
Supramolecular Peptide Assemblies as Antimicrobial Scaffolds


Abstract
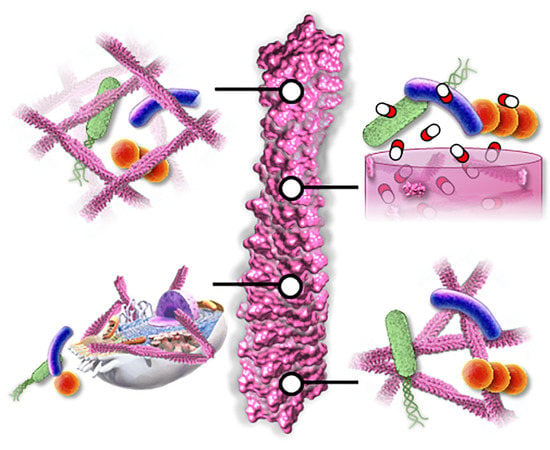
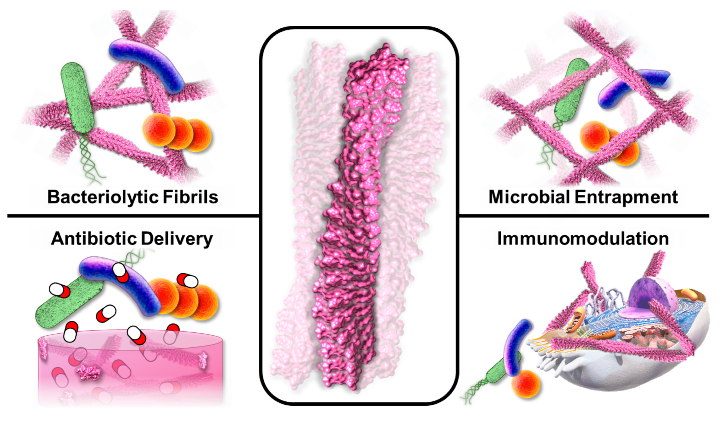
1. Introduction
2. Antimicrobial Supramolecular Peptide Assemblies
2.1. Monomorphic Nanofibers
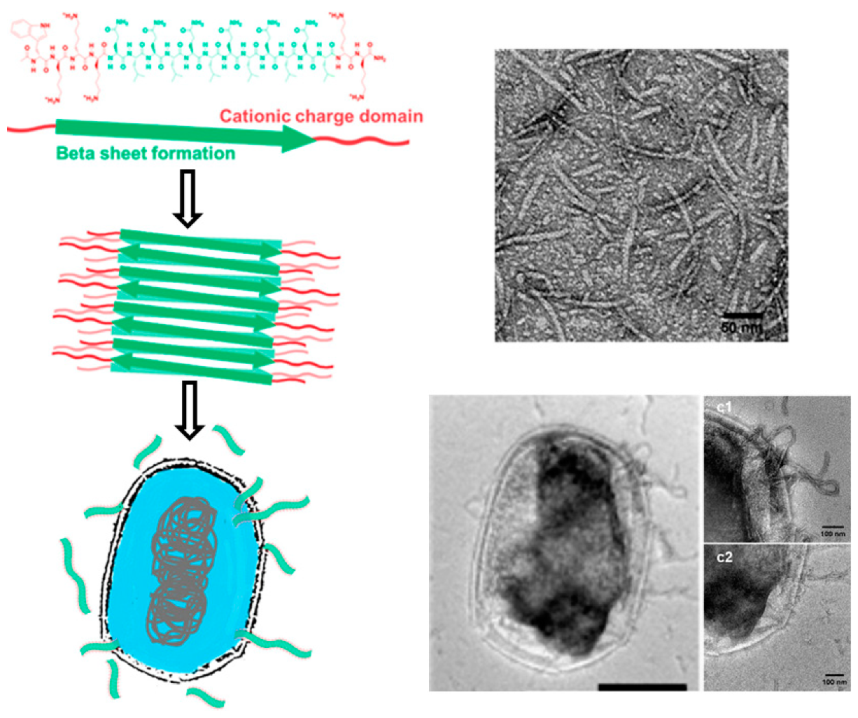
2.1.1. β-Amyloid Fibrils
2.1.2. Non β-Amyloid Fibril Assemblies
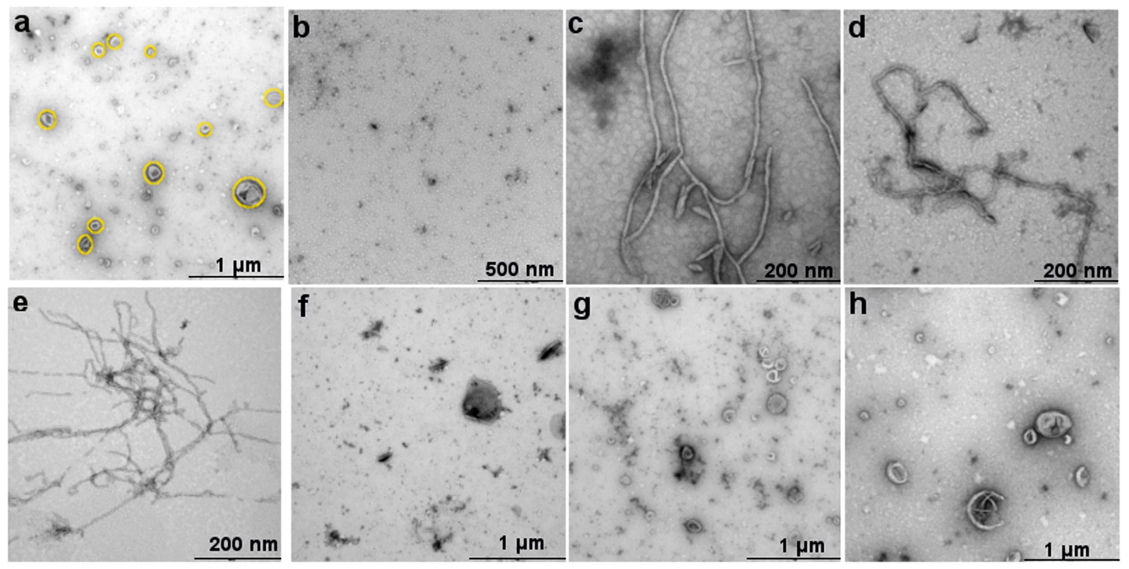
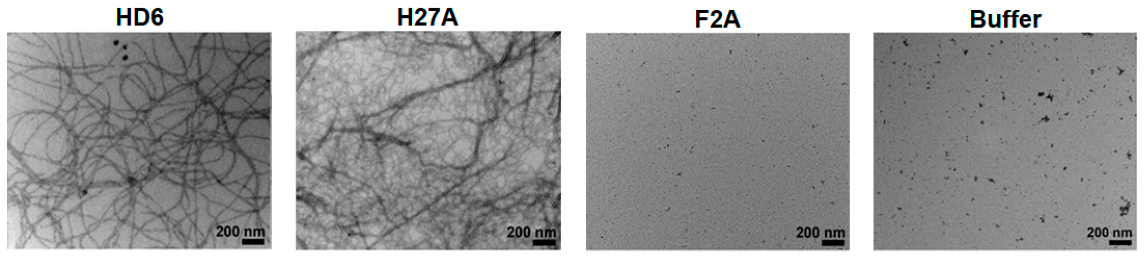
2.2. Nanofibrillar Nets
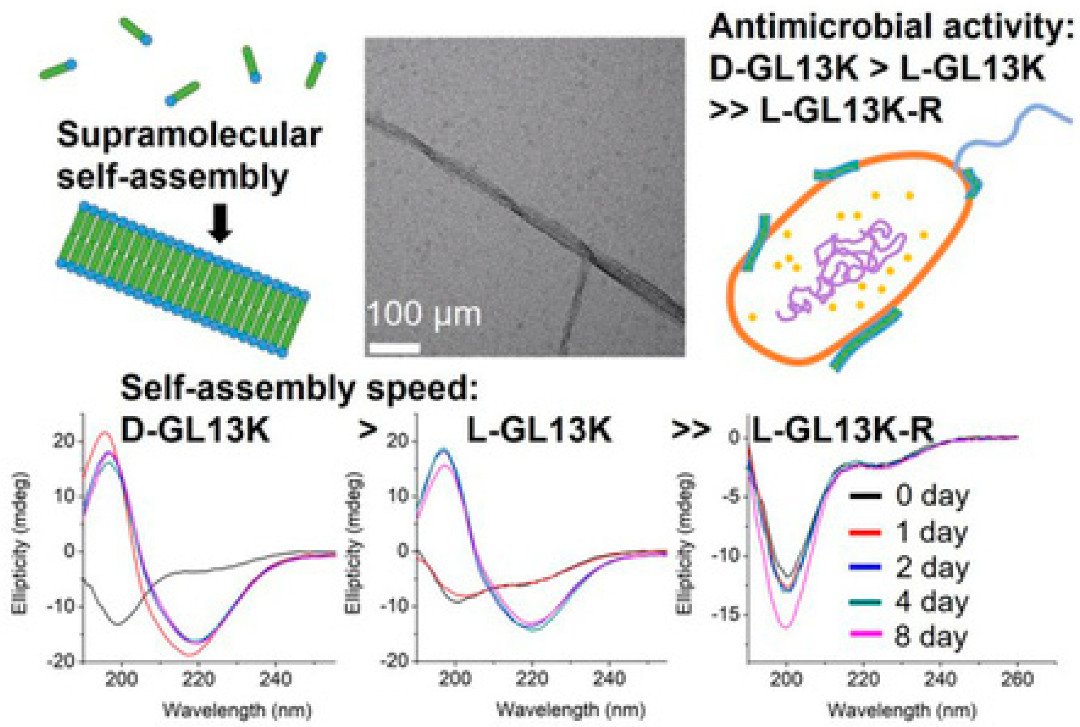
2.3. Nanoribbons
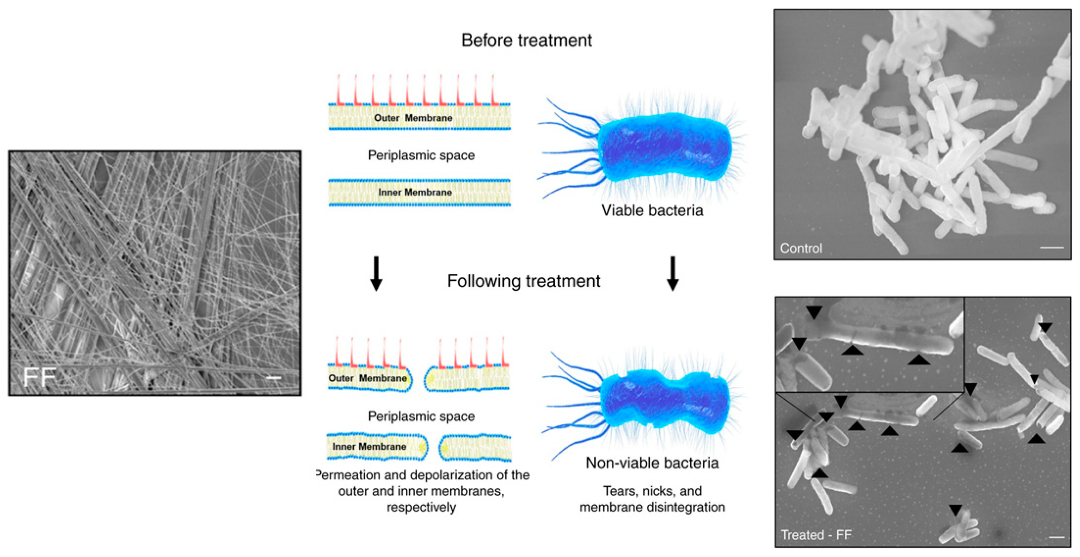
2.4. Nanotubes
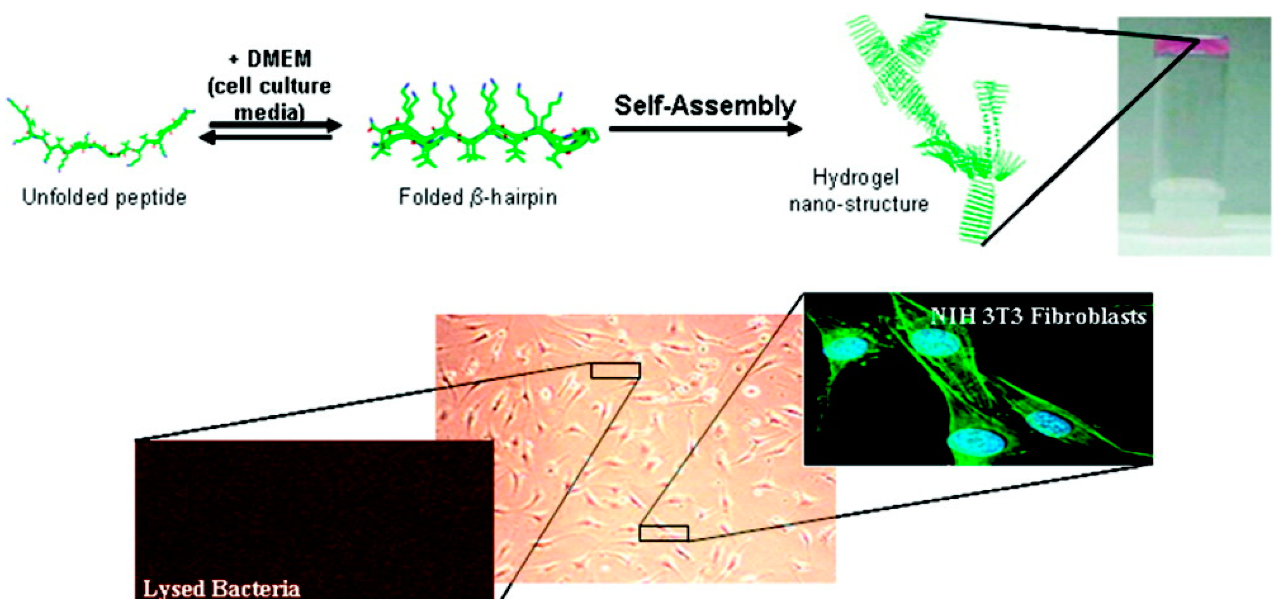
2.5. Macroscopic Fibril Gels
3. Combinatorial Strategies
3.1. AMP-Antimicrobial Hybrid Materials
3.2. Peptide-Excipient Co-Formulations
4. Peptide-Facilitated Antimicrobial Technologies
4.1. Bioresponsive Delivery
4.2. Immunomodulatory Materials
4.3. Active Targeting
5. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleming, A. Classics in Infectious Diseases: On the Antibacterial Action of Cultures of a Penicillium, With Special Reference to Their Use in the Isolation of B. Influenzae. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar]
- Gaynes, R. The discovery of penicillin—new insights after more than 75 years of clinical use. Emerg. Infect. Dis. 2017, 23, 849–853. [Google Scholar] [CrossRef]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells: Molecular mechanisms related to antibiotic tolerance. In Antibiotic Resistance; Springer: Berlin/Heidelberg, Germany, 2012; pp. 121–133. [Google Scholar]
- Hoffman, S.B. Mechanisms of Antibiotic Resistance. Compend. Contin. Educ. Pract. Vet. 2001, 23, 464–472. [Google Scholar]
- Lambert, P.A. Bacterial resistance to antibiotics: Modified target sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Dever, L.A.; Dermody, T.S. Mechanisms of Bacterial Resistance to Antibiotics. Arch. Intern. Med. 1991, 151, 886–895. [Google Scholar] [CrossRef]
- Soto, S.M. Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence 2013, 4, 223–229. [Google Scholar] [CrossRef]
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.; Martinez, J. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14. [Google Scholar] [CrossRef]
- van Duin, D.; Paterson, D.L. Multidrug-Resistant Bacteria in the Community: Trends and Lessons Learned. Infect. Dis. Clin. 2016, 30, 377–390. [Google Scholar] [CrossRef]
- Morley, V.J.; Woods, R.J.; Read, A.F. Bystander Selection for Antimicrobial Resistance: Implications for Patient Health. Trends Microbiol. 2019, 27, 864–877. [Google Scholar] [CrossRef]
- Norris, J.S.; Westwater, C.; Schofield, D. Prokaryotic gene therapy to combat multidrug resistant bacterial infection. Gene Ther. 2000, 7, 723–725. [Google Scholar] [CrossRef]
- Roach, D.R.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Silvestri, E.; Esposito, S. Advantages and Limitations of Bacteriophages for the Treatment of Bacterial Infections. Front. Pharmacol. 2019, 10, 513. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, 5480. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Zhang, L.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Izadpanah, A.; Gallo, R.L.; Diego, S. Antimicrobial peptides. J. Am. Acad. Dermatol. 2005, 52, 381–390. [Google Scholar] [CrossRef]
- Opal, S.M. Non-antibiotic treatments for bacterial diseases in an era of progressive antibiotic resistance. Crit. Care 2016, 20, 397. [Google Scholar] [CrossRef]
- Rex, J.H.; Fernandez Lynch, H.; Cohen, I.G.; Darrow, J.J.; Outterson, K. Designing development programs for non-traditional antibacterial agents. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Piddock, L.J. V Review Non-traditional Antibacterial Therapeutic Options and Challenges. Cell Host Microbe 2019, 26, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, N.V.; Rahme, L.; Cho, Y.H. Editorial: Beyond Antimicrobials: Non-traditional Approaches to Combating Multidrug-Resistant Bacteria. Front. Cell. Infect. Microbiol. 2019, 9, 343. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A.R.B. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Scott, M.G. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Guralp, S.A.; Murgha, Y.E.; Rouillard, J.-M.; Gulari, E. From Design to Screening: A New Antimicrobial Peptide Discovery Pipeline. PLoS ONE 2013, 8, e59305. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Tucker, A.T.; Leonard, S.P.; DuBois, C.D.; Knauf, G.A.; Cunningham, A.L.; Wilke, C.O.; Trent, M.S.; Davies, B.W. Discovery of Next-Generation Antimicrobials through Bacterial Self-Screening of Surface-Displayed Peptide Libraries. Cell 2018, 172, 618–628. [Google Scholar] [CrossRef]
- Kulkarni, K.; Habila, N.; Del Borgo, M.P.; Aguilar, M.I. Novel materials from the supramolecular self-assembly of short helical β3-peptide foldamers. Front. Chem. 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, K.; Xing, R.; Yan, X. Peptide self-assembly: Thermodynamics and kinetics. Chem. Soc. Rev. 2016, 45, 5589–5604. [Google Scholar] [CrossRef] [PubMed]
- Vanier, G.S. Microwave-assisted solid-phase peptide synthesis based on the fmoc protecting group strategy (CEM). Methods Mol. Biol. 2013, 1047, 235–249. [Google Scholar] [PubMed]
- Andersson, L.; Blomberg, L.; Flegel, M.; Lepsa, L.; Nilsson, B.; Verlander, M. Large-scale synthesis of peptides. Pept. Sci. 2000, 55, 227–250. [Google Scholar] [CrossRef]
- Bray, B.L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593. [Google Scholar] [CrossRef]
- Collier, J.H.; Segura, T. Evolving the use of peptides as components of biomaterials. Biomaterials 2011, 32, 4198–4204. [Google Scholar] [CrossRef]
- Spicer, C.D.; Pashuck, E.T.; Stevens, M.M. Achieving Controlled Biomolecule-Biomaterial Conjugation. Chem. Rev. 2018, 118, 7702–7743. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Becker, M.L. “Click” reactions: A versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev. 2014, 43, 7013–7039. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Lehrer, R. Cationic peptides: A new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Brogden, K.A.; Ackermann, M.; McCray, P.B.; Tack, B.F. Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob. Agents 2003, 22, 465–478. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar] [CrossRef]
- Van’T Hof, W.; Veerman, E.C.I.; Heimerhorst, E.J.; Nieuw Amerongen, A.V. Antimicrobial peptides: Properties and applicability. Biol. Chem. 2001, 382, 597–619. [Google Scholar] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolym. Pept. Sci. Sect. 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Wimley, W.C.; Hristova, K. Antimicrobial peptides: Successes, challenges and unanswered questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.P.S.; Hancock, R.E.W. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Kagan, B.L.; Jang, H.; Capone, R.; Teran Arce, F.; Ramachandran, S.; Lal, R.; Nussinov, R. Antimicrobial properties of amyloid peptides. Mol. Pharm. 2012, 9, 708–717. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, J.; Zheng, J. Molecular understanding of a potential functional link between antimicrobial and amyloid peptides. Soft Matter 2014, 10, 7425–7451. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Q.; Chen, J.C.; Cui, Y.X.; Zhou, B.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Antimicrobial activity of human islet amyloid polypeptides: An insight into amyloid peptides’ connection with antimicrobial peptides. Biol. Chem. 2012, 393, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Kagan, B.L. Antimicrobial amyloids? Biophys. J. 2011, 100, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Moir, R.D.; Wagner, S.L. Clearance of Alzheimer’s Aβ peptide: The many roads to perdition. Neuron 2004, 43, 605–608. [Google Scholar] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Parady, B. Innate Immune and Fungal Model of Alzheimer’s Disease. J. Alzheimer’s Dis. Reports 2018, 2, 139–152. [Google Scholar] [CrossRef]
- Spitzer, P.; Condic, M.; Herrmann, M.; Oberstein, T.J.; Scharin-Mehlmann, M.; Gilbert, D.F.; Friedrich, O.; Grömer, T.; Kornhuber, J.; Lang, R.; et al. Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Moir, R.D.; Tanzi, R.E. Low Evolutionary Selection Pressure in Senescence Does Not Explain the Persistence of Aβ in the Vertebrate Genome. Front. Aging Neurosci. 2019, 11, 70. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Aberrant action of amyloidogenic host defense peptides: A new paradigm to investigate neurodegenerative disorders? FASEB J. 2012, 26, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Kandel, N.; Zheng, T.; Huo, Q.; Tatulian, S.A. Membrane Binding and Pore Formation by a Cytotoxic Fragment of Amyloid β Peptide. J. Phys. Chem. B 2017, 121, 10293–10305. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.V.; Eimer, W.A.; Tanzi, R.E.; Moir, R.D. Alzheimer’s disease: The potential therapeutic role of the natural antibiotic amyloid-β peptide. Neurodegener. Dis. Manag. 2016, 6, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef]
- Peters, C.; Bascuñán, D.; Opazo, C.; Aguayo, L.G. Differential Membrane Toxicity of amyloid-β Fragments by Pore Forming Mechanisms. J. Alzheimer’s Dis. 2016, 51, 689–699. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, H.S.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Giridharan, V.V.; Masud, F.; Petronilho, F.; Dal-Pizzol, F.; Barichello, T. Infection-Induced Systemic Inflammation Is a Potential Driver of Alzheimer’s Disease Progression. Front. Aging Neurosci. 2019, 11, 122. [Google Scholar] [CrossRef]
- Lee, S.; Trinh, T.H.T.; Yoo, M.; Shin, J.; Lee, H.; Kim, J.; Hwang, E.; Lim, Y.B.; Ryou, C. Self-assembling peptides and their application in the treatment of diseases. Int. J. Mol. Sci. 2019, 20, 5850. [Google Scholar] [CrossRef]
- Schnaider, L.; Brahmachari, S.; Schmidt, N.W.; Mensa, B.; Shaham-Niv, S.; Bychenko, D.; Adler-Abramovich, L.; Shimon, L.J.W.; Kolusheva, S.; Degrado, W.F.; et al. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Hu, Y.; Lin, R.; Zhang, P.; Fern, J.; Cheetham, A.G.; Patel, K.; Schulman, R.; Kan, C.; Cui, H. Electrostatic-driven lamination and untwisting of β-sheet assemblies. ACS Nano 2016, 10, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Guo, Z.; Zhou, L.; Wang, Y.; Zhang, J.; Hu, J.; Zhang, Y. Biomembrane induced in situ self-assembly of peptide with enhanced antimicrobial activity. Biomater. Sci. 2020, 8, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Simonson, A.W.; Mongia, A.S.; Aronson, M.R.; Alumasa, J.N.; Chan, D.C.; Bolotsky, A.; Ebrahimi, A.; Proctor, E.A.; Keiler, K.C.; Medina, S.H. Pathogen-specific de novo antimicrobials engineered through membrane porin biomimicry. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Xu, L.; Chou, S.; Wang, J.; Shao, C.; Li, W.; Zhu, X.; Shan, A. Antimicrobial activity and membrane-active mechanism of tryptophan zipper-like β-hairpin antimicrobial peptides. Amino Acids 2015, 47, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Balandin, S.V.; Ovchinnikova, T.V. Effect of Arenicins and Other β-Hairpin Antimicrobial Peptides on Pseudomonas Aeruginosa PAO1 Biofilms. Pharm. Chem. J. 2017, 50, 715–720. [Google Scholar] [CrossRef]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: Chemical structures and biological activity. J. Biochem. 1989, 106, 663–668. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β-hairpin peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Wang, C.K.; King, G.J.; Conibear, A.C.; Ramos, M.C.; Chaousis, S.; Henriques, S.T.; Craik, D.J. Mirror images of antimicrobial peptides provide reflections on their functions and amyloidogenic properties. J. Am. Chem. Soc. 2016, 138, 5706–5713. [Google Scholar] [CrossRef]
- Wang, C.K.; Craik, D.J. Toward Structure Determination of Disulfide-Rich Peptides Using Chemical Shift-Based Methods. J. Phys. Chem. B 2019, 123, 1903–1912. [Google Scholar] [CrossRef]
- Wang, C.K.; Northfield, S.E.; Huang, Y.H.; Ramos, M.C.; Craik, D.J. Inhibition of tau aggregation using a naturally-occurring cyclic peptide scaffold. Eur. J. Med. Chem. 2016, 109, 342–349. [Google Scholar] [CrossRef]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van Der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Balandin, S.V.; Ivanov, V.T.; Ovchinnikova, T.V. A Therapeutic Potential of Animal β-hairpin Antimicrobial Peptides. Curr. Med. Chem. 2017, 24, 1724–1746. [Google Scholar] [CrossRef] [PubMed]
- Gour, S.; Kumar, V.; Singh, A.; Gadhave, K.; Goyal, P.; Pandey, J.; Giri, R.; Yadav, J.K. Mammalian antimicrobial peptide protegrin-4 self assembles and forms amyloid-like aggregates: Assessment of its functional relevance. J. Pept. Sci. 2019, 25, e3151. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Mustata, M.; Ramachandran, S.; Capone, R.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011, 100, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Chen, W.; Tobin-Miyaji, Y.J.; Sturge, C.R.; Yang, S.; Elmore, B.; Singh, A.; Pybus, C.; Greenberg, D.E.; Sellati, T.J.; et al. Fabrication and Microscopic and Spectroscopic Characterization of Cytocompatible Self-Assembling Antimicrobial Nanofibers. ACS Infect. Dis. 2018, 4, 1327–1335. [Google Scholar] [CrossRef]
- Xu, D.; Jiang, L.; Singh, A.; Dustin, D.; Yang, M.; Liu, L.; Lund, R.; Sellati, T.J.; Dong, H. Designed supramolecular filamentous peptides: Balance of nanostructure, cytotoxicity and antimicrobial activity. Chem. Commun. 2015, 51, 1289–1292. [Google Scholar] [CrossRef]
- Moore, A.N.; Hartgerink, J.D. Self-Assembling Multidomain Peptide Nanofibers for Delivery of Bioactive Molecules and Tissue Regeneration. Acc. Chem. Res. 2017, 50, 714–722. [Google Scholar] [CrossRef]
- Chen, W.; Yang, S.; Li, S.; Lang, J.C.; Mao, C.; Kroll, P.; Tang, L.; Dong, H. Self-Assembled Peptide Nanofibers Display Natural Antimicrobial Peptides to Selectively Kill Bacteria without Compromising Cytocompatibility. ACS Appl. Mater. Interfaces 2019, 11, 28681–28689. [Google Scholar] [CrossRef]
- Chen, W.; Li, S.; Renick, P.; Yang, S.; Pandy, N.; Boutte, C.; Nguyen, K.T.; Tang, L.; Dong, H. Bacterial acidity-triggered antimicrobial activity of self-assembling peptide nanofibers. J. Mater. Chem. B 2019, 7, 2915–2919. [Google Scholar] [CrossRef]
- Malishev, R.; Tayeb-Fligelman, E.; David, S.; Meijler, M.M.; Landau, M.; Jelinek, R. Reciprocal Interactions between Membrane Bilayers and S. aureus PSMα3 Cross-α Amyloid Fibrils Account for Species-Specific Cytotoxicity. J. Mol. Biol. 2018, 430, 1431–1441. [Google Scholar] [CrossRef]
- Tayeb-Fligelman, E.; Tabachnikov, O.; Moshe, A.; Goldshmidt-Tran, O.; Sawaya, M.R.; Coquelle, N.; Colletier, J.P.; Landau, M. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 2017, 355, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Salinas, N.; Colletier, J.-P.; Moshe, A.; Landau, M. Extreme amyloid polymorphism in Staphylococcus aureus virulent PSMα peptides. Nat. Commun. 2018, 9, 3512. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Xu, C.; Cheng, Q.; Liu, J.; Gao, W.; Yang, X.; Wong, K.; Chen, S.; Chan, K. Phenol-Soluble-Modulin-Inspired Amphipathic Peptides Have Bactericidal Activity against Multidrug-Resistant Bacteria. ChemMedChem 2019, 14, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Zeth, K.; Sancho-Vaello, E. The Human Antimicrobial Peptides Dermcidin and LL-37 Show Novel Distinct Pathways in Membrane Interactions. Front. Chem. 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Vaello, E.; François, P.; Bonetti, E.J.; Lilie, H.; Finger, S.; Gil-Ortiz, F.; Gil-Carton, D.; Zeth, K. Structural remodeling and oligomerization of human cathelicidin on membranes suggest fibril-like structures as active species. Sci. Rep. 2017, 7, 15371. [Google Scholar] [CrossRef] [PubMed]
- Aronson, M.R.; Simonson, A.W.; Orchard, L.M.; Llinás, M.; Medina, S.H. Lipopeptisomes: Anticancer peptide-assembled particles for fusolytic oncotherapy. Acta Biomater. 2018, 80, 269–277. [Google Scholar] [CrossRef]
- Sood, R.; Domanov, Y.; Pietiäinen, M.; Kontinen, V.P.; Kinnunen, P.K.J. Binding of LL-37 to model biomembranes: Insight into target vs host cell recognition. Biochim. Biophys. Acta Biomembr. 2008, 1778, 983–996. [Google Scholar] [CrossRef]
- Shahmiri, M.; Enciso, M.; Adda, C.G.; Smith, B.J.; Perugini, M.A.; Mechler, A. Membrane Core-Specific Antimicrobial Action of Cathelicidin LL-37 Peptide Switches between Pore and Nanofibre Formation. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- De Lorenzi, E.; Chiari, M.; Colombo, R.; Cretich, M.; Sola, L.; Vanna, R.; Gagni, P.; Bisceglia, F.; Morasso, C.; Lin, J.S.; et al. Evidence that the human innate immune peptide LL-37 may be a binding partner of amyloid-β and inhibitor of fibril assembly. J. Alzheimer’s Dis. 2017, 59, 1213–1226. [Google Scholar] [CrossRef]
- Chairatana, P.; Nolan, E.M. Molecular basis for self-assembly of a human host-defense peptide that entraps bacterial pathogens. J. Am. Chem. Soc. 2014, 136, 13267–13276. [Google Scholar] [CrossRef]
- Chairatana, P.; Nolan, E.M. Human α-Defensin 6: A Small Peptide That Self-Assembles and Protects the Host by Entangling Microbes. Acc. Chem. Res. 2017, 50, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Ehmann, D.; Precht, J.C.; Castillo, P.A.; Küchler, R.; Berger, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Paneth cell α-defensin 6 (HD-6) is an antimicrobial peptide. Mucosal Immunol. 2015, 8, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Roan, N.R.; Römling, U.; Bevins, C.L.; Münch, J. Amyloid formation: Functional friend or fearful foe? J. Intern. Med. 2016, 280, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Sankaran-Walters, S.; Hart, R.; Dills, C. Guardians of the gut: Enteric defensins. Front. Microbiol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Pazgier, M.; Jung, G.; Nuccio, S.P.; Castillo, P.A.; De Jong, M.F.; Winter, M.G.; Winter, S.E.; Wehkamp, J.; Shen, B.; et al. Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 2012, 337, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, D.; Wendler, J.; Koeninger, L.; Larsen, I.S.; Klag, T.; Berger, J.; Marette, A.; Schaller, M.; Stange, E.F.; Malek, N.P.; et al. Paneth cell α-defensins HD-5 and HD-6 display differential degradation into active antimicrobial fragments. Proc. Natl. Acad. Sci. USA 2019, 116, 3746–3751. [Google Scholar] [CrossRef]
- Raschig, J.; Mailänder-Sánchez, D.; Berscheid, A.; Berger, J.; Strömstedt, A.A.; Courth, L.F.; Malek, N.P.; Brötz-Oesterhelt, H.; Wehkamp, J. Ubiquitously expressed Human Beta Defensin 1 (hBD1) forms bacteria-entrapping nets in a redox dependent mode of action. PLoS Pathog. 2017, 13, e1006261. [Google Scholar] [CrossRef]
- Loth, K.; Vergnes, A.; Barreto, C.; Voisin, S.N.; Meudal, H.; Da Silva, J.; Bressan, A.; Belmadi, N.; Bachère, E.; Aucagne, V.; et al. The ancestral N-terminal domain of big defensins drives bacterially triggered assembly into antimicrobial nanonets. mBio 2019, 10, e01821-19. [Google Scholar] [CrossRef]
- Stambuk, F.; Ojeda, C.; Schmitt, P. Big Defensin ApBD1 from the scallop Argopecten purpuratus is an antimicrobial peptide which entraps bacteria through nanonets formation. bioRxiv 2020. [CrossRef]
- González, R.; Brokordt, K.; Cárcamo, C.B.; Coba de la Peña, T.; Oyanedel, D.; Mercado, L.; Schmitt, P. Molecular characterization and protein localization of the antimicrobial peptide big defensin from the scallop Argopecten purpuratus after Vibrio splendidus challenge. Fish. Shellfish Immunol. 2017, 68, 173–179. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, D.; Zhou, P.; Zhao, Y.; Sun, Y.; Wang, J. Influence of Conventional Surfactants on the Self-Assembly of a Bola Type Amphiphilic Peptide. Langmuir 2017, 33, 5446–5455. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yuan, C.; Shen, G.; Chang, R.; Xing, R.; Yan, X. Cyclic dipeptide nanoribbons formed by dye-mediated hydrophobic self-assembly for cancer chemotherapy. J. Colloid Interface Sci. 2019, 557, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Levin, A.; Adler-Abramovich, L.; Gazit, E. Fmoc-modified amino acids and short peptides: Simple bio-inspired building blocks for the fabrication of functional materials. Chem. Soc. Rev. 2016, 45, 3935–3953. [Google Scholar] [CrossRef] [PubMed]
- Abdolhosseini, M.; Nandula, S.R.; Song, J.; Hirt, H.; Gorr, S.U. Lysine substitutions convert a bacterial-agglutinating peptide into a bactericidal peptide that retains anti-lipopolysaccharide activity and low hemolytic activity. Peptides 2012, 35, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Harmouche, N.; Aisenbrey, C.; Porcelli, F.; Xia, Y.; Nelson, S.E.D.; Chen, X.; Raya, J.; Vermeer, L.; Aparicio, C.; Veglia, G.; et al. Solution and Solid-State Nuclear Magnetic Resonance Structural Investigations of the Antimicrobial Designer Peptide GL13K in Membranes. Biochemistry 2017, 56, 4269–4278. [Google Scholar] [CrossRef] [PubMed]
- Hirt, H.; Hall, J.W.; Larson, E.; Gorr, S.-U. A D-enantiomer of the antimicrobial peptide GL13K evades antimicrobial resistance in the Gram positive bacteria Enterococcus faecalis and Streptococcus gordonii. PLoS ONE 2018, 13, e0194900. [Google Scholar] [CrossRef]
- Ye, Z.; Zhu, X.; Acosta, S.; Kumar, D.; Sang, T.; Aparicio, C. Self-assembly dynamics and antimicrobial activity of all l- and d-amino acid enantiomers of a designer peptide. Nanoscale 2019, 11, 266–275. [Google Scholar] [CrossRef]
- Porter, S.L.; Coulter, S.M.; Pentlavalli, S.; Thompson, T.P.; Laverty, G. Self-assembling diphenylalanine peptide nanotubes selectively eradicate bacterial biofilm infection. Acta Biomater. 2018, 77, 96–105. [Google Scholar] [CrossRef]
- Yang, K.; Han, Q.; Chen, B.; Zheng, Y.; Zhang, K.; Li, Q.; Wang, J. Antimicrobial hydrogels: Promising materials for medical application. Int. J. Nanomed. 2018, 13, 2217–2263. [Google Scholar] [CrossRef]
- Ng, V.W.L.; Chan, J.M.W.; Sardon, H.; Ono, R.J.; García, J.M.; Yang, Y.Y.; Hedrick, J.L. Antimicrobial hydrogels: A new weapon in the arsenal against multidrug-resistant infections. Adv. Drug Deliv. Rev. 2014, 78, 46–62. [Google Scholar] [CrossRef]
- Salomé Veiga, A.; Schneider, J.P. Antimicrobial hydrogels for the treatment of infection. Biopolymers 2013, 100, 637–644. [Google Scholar] [CrossRef]
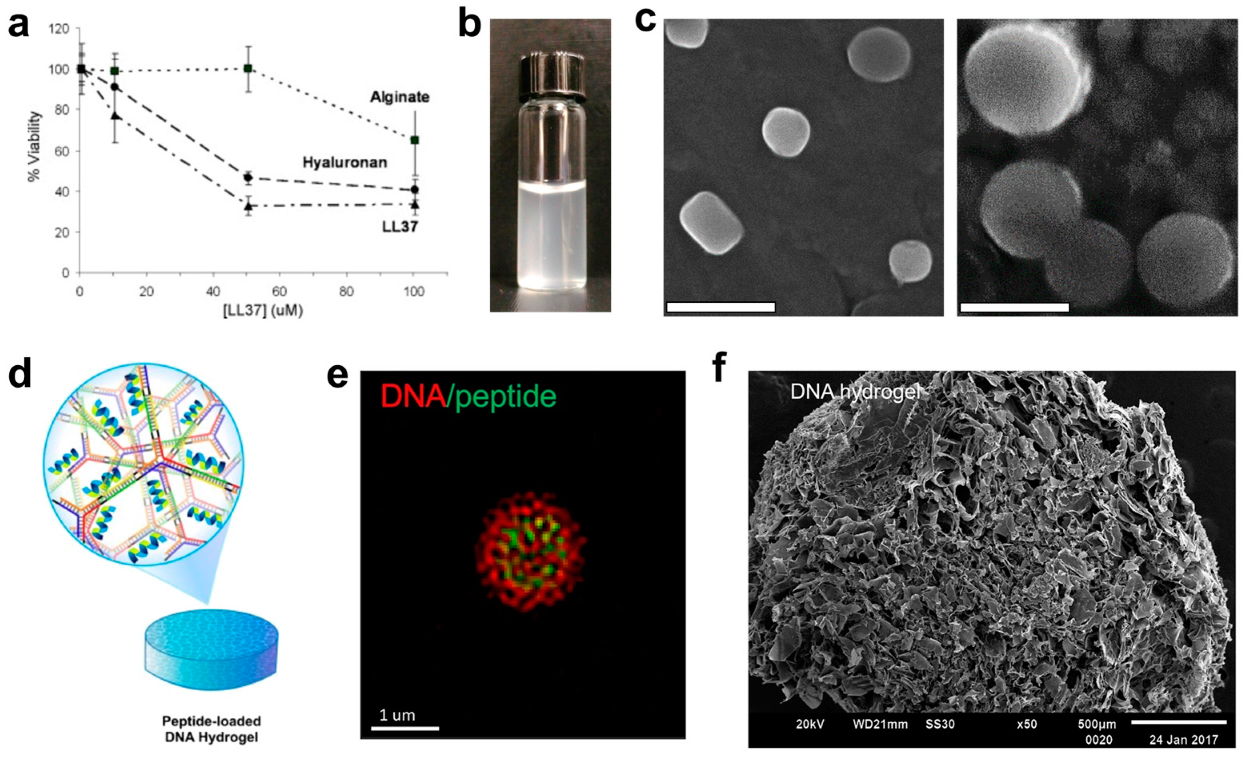
- Veiga, A.S.; Sinthuvanich, C.; Gaspar, D.; Franquelim, H.G.; Castanho, M.A.R.B.; Schneider, J.P. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials 2012, 33, 8907–8916. [Google Scholar] [CrossRef] [PubMed]
- Salick, D.A.; Kretsinger, J.K.; Pochan, D.J.; Schneider, J.P. Inherent antibacterial activity of a peptide-based β-hairpin hydrogel. J. Am. Chem. Soc. 2007, 129, 14793–14799. [Google Scholar] [CrossRef] [PubMed]
- Kretsinger, J.K.; Haines, L.A.; Ozbas, B.; Pochan, D.J.; Schneider, J.P. Cytocompatibility of self-assembled β-hairpin peptide hydrogel surfaces. Biomaterials 2005, 26, 5177–5186. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Smith, K.; Moore, E.; Schneider, J.; Tycko, R.; DeGrado, W.F. Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc. Natl. Acad. Sci. USA 2015, 112, 9816–9821. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Smith, K.; Beltramo, P.J.; Moore, E.; Tycko, R.; Furst, E.M.; Schneider, J.P. Molecular, Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey. ACS Cent. Sci. 2017, 3, 586–597. [Google Scholar] [CrossRef]
- Salick, D.A.; Pochan, D.J.; Schneider, J.P. Design of an Injectable β-Hairpin Peptide Hydrogel That Kills Methicillin-Resistant Staphylococcus aureus. Adv. Mater. 2009, 21, 4120–4123. [Google Scholar] [CrossRef]
- Yan, C.; Altunbas, A.; Yucel, T.; Nagarkar, R.P.; Schneider, J.P.; Pochan, D.J. Injectable solid hydrogel: Mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter 2010, 6, 5143–5156. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, D.; Sellati, T.J.; Dong He Dong, H. Self-assembly of cationic multidomain peptide hydrogels: Supramolecular nanostructure and rheological properties dictate antimicrobial activity. Nanoscale 2015, 7, 19160–19169. [Google Scholar] [CrossRef]
- Bai, J.; Chen, C.; Wang, J.; Zhang, Y.; Cox, H.; Zhang, J.; Wang, Y.; Penny, J.; Waigh, T.; Lu, J.R.; et al. Enzymatic Regulation of Self-Assembling Peptide A9K2 Nanostructures and Hydrogelation with Highly Selective Antibacterial Activities. ACS Appl. Mater. Interfaces 2016, 8, 15093–15102. [Google Scholar] [CrossRef]
- Sarkar, B.; Siddiqui, Z.; Nguyen, P.K.; Dube, N.; Fu, W.; Park, S.; Jaisinghani, S.; Paul, R.; Kozuch, S.D.; Deng, D.; et al. Membrane-Disrupting Nanofibrous Peptide Hydrogels. ACS Biomater. Sci. Eng. 2019, 5, 4657–4670. [Google Scholar] [CrossRef]
- McCloskey, A.P.; Draper, E.R.; Gilmore, B.F.; Laverty, G. Ultrashort self-assembling Fmoc-peptide gelators for anti-infective biomaterial applications. J. Pept. Sci. 2017, 23, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Raeburn, J.; Cardoso, A.Z.; Adams, D.J. The importance of the self-assembly process to control mechanical properties of low molecular weight hydrogels. Chem. Soc. Rev. 2013, 42, 5143–5156. [Google Scholar] [CrossRef] [PubMed]
- Mahler, A.; Reches, M.; Rechter, M.; Cohen, S.; Gazit, E. Rigid, Self-Assembled Hydrogel Composed of a Modified Aromatic Dipeptide. Adv. Mater. 2006, 18, 1365–1370. [Google Scholar] [CrossRef]
- Tang, C.; Smith, A.M.; Collins, R.F.; Ulijn, R.V.; Saiani, A. Fmoc-Diphenylalanine Self-Assembly Mechanism Induces Apparent pK a Shifts. Langmuir 2009, 25, 9447–9453. [Google Scholar] [CrossRef]
- Hughes, M.; Debnath, S.; Knapp, C.W.; Ulijn, R.V. Antimicrobial properties of enzymatically triggered self-assembling aromatic peptide amphiphiles. Biomater. Sci. 2013, 1, 1138–1142. [Google Scholar] [CrossRef]
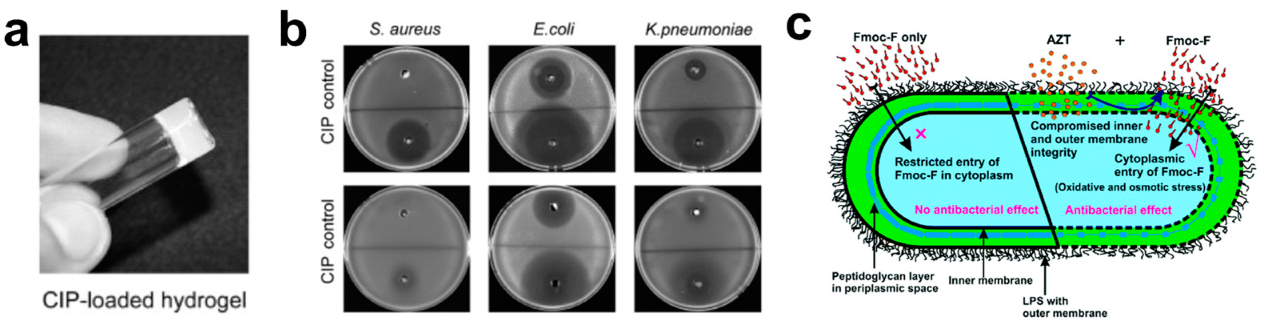
- Gahane, A.Y.; Ranjan, P.; Singh, V.; Sharma, R.K.; Sinha, N.; Sharma, M.; Chaudhry, R.; Thakur, A.K. Fmoc-phenylalanine displays antibacterial activity against Gram-positive bacteria in gel and solution phases. Soft Matter 2018, 14, 2234–2244. [Google Scholar] [CrossRef]
- Xie, Y.Y.; Zhang, Y.W.; Qin, X.T.; Liu, L.P.; Wahid, F.; Zhong, C.; Jia, S.R. Structure-Dependent Antibacterial Activity of Amino Acid-Based Supramolecular Hydrogels. Colloids Surfaces B Biointerfaces 2020, 193, 111099. [Google Scholar] [CrossRef]
- Shi, J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef]
- Pizzolato-Cezar, L.R.; Okuda-Shinagawa, N.M.; Machini, M.T. Combinatory Therapy Antimicrobial Peptide-Antibiotic to Minimize the Ongoing Rise of Resistance. Front. Microbiol. 2019, 10, 1703. [Google Scholar] [CrossRef]
- Grassi, L.; Maisetta, G.; Esin, S.; Batoni, G. Combination Strategies to Enhance the Efficacy of Antimicrobial Peptides against Bacterial Biofilms. Front. Microbiol. 2017, 8, 2409. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Z.; Li, X.; Tian, Y.; Fan, Y.; Yu, C.; Zhou, B.; Liu, Y.; Xiang, R.; Yang, L. Synergistic effects of antimicrobial peptide DP7 combined with antibiotics against multidrug-resistant bacteria. Drug Des. Devel. Ther. 2017, 11, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Xu, C.; Liu, C.; Liu, J.; Cheng, Q.; Gao, W.; Yang, X.; Chen, S.; Chan, K.F.; Wong, K.Y. De Novo Designed Hexadecapeptides Synergize Glycopeptide Antibiotics Vancomycin and Teicoplanin against Pathogenic Klebsiella pneumoniae via Disruption of Cell Permeability and Potential. ACS Appl. Bio Mater. 2020, 3, 1738–1752. [Google Scholar] [CrossRef]
- Pletzer, D.; Mansour, S.C.; Hancock, R.E.W. Synergy between conventional antibiotics and anti-biofilm peptides in a murine, sub-cutaneous abscess model caused by recalcitrant ESKAPE pathogens. PLoS Pathog. 2018, 14, e1007084. [Google Scholar] [CrossRef]
- Aqeele, E.; Abkooh, E. Determination of the Effective Dose of Curcumin alone and in Combination with Antimicrobial Peptide CM11 on Promastigote Forms of Iranian Strain of L. major (MRHO/IR/75/ER). Arch. Razi Inst. 2019, 74, 413–422. [Google Scholar]
- Marchesan, S.; Qu, Y.; Waddington, L.J.; Easton, C.D.; Glattauer, V.; Lithgow, T.J.; McLean, K.M.; Forsythe, J.S.; Hartley, P.G. Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel. Biomaterials 2013, 34, 3678–3687. [Google Scholar] [CrossRef]
- Gahane, A.Y.; Singh, V.; Kumar, A.; Kumar Thakur, A. Development of mechanism-based antibacterial synergy between Fmoc-phenylalanine hydrogel and aztreonam. Biomater. Sci. 2020, 8, 1996–2006. [Google Scholar] [CrossRef]
- Vegners, R.; Shestakova, I.; Kalvinsh, I.; Ezzell, R.M.; Janmey, P.A. Use of a gel-forming dipeptide derivative as a carrier for antigen presentation. J. Pept. Sci. 1995, 1, 371–378. [Google Scholar] [CrossRef]
- Thota, C.K.; Yadav, N.; Chauhan, V.S. A novel highly stable and injectable hydrogel based on a conformationally restricted ultrashort peptide. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Guler, M.O.; Claussen, R.C.; Stupp, S.I. Encapsulation of pyrene within self-assembled peptide amphiphile nanofibers. J. Mater. Chem. 2005, 15, 4507–4512. [Google Scholar] [CrossRef]
- Nagai, Y.; Unsworth, L.D.; Koutsopoulos, S.; Zhang, S. Slow release of molecules in self-assembling peptide nanofiber scaffold. J. Control. Release 2006, 115, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Altunbas, A.; Lee, S.J.; Rajasekaran, S.A.; Schneider, J.P.; Pochan, D.J. Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles. Biomaterials 2011, 32, 5906–5914. [Google Scholar] [CrossRef]
- McCloskey, A.P.; Gilmore, S.M.; Zhou, J.; Draper, E.R.; Porter, S.; Gilmore, B.F.; Xu, B.; Laverty, G. Self-assembling ultrashort NSAID-peptide nanosponges: Multifunctional antimicrobial and anti-inflammatory materials. RSC Adv. 2016, 6, 114738–114749. [Google Scholar] [CrossRef]
- Li, X.; Fan, R.; Tong, A.; Yang, M.; Deng, J.; Zhou, L.; Zhang, X.; Guo, G. In situ gel-forming AP-57 peptide delivery system for cutaneous wound healing. Int. J. Pharm. 2015, 495, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Augst, A.D.; Kong, H.J.; Mooney, D.J. Alginate Hydrogels as Biomaterials. Macromol. Biosci. 2006, 6, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, G.; Bouropoulos, N. Swelling studies and in vitro release of verapamil from calcium alginate and calcium alginate-chitosan beads. Int. J. Pharm. 2006, 323, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Mikula, K.; Skrzypczak, D.; Ligas, B.; Witek-Krowiak, A. Preparation of hydrogel composites using Ca2+ and Cu2+ ions as crosslinking agents. SN Appl. Sci. 2019, 1, 1–15. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Chang, H.-Y.; Lu, J.-K.; Huang, Y.-C.; Harroun, S.G.; Tseng, Y.-T.; Li, Y.-J.; Huang, C.-C.; Chang, H.-T. Self-Assembly of Antimicrobial Peptides on Gold Nanodots: Against Multidrug-Resistant Bacteria and Wound-Healing Application. Adv. Funct. Mater. 2015, 25, 7189–7199. [Google Scholar] [CrossRef]
- Rai, A.; Pinto, S.; Velho, T.R.; Ferreira, A.F.; Moita, C.; Trivedi, U.; Evangelista, M.; Comune, M.; Rumbaugh, K.P.; Simões, P.N.; et al. One-step synthesis of high-density peptide-conjugated gold nanoparticles with antimicrobial efficacy in a systemic infection model. Biomaterials 2016, 85, 99–110. [Google Scholar] [CrossRef]
- Huang, C.-C.; Yang, Z.; Lee, K.-H.; Chang, H.-T. Synthesis of Highly Fluorescent Gold Nanoparticles for Sensing Mercury(II). Angew. Chemie Int. Ed. 2007, 46, 6824–6828. [Google Scholar] [CrossRef]
- Yang, T.-Q.; Peng, B.; Shan, B.-Q.; Zong, Y.-X.; Jiang, J.-G.; Wu, P.; Zhang, K. Origin of the Photoluminescence of Metal Nanoclusters: From Metal-Centered Emission to Ligand-Centered Emission. Nanomaterials 2020, 10, 261. [Google Scholar] [CrossRef] [PubMed]
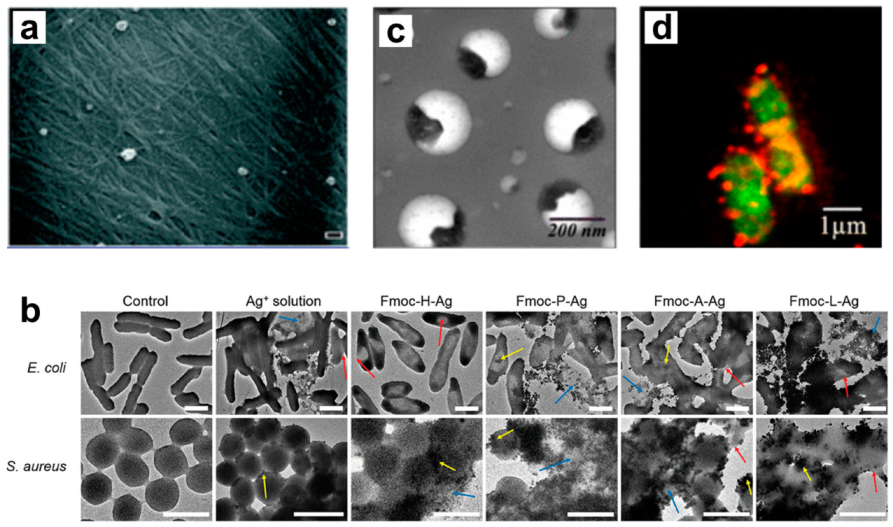
- Simon, T.; Wu, C.S.; Liang, J.C.; Cheng, C.; Ko, F.H. Facile synthesis of a biocompatible silver nanoparticle derived tripeptide supramolecular hydrogel for antibacterial wound dressings. New J. Chem. 2016, 40, 2036–2043. [Google Scholar] [CrossRef]
- Song, J.; Yuan, C.; Jiao, T.; Xing, R.; Yang, M.; Adams, D.J.; Yan, X. Multifunctional Antimicrobial Biometallohydrogels Based on Amino Acid Coordinated Self-Assembly. Small 2020, 16, 1907309. [Google Scholar] [CrossRef] [PubMed]
- Panja, S.; Bharti, R.; Dey, G.; Lynd, N.A.; Chattopadhyay, S. Coordination-assisted self-assembled polypeptide nanogels to selectively combat bacterial infection. ACS Appl. Mater. Interfaces 2019, 11, 33599–33611. [Google Scholar] [CrossRef]
- Pasquet, J.; Chevalier, Y.; Pelletier, J.; Couval, E.; Bouvier, D.; Bolzinger, M.A. The contribution of zinc ions to the antimicrobial activity of zinc oxide. Colloids Surfaces A Physicochem. Eng. Asp. 2014, 457, 263–274. [Google Scholar] [CrossRef]
- Chan, C.; Burrows, L.L.; Deber, C.M. Helix induction in antimicrobial peptides by alginate in biofilms. J. Biol. Chem. 2004, 279, 38749–38754. [Google Scholar] [CrossRef]
- Lin, Z.; Wu, T.; Wang, W.; Li, B.; Wang, M.; Chen, L.; Xia, H.; Zhang, T. Biofunctions of antimicrobial peptide-conjugated alginate/hyaluronic acid/collagen wound dressings promote wound healing of a mixed-bacteria-infected wound. Int. J. Biol. Macromol. 2019, 140, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Toppazzini, M.; Coslovi, A.; Boschelle, M.; Marsich, E.; Benincasa, M.; Gennaro, R.; Paoletti, S. Can the interaction between the antimicrobial peptide LL-37 and alginate be exploited for the formulation of new biomaterials with antimicrobial properties? Carbohydr. Polym. 2011, 83, 578–585. [Google Scholar] [CrossRef]
- Mateescu, M.; Baixe, S.; Garnier, T.; Jierry, L.; Ball, V.; Haikel, Y.; Metz-Boutigue, M.H.; Nardin, M.; Schaaf, P.; Etienne, O.; et al. Antibacterial peptide-based gel for prevention of medical implanted-device infection. PLoS ONE 2015, 10, e0145143. [Google Scholar] [CrossRef]
- Pereira, P.; Pedrosa, S.S.; Correia, A.; Lima, C.F.; Olmedo, M.P.; González-Fernández, Á.; Vilanova, M.; Gama, F.M. Biocompatibility of a self-assembled glycol chitosan nanogel. Toxicol. In Vitro 2015, 29, 638–646. [Google Scholar] [CrossRef]
- Wu, C.; Wu, T.; Fang, Z.; Zheng, J.; Xu, S.; Chen, S.; Hu, Y.; Ye, X. Formation, characterization and release kinetics of chitosan/γ-PGA encapsulated nisin nanoparticles. RSC Adv. 2016, 6, 46686–46695. [Google Scholar] [CrossRef]
- He, Y.; Jin, Y.; Wang, X.; Yao, S.; Li, Y.; Wu, Q.; Ma, G.; Cui, F.; Liu, H. An antimicrobial peptide-loaded gelatin/chitosan nanofibrous membrane fabricated by sequential layer-by-layer electrospinning and electrospraying techniques. Nanomaterials 2018, 8, 327. [Google Scholar] [CrossRef] [PubMed]
- Water, J.J.; Kim, Y.; Maltesen, M.J.; Franzyk, H.; Foged, C.; Nielsen, H.M. Hyaluronic acid-based nanogels produced by microfluidics-facilitated self-assembly improves the safety profile of the cationic host defense peptide novicidin. Pharm. Res. 2015, 32, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Gonçalves, C.; Costa, C.; Sousa, J.; Silva-Gomes, R.; Castro, A.G.; Pedrosa, J.; Appelberg, R.; Gama, F.M. Delivery of LLKKK18 loaded into self-assembling hyaluronic acid nanogel for tuberculosis treatment. J. Control. Release 2016, 235, 112–124. [Google Scholar] [CrossRef]
- Simonson, A.W.; Lawanprasert, A.; Goralski, T.D.P.; Keiler, K.C.; Medina, S.H. Bioresponsive peptide-polysaccharide nanogels—A versatile delivery system to augment the utility of bioactive cargo. Nanomedicine 2019, 17, 391–400. [Google Scholar] [CrossRef]
- Huang, R.; Qi, W.; Feng, L.; Su, R.; He, Z. Self-assembling peptide-polysaccharide hybrid hydrogel as a potential carrier for drug delivery. Soft Matter 2011, 7, 6222–6230. [Google Scholar] [CrossRef]
- Khoushab, F.; Jaruseranee, N.; Tanthanuch, W.; Yamabhai, M. Formation of chitin-based nanomaterials using a chitin-binding peptide selected by phage-display. Int. J. Biol. Macromol. 2012, 50, 1267–1274. [Google Scholar] [CrossRef]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar] [CrossRef]
- Lequeux, I.; Ducasse, E.; Jouenne, T.; Thebault, P. Addition of antimicrobial properties to hyaluronic acid by grafting of antimicrobial peptide. Eur. Polym. J. 2014, 51, 182–190. [Google Scholar] [CrossRef]
- Chu, T.W.; Feng, J.; Yang, J.; Kopeček, J. Hybrid polymeric hydrogels via peptide nucleic acid (PNA)/DNA complexation. J. Control. Release 2015, 220, 608–616. [Google Scholar] [CrossRef]
- Lee, E.Y.; Takahashi, T.; Curk, T.; Dobnikar, J.; Gallo, R.L.; Wong, G.C.L. Crystallinity of Double-Stranded RNA-Antimicrobial Peptide Complexes Modulates Toll-Like Receptor 3-Mediated Inflammation. ACS Nano 2017, 11, 12145–12155. [Google Scholar] [CrossRef] [PubMed]
- Macleod, T.; Ward, J.; Alase, A.A.; Bridgewood, C.; Wittmann, M.; Stonehouse, N.J. Antimicrobial Peptide LL-37 Facilitates Intracellular Uptake of RNA Aptamer Apt 21-2 Without Inducing an Inflammatory or Interferon Response. Front. Immunol. 2019, 10, 857. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Adler-Abramovich, L.; Lampel, A.; Bram, Y.; Lipstman, S.; Gazit, E. Formation of functional super-helical assemblies by constrained single heptad repeat. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C.; Sleiman, H.F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 1–23. [Google Scholar]
- Li, Z.; Wang, J.; Li, Y.; Liu, X.; Yuan, Q. Self-assembled DNA nanomaterials with highly programmed structures and functions. Mater. Chem. Front. 2018, 2, 423–436. [Google Scholar] [CrossRef]
- Senyürek, I.; Klein, G.; Kalbacher, H.; Deeg, M.; Schittek, B. Peptides derived from the human laminin α4 and α5 chains exhibit antimicrobial activity. Peptides 2010, 31, 1468–1472. [Google Scholar] [CrossRef]
- Obuobi, S.; Tay, H.K.L.; Tram, N.D.T.; Selvarajan, V.; Khara, J.S.; Wang, Y.; Ee, P.L.R. Facile and efficient encapsulation of antimicrobial peptides via crosslinked DNA nanostructures and their application in wound therapy. J. Control. Release 2019, 313, 120–130. [Google Scholar] [CrossRef]
- Müller, R.H.; Runge, S.; Ravelli, V.; Mehnert, W.; Thünemann, A.F.; Souto, E.B. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89. [Google Scholar] [CrossRef]
- Avrahami, D.; Shai, Y. Bestowing Antifungal and Antibacterial Activities by Lipophilic Acid Conjugation to D, L-Amino Acid-Containing Antimicrobial Peptides: A Plausible Mode of Action. Biochemistry 2003, 42, 14946–14956. [Google Scholar] [CrossRef]
- Dong, W.; Liu, Z.; Sun, L.; Wang, C.; Guan, Y.; Mao, X.; Shang, D. Antimicrobial activity and self-assembly behavior of antimicrobial peptide chensinin-1b with lipophilic alkyl tails. Eur. J. Med. Chem. 2018, 150, 546–558. [Google Scholar] [CrossRef]
- Cummings, J.E.; Vanderlick, T.K. Aggregation and hemi-fusion of anionic vesicles induced by the antimicrobial peptide cryptdin-4. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.M.; Alpar, H.O.; Brown, M.R.W. Antimicrobial properties of liposomal polymyxin B. J. Antimicrob. Chemother. 1999, 43, 203–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
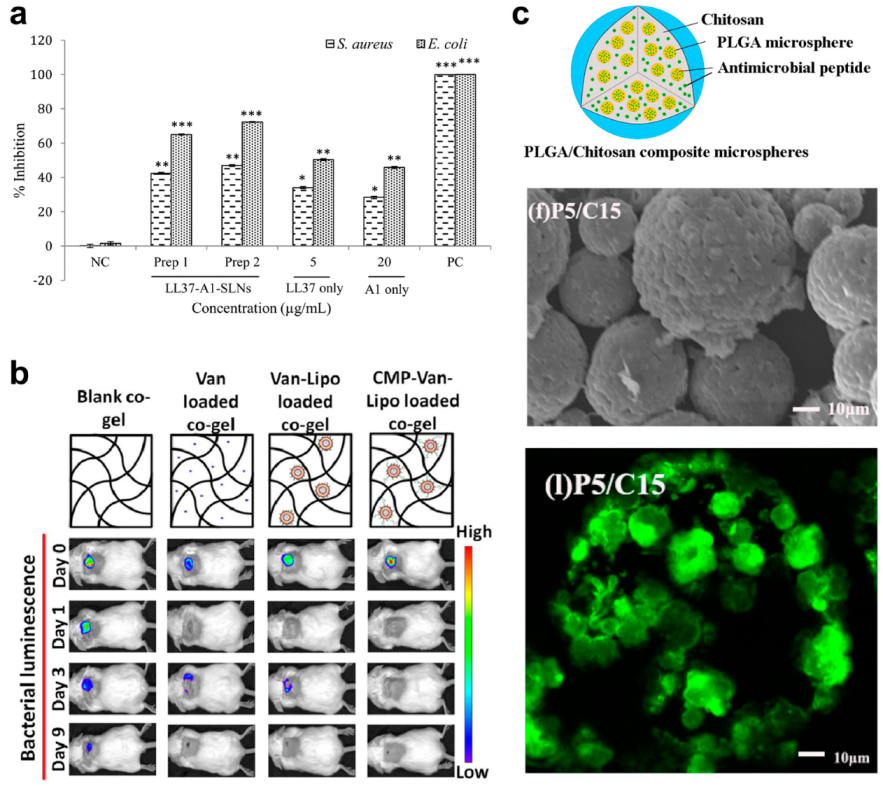
- Fumakia, M.; Ho, E.A. Nanoparticles encapsulated with LL37 and serpin A1 promotes wound healing and synergistically enhances antibacterial activity. Mol. Pharm. 2016, 13, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Kiick, K.L.; Sullivan, M.O. Encapsulation of collagen mimetic peptide-tethered vancomycin liposomes in collagen-based scaffolds for infection control in wounds. Acta Biomater. 2020, 103, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, J.; Ran, R.; Liu, Y.; Zhang, Z.; Gao, H.; He, Q. Development of an anti-microbial peptide-mediated liposomal delivery system: A novel approach towards pH-responsive anti-microbial peptides. Drug Deliv. 2016, 23, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Velasquez Guzman, J.C.; Basu, S.; Rabara, R.; Huynh, L.K.; Basu, G.C.; Nguyen, H.B.; Gupta, G. Liposome Delivery System of Antimicrobial Peptides against Huanglongbing (HLB) Citrus Disease. Biophys. J. 2018, 114, 266a. [Google Scholar] [CrossRef]
- Mowery, B.P.; Lee, S.E.; Kissounko, D.A.; Epand, R.F.; Epand, R.M.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Mimicry of Antimicrobial Host-Defense Peptides by Random Copolymers The eukaryotic innate immune response to bacterial infection includes the production of peptides that kill prokaryotic invaders. J. Am. Chem. Soc. 2007, 129, 15474–15476. [Google Scholar] [CrossRef]
- Li, Y.; Na, R.; Wang, X.; Liu, H.; Zhao, L.; Sun, X.; Ma, G.; Cui, F. Fabrication of Antimicrobial Peptide-Loaded PLGA/Chitosan Composite Microspheres for Long-Acting Bacterial Resistance. Molecules 2017, 22, 1637. [Google Scholar] [CrossRef]
- Cleophas, R.T.C.; Sjollema, J.; Busscher, H.J.; Kruijtzer, J.A.W.; Liskamp, R.M.J. Characterization and activity of an immobilized antimicrobial peptide containing bactericidal PEG-hydrogel. Biomacromolecules 2014, 15, 3390–3395. [Google Scholar] [CrossRef]
- Gabriel, M.; Nazmi, K.; Veerman, E.C.; Amerongen, A.V.N.; Zentner, A. Preparation of LL-37-grafted titanium surfaces with bactericidal activity. Bioconjug. Chem. 2006, 17, 548–550. [Google Scholar] [CrossRef]
- Lu, Y.; Aimetti, A.A.; Langer, R.; Gu, Z. Bioresponsive materials. Nat. Rev. Mater. 2016, 2, 1–17. [Google Scholar] [CrossRef]
- Zhao, F.; Ma, M.L.; Xu, B. Molecular hydrogels of therapeutic agents. Chem. Soc. Rev. 2009, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Mart, R.J.; Osborne, R.D.; Stevens, M.M.; Ulijn, R.V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. [Google Scholar] [CrossRef]
- Webber, M.J.; Matson, J.B.; Tamboli, V.K.; Stupp, S.I. Controlled release of dexamethasone from peptide nanofiber gels to modulate inflammatory response. Biomaterials 2012, 33, 6823–6832. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Guo, X.; Zou, M.; Zheng, Z.; Li, Y.; Li, X.; Li, L.; Wang, H. Bacteria-Instructed In Situ Aggregation of AuNPs with Enhanced Photoacoustic Signal for Bacterial Infection Bioimaging. Adv. Healthc. Mater. 2020, 9, 1901229. [Google Scholar] [CrossRef] [PubMed]
- Haines-Butterick, L.; Rajagopal, K.; Branco, M.; Salick, D.; Rughani, R.; Pilarz, M.; Lamm, M.S.; Pochan, D.J.; Schneider, J.P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. USA 2007, 104, 7791–7796. [Google Scholar] [CrossRef]
- Shlar, I.; Droby, S.; Choudhary, R.; Rodov, V. The mode of antimicrobial action of curcumin depends on the delivery system: Monolithic nanoparticles vs. supramolecular inclusion complex. RSC Adv. 2017, 7, 42559–42569. [Google Scholar] [CrossRef]
- Li, Z.; He, C.; Yuan, B.; Dong, X.; Chen, X. Injectable Polysaccharide Hydrogels as Biocompatible Platforms for Localized and Sustained Delivery of Antibiotics for Preventing Local Infections. Macromol. Biosci. 2017, 17, 1600347. [Google Scholar] [CrossRef]
- Pranantyo, D.; Liu, P.; Zhong, W.; Kang, E.T.; Chan-Park, M.B. Antimicrobial Peptide-Reduced Gold Nanoclusters with Charge-Reversal Moieties for Bacterial Targeting and Imaging. Biomacromolecules 2019, 20, 2922–2933. [Google Scholar] [CrossRef]
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; De Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the Potency of Antimicrobial Peptides through Molecular Engineering and Self-Assembly. Biomacromolecules 2019, 20, 1362–1374. [Google Scholar] [CrossRef]
- Wen, Y.; Collier, J.H. Supramolecular peptide vaccines: Tuning adaptive immunity. Curr. Opin. Immunol. 2015, 35, 73–79. [Google Scholar] [CrossRef]
- Lee, E.Y.; Zhang, C.; Di Domizio, J.; Jin, F.; Connell, W.; Hung, M.; Malkoff, N.; Veksler, V.; Gilliet, M.; Ren, P.; et al. Helical antimicrobial peptides assemble into protofibril scaffolds that present ordered dsDNA to TLR9. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, J.; Lu, S.; Igweze, J.; Xu, W.; Kuang, D.; Zealey, C.; Liu, D.; Gregor, A.; Bozorgzad, A.; et al. Self-assembling peptide for co-delivery of HIV-1 CD8+ T cells epitope and Toll-like receptor 7/8 agonists R848 to induce maturation of monocyte derived dendritic cell and augment polyfunctional cytotoxic T lymphocyte (CTL) response. J. Control. Release 2016, 236, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.H.; Messersmith, P.B. Enzymatic modification of self-assembled peptide structures with tissue transglutaminase. Bioconjug. Chem. 2003, 14, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Hudalla, G.A.; Modica, J.A.; Tian, Y.F.; Rudra, J.S.; Chong, A.S.; Sun, T.; Mrksich, M.; Collier, J.H. A Self-Adjuvanting Supramolecular Vaccine Carrying a Folded Protein Antigen. Adv. Healthc. Mater. 2013, 2, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Wen, Y.; Chen, J.; Pompano, R.R.; Han, H.; Collier, J.H.; Chong, A.S. MyD88 in antigen-presenting cells is not required for CD4+ T-cell responses during peptide nanofiber vaccination. Medchemcomm 2018, 9, 138–148. [Google Scholar] [CrossRef]
- Si, Y.; Wen, Y.; Kelly, S.H.; Chong, A.S.; Collier, J.H. Intranasal delivery of adjuvant-free peptide nanofibers elicits resident CD8+ T cell responses. J. Control. Release 2018, 282, 120–130. [Google Scholar] [CrossRef]
- Rudra, J.S.; Tian, Y.F.; Jung, J.P.; Collier, J.H. A self-assembling peptide acting as an immune adjuvant. Proc. Natl. Acad. Sci. USA 2010, 107, 622–627. [Google Scholar] [CrossRef]
- Kelly, S.H.; Wu, Y.; Varadhan, A.K.; Curvino, E.J.; Chong, A.S.; Collier, J.H. Enabling sublingual peptide immunization with molecular self-assemblies. Biomaterials 2020, 241, 119903. [Google Scholar] [CrossRef]
- Sreejit, G.; Ahmed, A.; Parveen, N.; Jha, V.; Valluri, V.L.; Ghosh, S.; Mukhopadhyay, S. The ESAT-6 Protein of Mycobacterium tuberculosis Interacts with Beta-2-Microglobulin (β2M) Affecting Antigen Presentation Function of Macrophage. PLoS Pathog. 2014, 10, e1004446. [Google Scholar] [CrossRef]
- Gruber, C.W.; Muttenthaler, M.; Freissmuth, M. Ligand-Based Peptide Design and Combinatorial Peptide Libraries to Target G Protein-Coupled Receptors. Curr. Pharm. Des. 2010, 16, 3071. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.H.; Michie, M.S.; Miller, S.E.; Schnermann, M.J.; Schneider, J.P. Fluorous Phase-Directed Peptide Assembly Affords Nano-Peptisomes Capable of Ultrasound-Triggered Cellular Delivery. Angew. Chemie Int. Ed. 2017, 56, 11404–11408. [Google Scholar] [CrossRef] [PubMed]
- Sloand, J.N.; Nguyen, T.T.; Zinck, S.A.; Cook, E.C.; Zimudzi, T.J.; Showalter, S.A.; Glick, A.B.; Simon, J.C.; Medina, S.H. Ultrasound-Guided Cytosolic Protein Delivery via Transient Fluorous Masks. ACS Nano 2020, 14, 4061–4073. [Google Scholar] [CrossRef] [PubMed]
- Asati, S.; Pandey, V.; Soni, V. RGD Peptide as a Targeting Moiety for Theranostic Purpose: An Update Study. Int. J. Pept. Res. Ther. 2019, 25, 49–65. [Google Scholar] [CrossRef]
- Di Pietro, P.; Zaccaro, L.; Comegna, D.; Del Gatto, A.; Saviano, M.; Snyders, R.; Cossement, D.; Satriano, C.; Rizzarelli, E. Silver nanoparticles functionalized with a fluorescent cyclic RGD peptide: A versatile integrin targeting platform for cells and bacteria. RSC Adv. 2016, 6, 112381–112392. [Google Scholar] [CrossRef]
- Pushpanathan, M.; Rajendhran, J.; Jayashree, S.; Sundarakrishnan, B.; Jayachandran, S.; Gunasekaran, P. Identification of a Novel Antifungal Peptide with Chitin-Binding Property from Marine Metagenome. Protein Pept. Lett. 2012, 19, 1289–1296. [Google Scholar] [CrossRef]
- Braun, K.; Pochert, A.; Lindén, M.; Davoudi, M.; Schmidtchen, A.; Nordström, R.; Malmsten, M. Membrane interactions of mesoporous silica nanoparticles as carriers of antimicrobial peptides. J. Colloid Interface Sci. 2016, 475, 161–170. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (Sequence) | Morphology | Mechanism | Bioactive Conc. | Ref. |
|---|---|---|---|---|
| FF8 (KRRFFRRK) | β-Amyloid fibril | Bacteriolysis | 25.6 μM | [73] |
| MAD1 (KRWHWWRRHWVVW-NH2) | β-Amyloid fibril | Bacteriolysis | 2.5 μM | [74] |
| zp3 (GIIAGIIIKIKK-NH2) | Non β-Amyloid fibril | Bacteriolysis | 8 μM | [94] |
| Cg-BigDef1 (GenBank: AEE92768.1) | Nanofibrillar net | Entrapment | 0.3 μM | [109] |
| GL13K (GKIIKLKASLKLL-NH2) | Nanoribbon | Non-lytic/unknown | 5 μg/mL | [115] |
| Diphenylalanine (FF) | Nanotube | Permeation/ depolarization | 125 μg/mL | [71] |
| MARG1 (VKVKVRVKVDPPTKVKVRVKV-NH2) | Macroscopic gel | Electrostatic disruption | 2 wt% | [128] |
| Peptide | Co-Assembled Agent | Mechanism | Bioactive Response | Ref. |
| DLeu–Phe–Phe | Ciprofloxacin | Combinatorial synergy | 500–1000 fold enhanced toxicity | [148] |
| Poly(arginine-r-valine)-mannose | Zn2+ | Combinatorial synergy | Conserved bacterial toxicity. Reduced toxicity | [165] |
| LL-37 | Alginate | Enhanced delivery | Enhanced selectivity index. Prolonged release kinetics | [169] |
| L12 | DNA scaffold and linker | Enhanced delivery | Induced selectivity | [188] |
| Collagen mimetic | Loaded liposome Collagen/fibrin | Enhanced delivery and retention | Improved in vivo efficacy | [195] |
| CysHHC10 | Au nanoclusters | Autocleavable motif | pH responsive bioactivity. Minimized off target effects | [210] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simonson, A.W.; Aronson, M.R.; Medina, S.H. Supramolecular Peptide Assemblies as Antimicrobial Scaffolds. Molecules 2020, 25, 2751. https://doi.org/10.3390/molecules25122751
Simonson AW, Aronson MR, Medina SH. Supramolecular Peptide Assemblies as Antimicrobial Scaffolds. Molecules. 2020; 25(12):2751. https://doi.org/10.3390/molecules25122751
Chicago/Turabian StyleSimonson, Andrew W., Matthew R. Aronson, and Scott H. Medina. 2020. "Supramolecular Peptide Assemblies as Antimicrobial Scaffolds" Molecules 25, no. 12: 2751. https://doi.org/10.3390/molecules25122751
APA StyleSimonson, A. W., Aronson, M. R., & Medina, S. H. (2020). Supramolecular Peptide Assemblies as Antimicrobial Scaffolds. Molecules, 25(12), 2751. https://doi.org/10.3390/molecules25122751