
Curcumin-1,2,3-Triazole Conjugation for Targeting the Cancer Apoptosis Machinery

 , , ,
, , ,  , ,
, ,  ,
,  ,
, 
 , ,
, ,  and
and 
Abstract
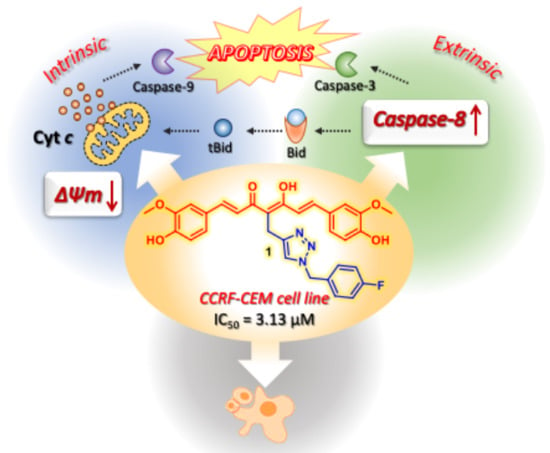
1. Introduction
2. Results
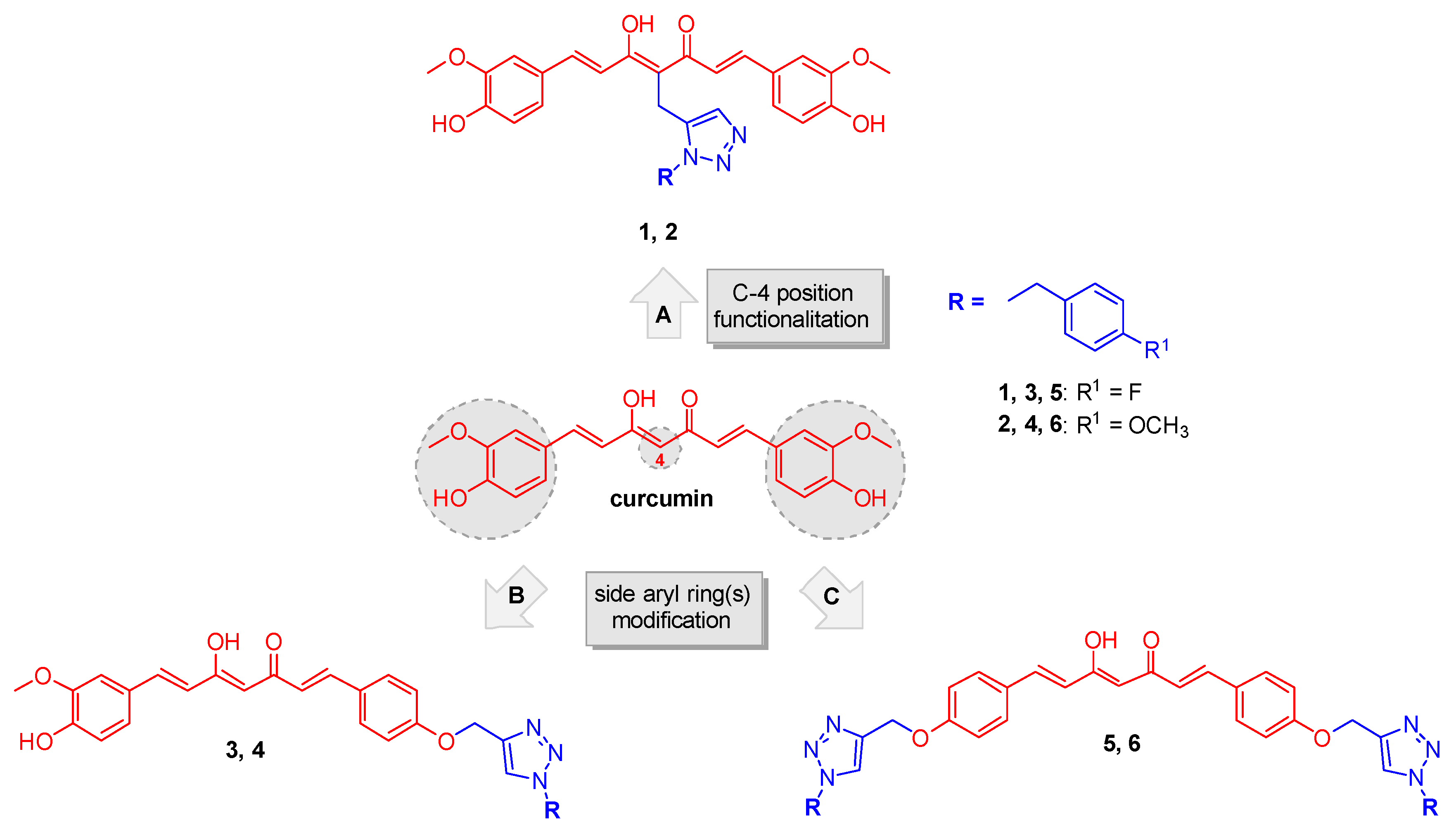
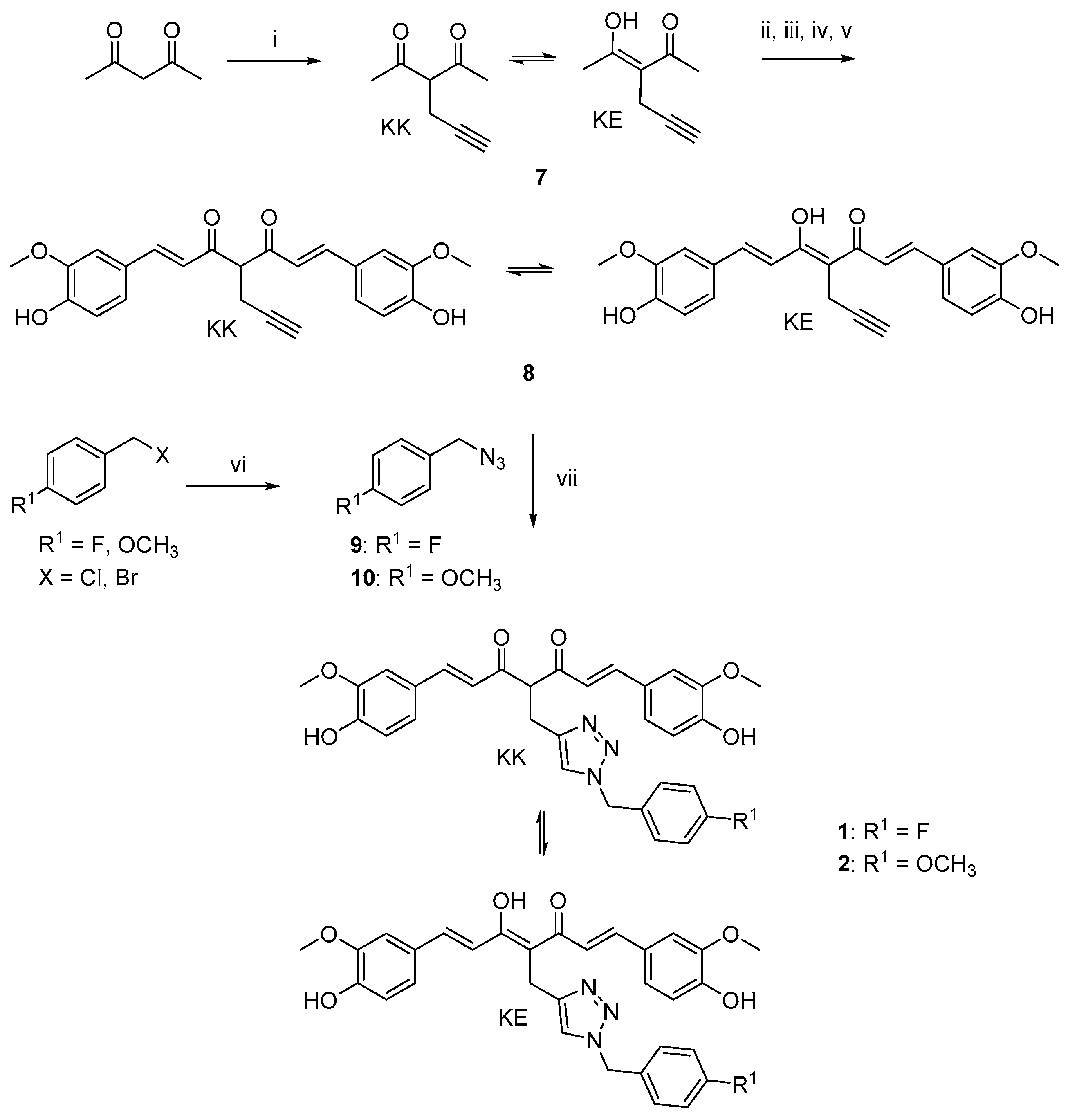
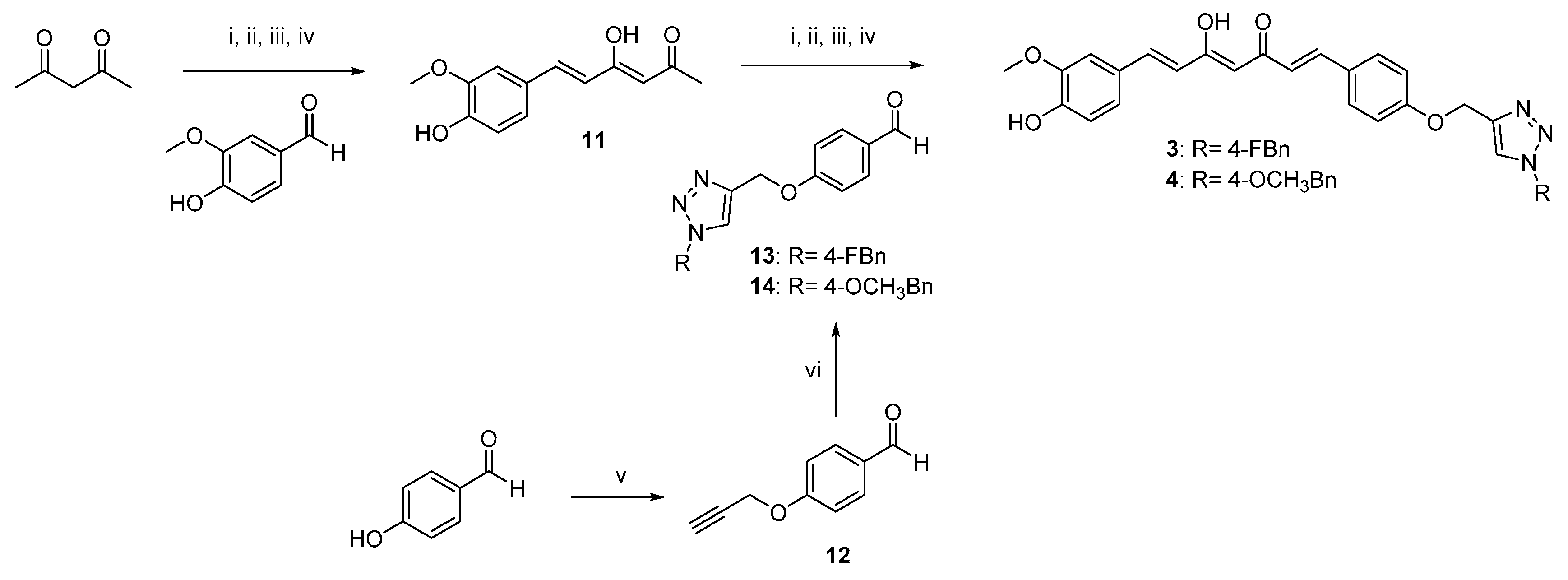
2.1. Synthesis
2.2. Biologic Evaluation
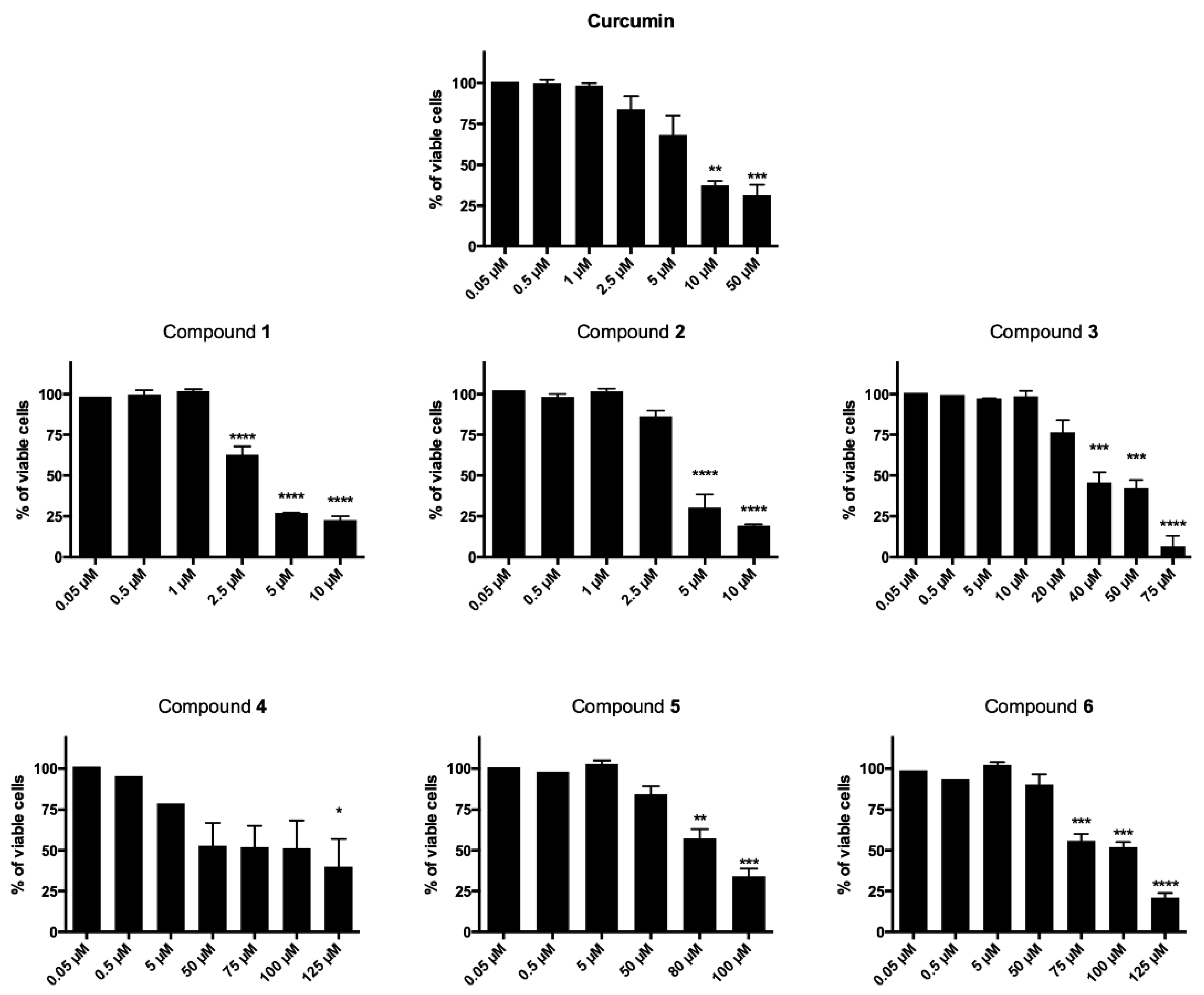
2.2.1. Cytotoxicity
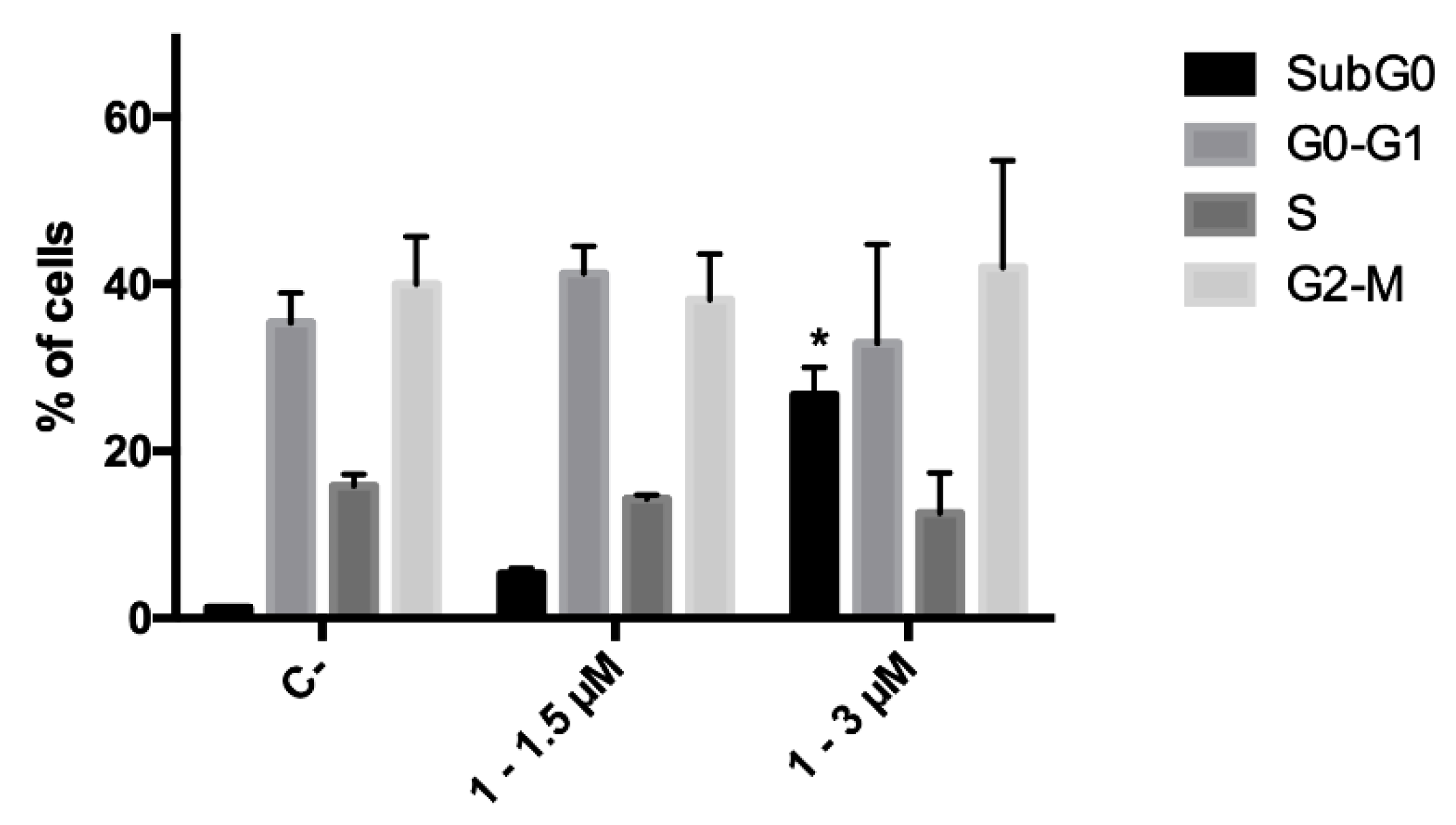
2.2.2. Cell-Cycle Progression
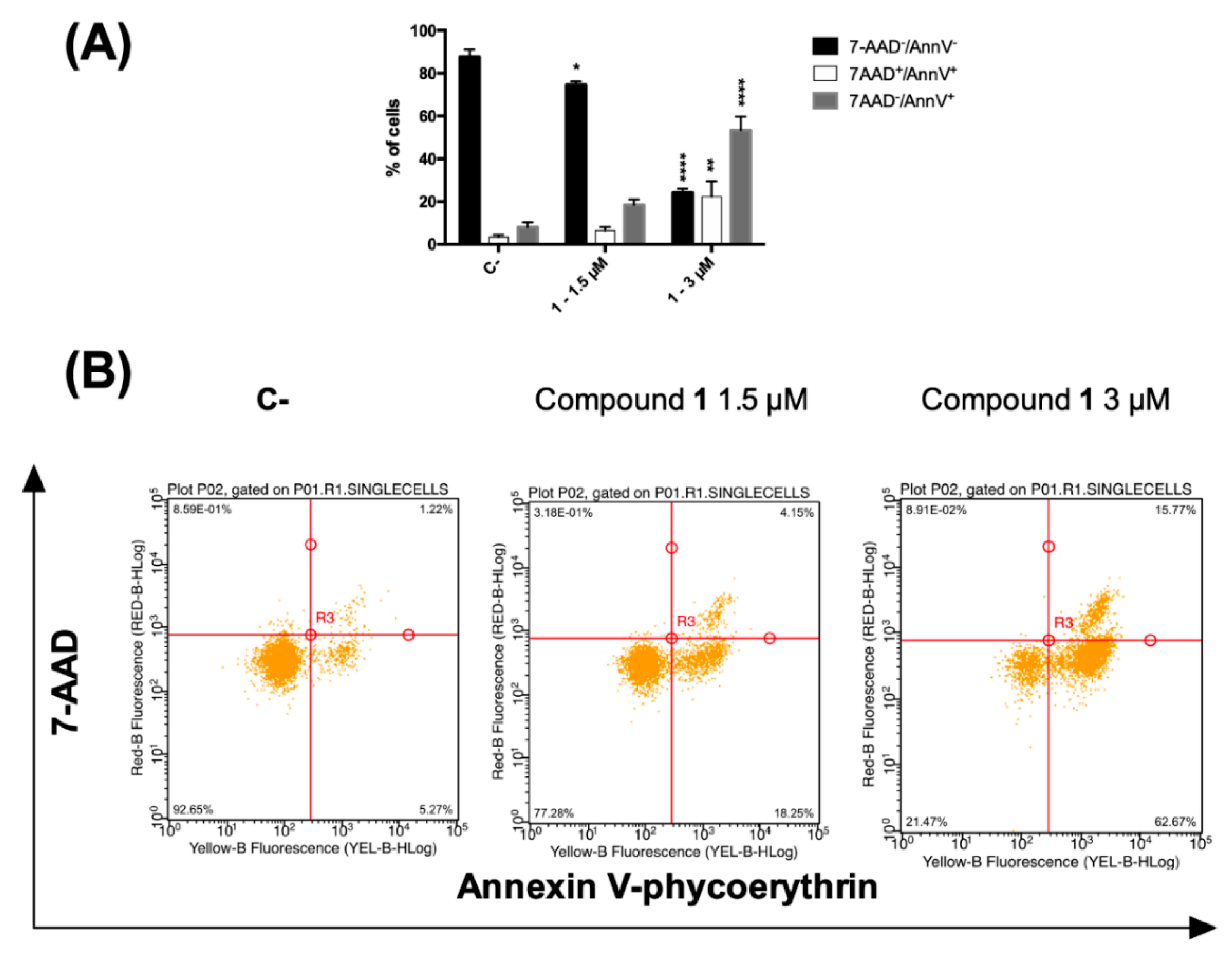
2.2.3. Phosphatidylserine Exposure and Cell Death Analysis
2.2.4. Mitochondrial Transmembrane Potential
2.2.5. Caspase-8
2.2.6. Physicochemical Properties Prediction
2.2.7. FACS analysis to Study PAINS-Like Behavior
2.2.8. Chemical Stability Study
3. Discussion
4. Materials and Methods
4.1. Chemistry
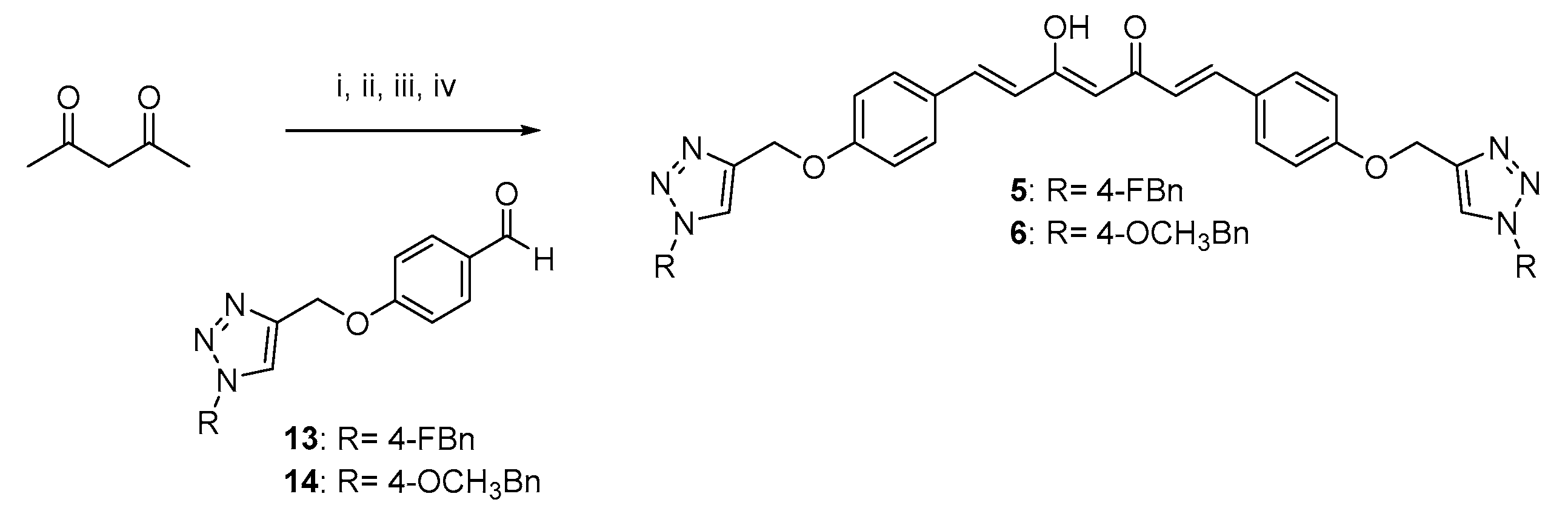
4.1.1. Huisgen 1,3-Dipolar Cycloaddition. General Procedure for the Synthesis of Curcumin-Triazole Conjugates 1, 2 and Intermediates 13, 14
- (1E,4Z,6E)-4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one (1-KE); (1E,6E)-4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (1-KK).
- (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-4-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)hepta-1,4,6-trien-3-one (2-KE); (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-4-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)hepta-1,6-diene-3,5-dione (2-KK).
- 4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13).
- 4-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (14).
4.1.2. Pabon Reaction. General Procedure for the Synthesis of Curcumin-Triazole Conjugates 3–6 and Intermediates 8 and 11
- (1E,4Z,6E)-1-(4-((1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-hydroxy-7-(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one (3).
- (1E,4Z,6E)-5-hydroxy-7-(4-hydroxy-3-methoxyphenyl)-1-(4-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)hepta-1,4,6-trien-3-one (4).
- (1E,4Z,6E)-1,7-bis(4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-hydroxyhepta-1,4,6-trien-3-one (5).
- (1E,4Z,6E)-5-hydroxy-1,7-bis(4-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)hepta-1,4,6-trien-3-one (6).
- (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-4-(prop-2-yn-1-yl)hepta-1,4,6-trien-3-one (8-KE); (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-4-(prop-2-yn-1-yl)hepta-1,6-diene-3,5-dione (8-KK).
- General Procedure for the Synthesis of Azido Intermediates 9 and 10.
- 1-(azidomethyl)-4-fluorobenzene (9)
- 1-(azidomethyl)-4-methoxybenzene (10)
- 4-(Prop-2-yn-1-yloxy)benzaldehyde (12) [46]
- (Z)-3-(1-hydroxyethylidene)hex-5-yn-2-one (7-KE); 3-(prop-2-yn-1-yl)pentane-2,4-dione (7-KK).
4.2. Cell Cultures and Treatments
4.3. Analysis of Cell Viability
4.4. Analysis of Cell Cycle
4.5. Analysis of Cell Death
4.6. Analysis of Mitochondrial Transmembrane Potential
4.7. Evaluation of Caspase-8 Activity
4.8. Flow Cytometry
4.9. Statistical Analysis
4.10. Chemical Stability Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.; Seevasant, I.; Mohamad, J.; Mukheem, A.; Huri, H.Z.; Kamarul, T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis. 2016, 7, e2058. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. BioMed Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Targeting apoptosis pathways in cancer therapy. Curr. Cancer Drug Targets 2004, 4, 569–576. [Google Scholar] [CrossRef]
- Lim, B.; Greer, Y.; Lipkowitz, S.; Takebe, N. Novel Apoptosis-Inducing Agents for the Treatment of Cancer, a New Arsenal in the Toolbox. Cancers 2019, 11, 1087. [Google Scholar] [CrossRef]
- Catanzaro, E.; Seghetti, F.; Calcabrini, C.; Rampa, A.; Gobbi, S.; Sestili, P.; Turrini, E.; Maffei, F.; Hrelia, P.; Bisi, A.; et al. Identification of a new tamoxifen-xanthene hybrid as pro-apoptotic anticancer agent. Bioorg. Chem. 2019, 86, 538–549. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef]
- Amslinger, S. The tunable functionality of alpha,beta-unsaturated carbonyl compounds enables their differential application in biological systems. Chem. Med. Chem. 2010, 5, 351–356. [Google Scholar] [CrossRef]
- Di Martino, R.M.; Luppi, B.; Bisi, A.; Gobbi, S.; Rampa, A.; Abruzzo, A.; Belluti, F. Recent progress on curcumin-based therapeutics: A patent review (2012–2016). Part I: Curcumin. Expert Opin. Ther. Pat. 2017, 27, 579–590. [Google Scholar] [CrossRef]
- Willenbacher, E.; Khan, S.Z.; Mujica, S.C.A.; Trapani, D.; Hussain, S.; Wolf, D.; Willenbacher, W.; Spizzo, G.; Seeber, A. Curcumin: New Insights into an Ancient Ingredient against Cancer. Int. J. Mol. Sci 2019, 20, 1808. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Bahadori, F.; Demiray, M. A Realistic View on “The Essential Medicinal Chemistry of Curcumin”. ACS Med. Chem. Lett. 2017, 8, 893–896. [Google Scholar] [CrossRef]
- Bajorath, J. Activity artifacts in drug discovery and different facets of compound promiscuity. F1000Research 2014, 3, 233. [Google Scholar] [CrossRef]
- Jasial, S.; Hu, Y.; Bajorath, J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; Bisi, A.; Rampa, A.; Gobbi, S.; Belluti, F. Recent progress on curcumin-based therapeutics: A patent review (2012–2016). Part II: Curcumin derivatives in cancer and neurodegeneration. Expert Opin. Ther. Pat. 2017, 27, 953–965. [Google Scholar] [CrossRef]
- Bisceglia, F.; Seghetti, F.; Serra, M.; Zusso, M.; Gervasoni, S.; Verga, L.; Vistoli, G.; Lanni, C.; Catanzaro, M.; De Lorenzi, E.; et al. Prenylated Curcumin Analogues as Multipotent Tools To Tackle Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 1420–1433. [Google Scholar] [CrossRef]
- Zusso, M.; Mercanti, G.; Belluti, F.; Di Martino, R.M.C.; Pagetta, A.; Marinelli, C.; Brun, P.; Ragazzi, E.; Lo, R.; Stifani, S.; et al. Phenolic 1,3-diketones attenuate lipopolysaccharide-induced inflammatory response by an alternative magnesium-mediated mechanism. Br. J. Pharmacol. 2017, 174, 1090–1103. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; De Simone, A.; Andrisano, V.; Bisignano, P.; Bisi, A.; Gobbi, S.; Rampa, A.; Fato, R.; Bergamini, C.; Perez, D.I.; et al. Versatility of the Curcumin Scaffold: Discovery of Potent and Balanced Dual BACE-1 and GSK-3 beta Inhibitors. J. Med. Chem. 2016, 59, 531–544. [Google Scholar] [CrossRef]
- Tedesco, S.; Zusso, M.; Facci, L.; Trenti, A.; Boscaro, C.; Belluti, F.; Fadini, G.P.; Skaper, S.D.; Giusti, P.; Bolego, C.; et al. Bisdemethoxycurcumin and Its Cyclized Pyrazole Analogue Differentially Disrupt Lipopolysaccharide Signalling in Human Monocyte-Derived Macrophages. Mediat. Inflamm. 2018, 2018, 2868702. [Google Scholar] [CrossRef] [PubMed]
- Lagoutte, R.; Patouret, R.; Winssinger, N. Covalent inhibitors: An opportunity for rational target selectivity. Curr. Opin Chem. Biol. 2017, 39, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Uliassi, E.; Piazzi, L.; Belluti, F.; Mazzanti, A.; Kaiser, M.; Brun, R.; Moraes, C.B.; Freitas, L.H.; Gul, S.; Kuzikov, M.; et al. Development of a Focused Library of Triazole-Linked Privileged-Structure-Based Conjugates Leading to the Discovery of Novel Phenotypic Hits against Protozoan Parasitic Infections. Chemmedchem 2018, 13, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Spear, K.L.; Brown, S.P. The evolution of library design: Crafting smart compound collections for phenotypic screens. Drug Discov. Today Technol. 2017, 23, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Bonvicini, F.; Gentilomi, G.A.; Bressan, F.; Gobbi, S.; Rampa, A.; Bisi, A.; Belluti, F. Functionalization of the Chalcone Scaffold for the Discovery of Novel Lead Compounds Targeting Fungal Infections. Molecules 2019, 24, 372. [Google Scholar] [CrossRef]
- Ortalli, M.; Ilari, A.; Colotti, G.; De Ionna, I.; Battista, T.; Bisi, A.; Gobbi, S.; Rampa, A.; Di Martino, R.M.C.; Gentilomi, G.A.; et al. Identification of chalcone-based antileishmanial agents targeting trypanothione reductase. Eur. J. Med. Chem. 2018, 152, 527–541. [Google Scholar] [CrossRef]
- Pabon, H.J. A synthesis of curcumin and related compounds. Recl. Trav. Chim. Pays-Bas. 1964, 83, 379–386. [Google Scholar] [CrossRef]
- Agnati, L.F.; Guidolin, D.; Cervetto, C.; Borroto-Escuela, D.O.; Fuxe, K. Role of iso-receptors in receptor-receptor interactions with a focus on dopamine iso-receptor complexes. Rev. Neurosci. 2016, 27, 1–25. [Google Scholar] [CrossRef]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, M.; Philippé, J.; De Sitter, S.; De Ridder, L. Annexin V expression in apoptotic peripheral blood lymphocytes: An electron microscopic evaluation. Apoptosis 2002, 7, 41–47. [Google Scholar] [PubMed]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Diez-Fraile, A.; Criel, G.; Svistunov, A.A.; Vandenabeele, P.; D’Herde, K. Life and death of female gametes during oogenesis and folliculogenesis. Apoptosis 2008, 13, 1065–1087. [Google Scholar] [CrossRef] [PubMed]
- Kloditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Demuynck, R.; Efimova, I.; Lin, A.; Declercq, H.; Krysko, D.V. A 3D Cell Death Assay to Quantitatively Determine Ferroptosis in Spheroids. Cells 2020, 9, 703. [Google Scholar] [CrossRef]
- Grootjans, S.; Hassannia, B.; Delrue, I.; Goossens, V.; Wiernicki, B.; Dondelinger, Y.; Bertrand, M.J.; Krysko, D.V.; Vuylsteke, M.; Vandenabeele, P.; et al. A real-time fluorometric method for the simultaneous detection of cell death type and rate. Nat. Protoc. 2016, 11, 1444–1454. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. ACS Med. Chem. Lett. 2017, 8, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef]
- Sala de Oyanguren, F.J.; Rainey, N.E.; Moustapha, A.; Saric, A.; Sureau, F.; O’Connor, J.E.; Petit, P.X. Highlighting Curcumin-Induced Crosstalk between Autophagy and Apoptosis as Supported by Its Specific Subcellular Localization. Cells 2020, 9, 361. [Google Scholar] [CrossRef]
- Giguere, J.B.; Thibeault, D.; Cronier, F.; Marois, J.S.; Auger, M.; Morin, J.F. Synthesis of 2- and 3 rotaxanes through Sonogashira coupling. Tetrahedron Lett. 2009, 50, 5497–5500. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Soleimani, V.; Sahebkar, A.; Hosseinzadeh, H. Turmeric (Curcuma longa) and its major constituent (curcumin) as nontoxic and safe substances: Review. Phytother Res. 2018, 32, 985–995. [Google Scholar] [CrossRef]
- Syng-Ai, C.; Kumari, A.L.; Khar, A. Effect of curcumin on normal and tumor cells: Role of glutathione and bcl-2. Mol. Cancer Ther. 2004, 3, 1101–1108. [Google Scholar]
- Rezaee, R.; Momtazi, A.A.; Monemi, A.; Sahebkar, A. Curcumin: A potentially powerful tool to reverse cisplatin-induced toxicity. Pharm. Res. 2017, 117, 218–227. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound 1 is available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | CCRF–CEM Cell Line IC50 (µM) a | |
|---|---|---|---|
| 24 h | 48 h | ||
| 1 |  | 68.30 | 3.13 |
| 2 |  | 55.63 | 3.95 |
| 3 |  | 38.68 | 38.00 |
| 4 |  | >150 | 69.07 |
| 5 |  | >150 | 88.94 |
| 6 |  | 122.1 | 93.40 |
| curcumin | / | >150 | 10.51 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seghetti, F.; Di Martino, R.M.C.; Catanzaro, E.; Bisi, A.; Gobbi, S.; Rampa, A.; Canonico, B.; Montanari, M.; Krysko, D.V.; Papa, S.; et al. Curcumin-1,2,3-Triazole Conjugation for Targeting the Cancer Apoptosis Machinery. Molecules 2020, 25, 3066. https://doi.org/10.3390/molecules25133066
Seghetti F, Di Martino RMC, Catanzaro E, Bisi A, Gobbi S, Rampa A, Canonico B, Montanari M, Krysko DV, Papa S, et al. Curcumin-1,2,3-Triazole Conjugation for Targeting the Cancer Apoptosis Machinery. Molecules. 2020; 25(13):3066. https://doi.org/10.3390/molecules25133066
Chicago/Turabian StyleSeghetti, Francesca, Rita Maria Concetta Di Martino, Elena Catanzaro, Alessandra Bisi, Silvia Gobbi, Angela Rampa, Barbara Canonico, Mariele Montanari, Dmitri V. Krysko, Stefano Papa, and et al. 2020. "Curcumin-1,2,3-Triazole Conjugation for Targeting the Cancer Apoptosis Machinery" Molecules 25, no. 13: 3066. https://doi.org/10.3390/molecules25133066
APA StyleSeghetti, F., Di Martino, R. M. C., Catanzaro, E., Bisi, A., Gobbi, S., Rampa, A., Canonico, B., Montanari, M., Krysko, D. V., Papa, S., Fimognari, C., & Belluti, F. (2020). Curcumin-1,2,3-Triazole Conjugation for Targeting the Cancer Apoptosis Machinery. Molecules, 25(13), 3066. https://doi.org/10.3390/molecules25133066