme catalyzed Diels-Alder reaction: An unexpected inversion of enantioselectivity modulated by K+ and NH4+ ions. Chem. Commun. 2015, 51, 13174–13177. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Li, Y.; Zhou, J.; Jia, G.; Lu, S.-M.; Yang, Y.; Li, C. Enantioselective sulfoxidation reaction catalyzed by a G-quadruplex DNA metalloenzyme. Chem. Commun. 2016, 52, 9644–9647. [Google Scholar] [CrossRef]
- Xu, X.; Mao, W.; Lin, F.; Hu, J.; He, Z.; Weng, X.; Wang, C.-J.; Zhou, X. Enantioselective Diels–Alder reactions using a G-triplex DNA-based catalyst. Catal. Commun. 2016, 74, 16–18. [Google Scholar] [CrossRef]
- Caprioara, M.; Fiammengo, R.; Engeser, M.; Jäschke, A. DNA-Based Phosphane Ligands. Chem.—Eur. J. 2007, 13, 2089–2095. [Google Scholar] [CrossRef]
- Jakobsen, U.; Rohr, K.; Vogel, S. Toward a Catalytic Site in DNA: Polyaza Crown Ether as Non-Nucleosidic Building Blocks in DNA Conjugates. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1419–1422. [Google Scholar] [CrossRef]
- Oltra, N.S.; Roelfes, G. Modular assembly of novel DNA-based catalysts. Chem. Commun. 2008, 6039–6041. [Google Scholar] [CrossRef]
- Tang, Z.; Gonçalves, D.P.N.; Wieland, M.; Marx, A.; Hartig, J.S. Novel DNA Catalysts Based on G-Quadruplex Recognition. Chembiochem 2008, 9, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Fiammengo, R.; Jäschke, A. Allylic Amination by a DNA-Diene-Iridium(I) Hybrid Catalyst. Angew. Chem. Int. Ed. 2009, 48, 4426–4429. [Google Scholar] [CrossRef] [PubMed]
- Gjonaj, L.; Roelfes, G. Novel Catalyst Design by using Cisplatin To Covalently Anchor Catalytically Active Copper Complexes to DNA. ChemCatChem 2013, 5, 1718–1721. [Google Scholar] [CrossRef]
- Park, S.; Zheng, L.; Kumakiri, S.; Sakashita, S.; Otomo, H.; Ikehata, K.; Sugiyama, H. Development of DNA-Based Hybrid Catalysts through Direct Ligand Incorporation: Toward Understanding of DNA-Based Asymmetric Catalysis. ACS Catalysis 2014, 4, 4070–4073. [Google Scholar] [CrossRef]
- Park, S.; Okamura, I.; Sakashita, S.; Yum, J.H.; Acharya, C.; Gao, L.; Sugiyama, H. Development of DNA Metalloenzymes Using a Rational Design Approach and Application in the Asymmetric Diels–Alder Reaction. ACS Catalysis 2015, 5, 4708–4712. [Google Scholar] [CrossRef]
- Mansot, J.; Aubert, S.; Duchemin, N.; Vasseur, J.-J.; Arseniyadis, S.; Smietana, M. A rational quest for selectivity through precise ligand-positioning in tandem DNA-catalysed Friedel–Crafts alkylation/asymmetric protonation. Chem. Sci. 2019, 10, 2875–2881. [Google Scholar] [CrossRef] [PubMed]
- Yum, J.H.; Park, S.; Hiraga, R.; Okamura, I.; Notsu, S.; Sugiyama, H. Modular DNA-based hybrid catalysts as a toolbox for enantioselective hydration of α,β-unsaturated ketones. Org. Biomol. Chem. 2019, 17, 2548–2553. [Google Scholar] [CrossRef]
- Park, S.; Matsui, H.; Fukumoto, K.; Yum, J.H.; Sugiyama, H. Histidine-conjugated DNA as a biomolecular depot for metal ions. RSC Advances 2020, 10, 9717–9722. [Google Scholar] [CrossRef]
- Dey, S.; Jäschke, A. Tuning the Stereoselectivity of a DNA-Catalyzed Michael Addition through Covalent Modification. Angew. Chem. Int. Ed. 2015, 54, 11279–11282. [Google Scholar] [CrossRef]
- Dey, S.; Rühl, C.L.; Jäschke, A. Catalysis of Michael Additions by Covalently Modified G-Quadruplex DNA. Chem.—Eur. J. 2017, 23, 12162–12170. [Google Scholar] [CrossRef]
- Wei, D.G.; Parkinson, G.N.; Reszka, A.P.; Neidle, S. Crystal structure of a c-kit promoter quadruplex reveals the structural role of metal ions and water molecules in maintaining loop conformation. Nucleic Acids Res. 2012, 40, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Burge, S.; Neidle, S.; Patel, D.J. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J. Am. Chem. Soc. 2007, 129, 4386–4392. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.J.; Seaman, S.E.; Mazura, J.C.; Tor, Y. Fluorescent 1,10-Phenanthroline-Containing Oligonucleotides Distinguish between Perfect and Mismatched Base Pairing. Org. Lett. 2002, 4, 2305–2308. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.J.; Tor, Y. Ru(II) and Os(II) Nucleosides and Oligonucleotides: Synthesis and Properties. J. Am. Chem. Soc. 2002, 124, 3749–3762. [Google Scholar] [CrossRef]
- Kalachova, L.; Pohl, R.; Hocek, M. Synthesis of 2′-Deoxyuridine and 2′-Deoxycytidine Nucleosides Bearing Bipyridine and Terpyridine Ligands at Position 5. Synthesis 2009, 105–112. [Google Scholar] [CrossRef]
- Kalachova, L.; Pohl, R.; Hocek, M. Synthesis of nucleoside mono- and triphosphates bearing oligopyridine ligands, their incorporation into DNA and complexation with transition metals. Org. Biomol. Chem. 2012, 10, 49–55. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}

| Entry | DNA/Linker | n | dU12-modified DNA | dU10-modified DNA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GQ-DNA | ds-DNA | GQ-DNA | ds-DNA | |||||||
| conv (%) b | ee (%) b, c | conv (%) b | ee (%) b, c | conv (%) b | ee (%) b, c | conv (%) b | ee (%) b, c | |||
| 1 | c-kit(wt) | - | 34 | <−5 | 28 | <+5 | - | - | - | - |
| 2 | c-kit-T10 | - | - | - | - | - | 45 | <+5 | 21 | <+5 |
| 3 | Propargyl-bpy | 1 | 64 | −23 | 27 | +62 | 44 | +17 | 23 | +37 |
| 4 | Butynyl-bpy | 2 | 78 | −35 | 48 | +16 | 64 | +45 | 36 | +21 |
| 5 | Pentynyl-bpy | 3 | 99 | −65 | 63 | +25 | 68 | +14 | 49 | <+5 |
| 6 | Hexynyl-bpy | 4 | 99 | −19 | 79 | +8 | 65 | +16 | 61 | +31 |
| 7 | Heptynyl-bpy | 5 | 99 | −22 | 99 | −8 | 67 | +10 | 88 | +33 |
| 8 | Octynyl-bpy | 6 | 99 | +29 | 99 | +7 | 66 | +21 | 89 | +40 |
| 9 | Nonynyl-bpy | 7 | 99 | −13 | 99 | −23 | 64 | +8 | 89 | +53 |
| 10 | Decynyl-bpy | 8 | 99 | +8 | 99 | −11 | 65 | +7 | 91 | +38 |
| 11 | Ethynyl-bpy | - | 68 | +45 | 28 | <−5 | 45 | +30 | 21 | −19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, S.; Jäschke, A. Covalently Functionalized DNA Duplexes and Quadruplexes as Hybrid Catalysts in an Enantioselective Friedel–Crafts Reaction. Molecules 2020, 25, 3121. https://doi.org/10.3390/molecules25143121
Dey S, Jäschke A. Covalently Functionalized DNA Duplexes and Quadruplexes as Hybrid Catalysts in an Enantioselective Friedel–Crafts Reaction. Molecules. 2020; 25(14):3121. https://doi.org/10.3390/molecules25143121
Chicago/Turabian StyleDey, Surjendu, and Andres Jäschke. 2020. "Covalently Functionalized DNA Duplexes and Quadruplexes as Hybrid Catalysts in an Enantioselective Friedel–Crafts Reaction" Molecules 25, no. 14: 3121. https://doi.org/10.3390/molecules25143121
APA StyleDey, S., & Jäschke, A. (2020). Covalently Functionalized DNA Duplexes and Quadruplexes as Hybrid Catalysts in an Enantioselective Friedel–Crafts Reaction. Molecules, 25(14), 3121. https://doi.org/10.3390/molecules25143121