
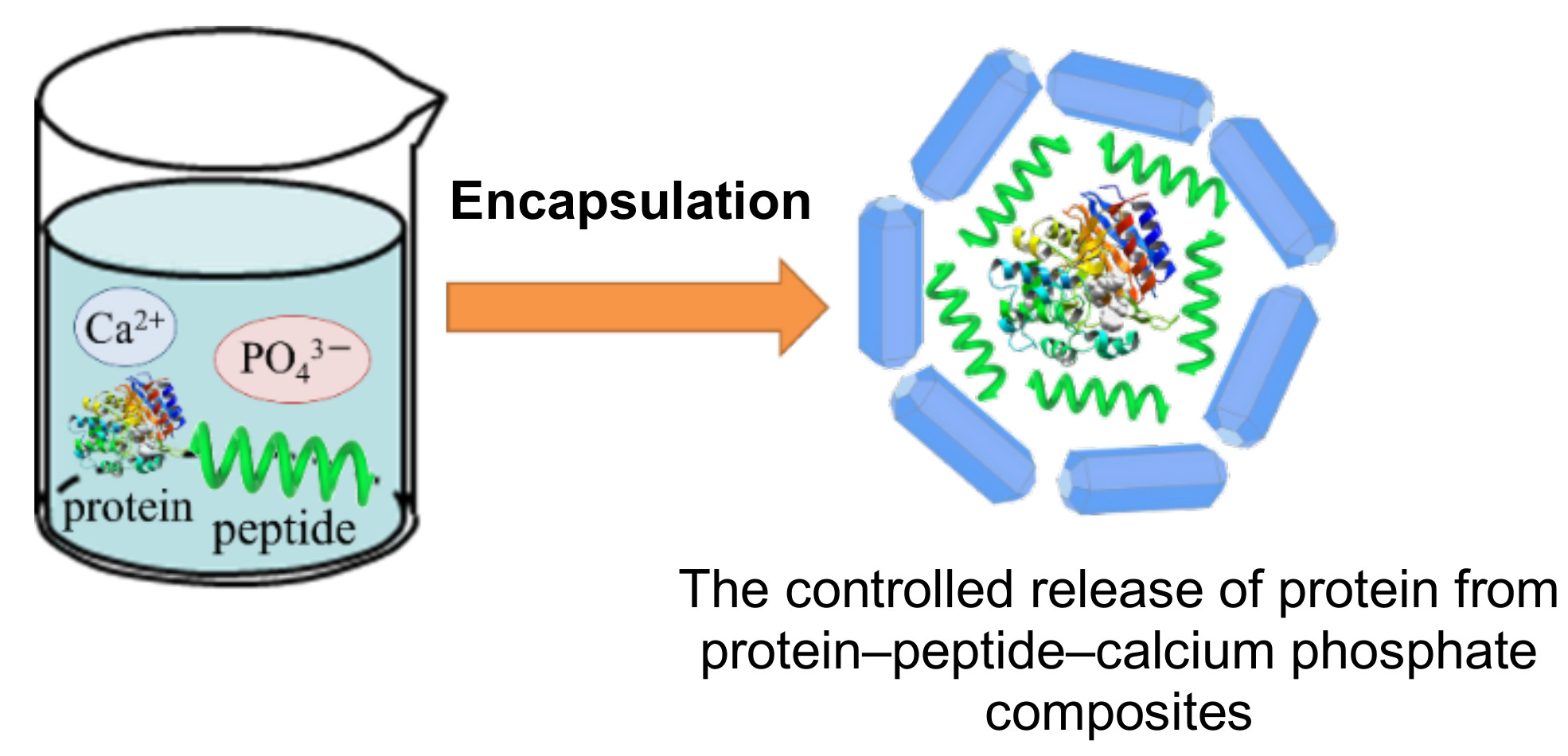
Preparation of Protein–Peptide–Calcium Phosphate Composites for Controlled Protein Release


Abstract

1. Introduction
2. Results and Discussion
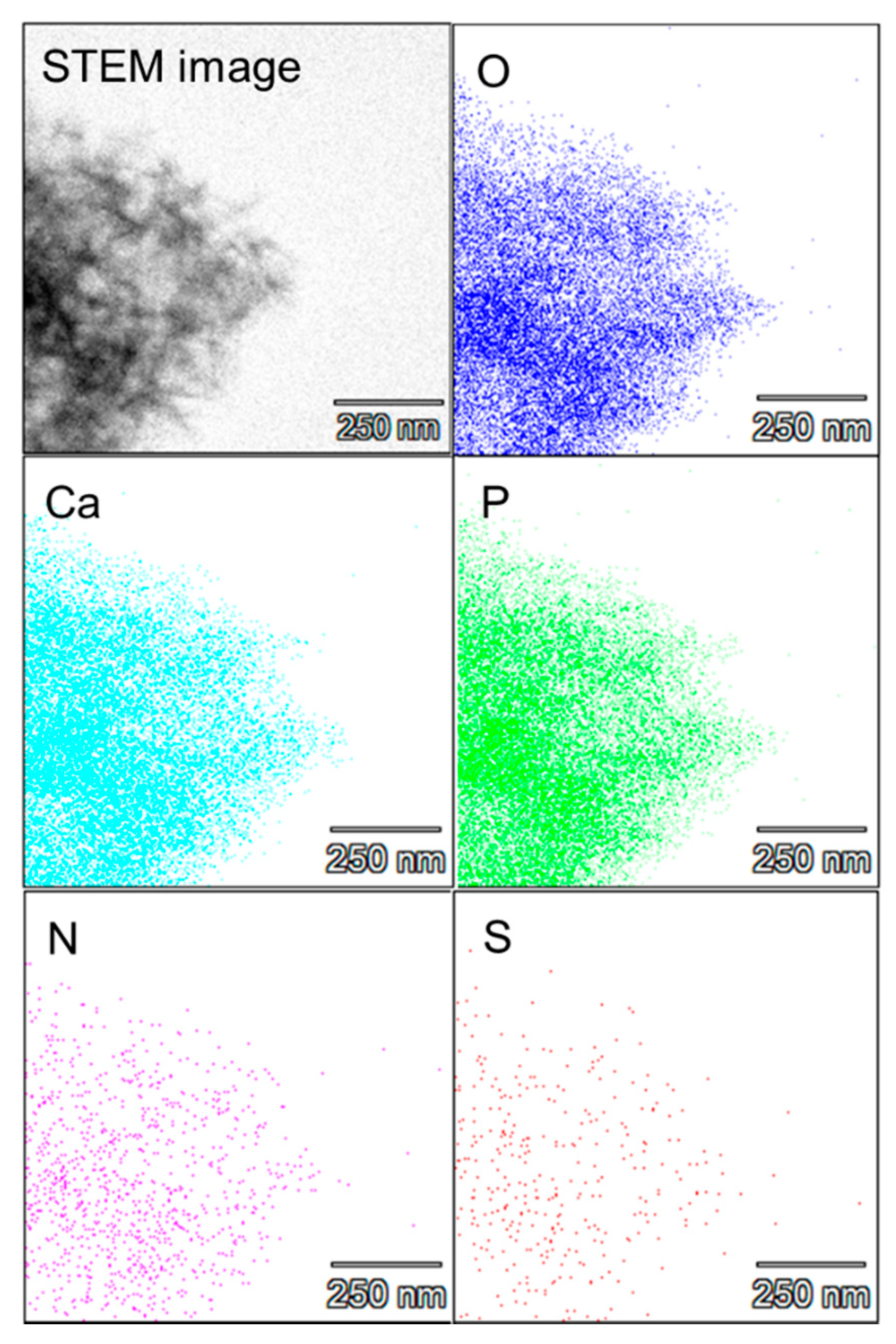
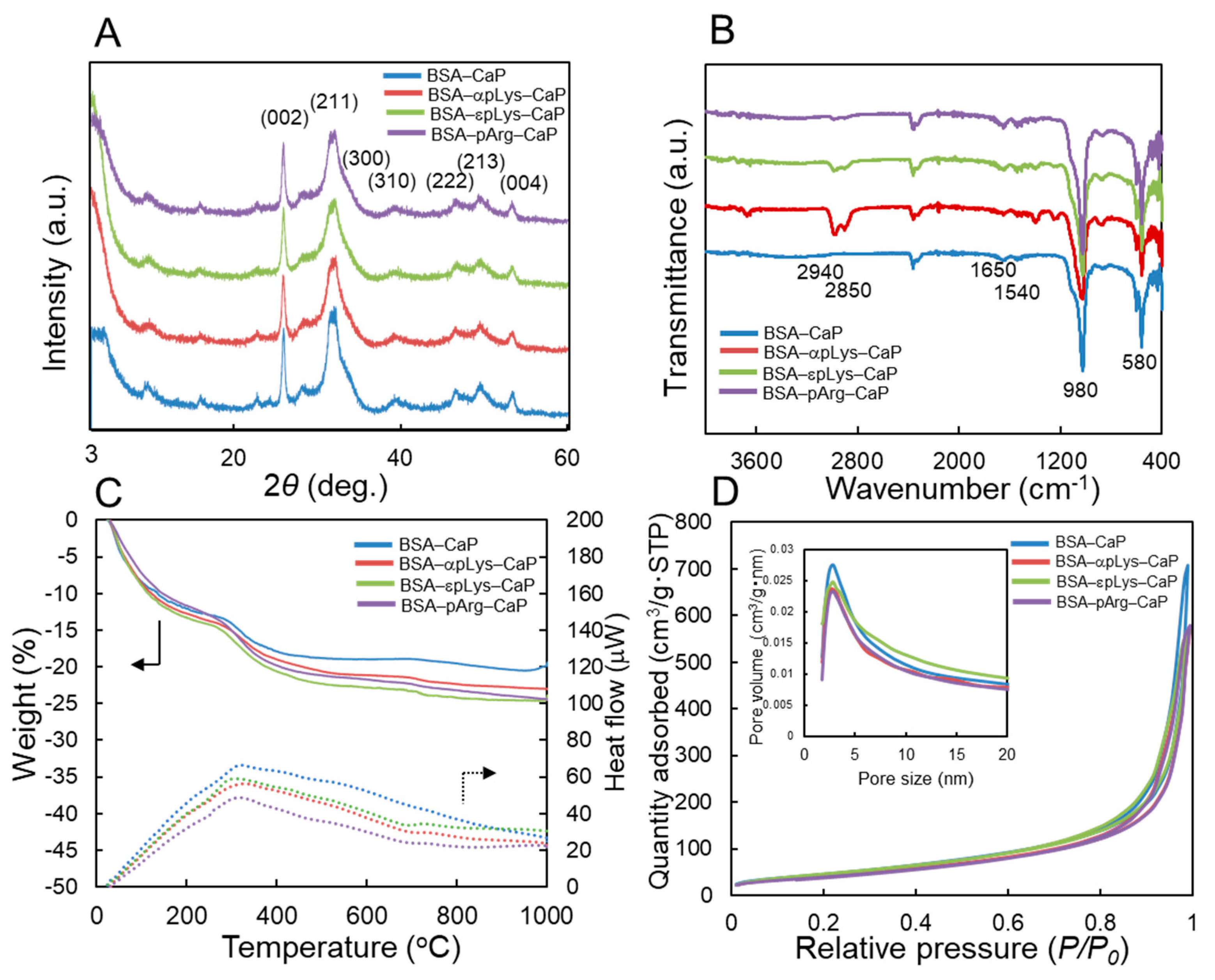
2.1. Preparation of the Protein–Peptide–CaP Composites and Their Characterization
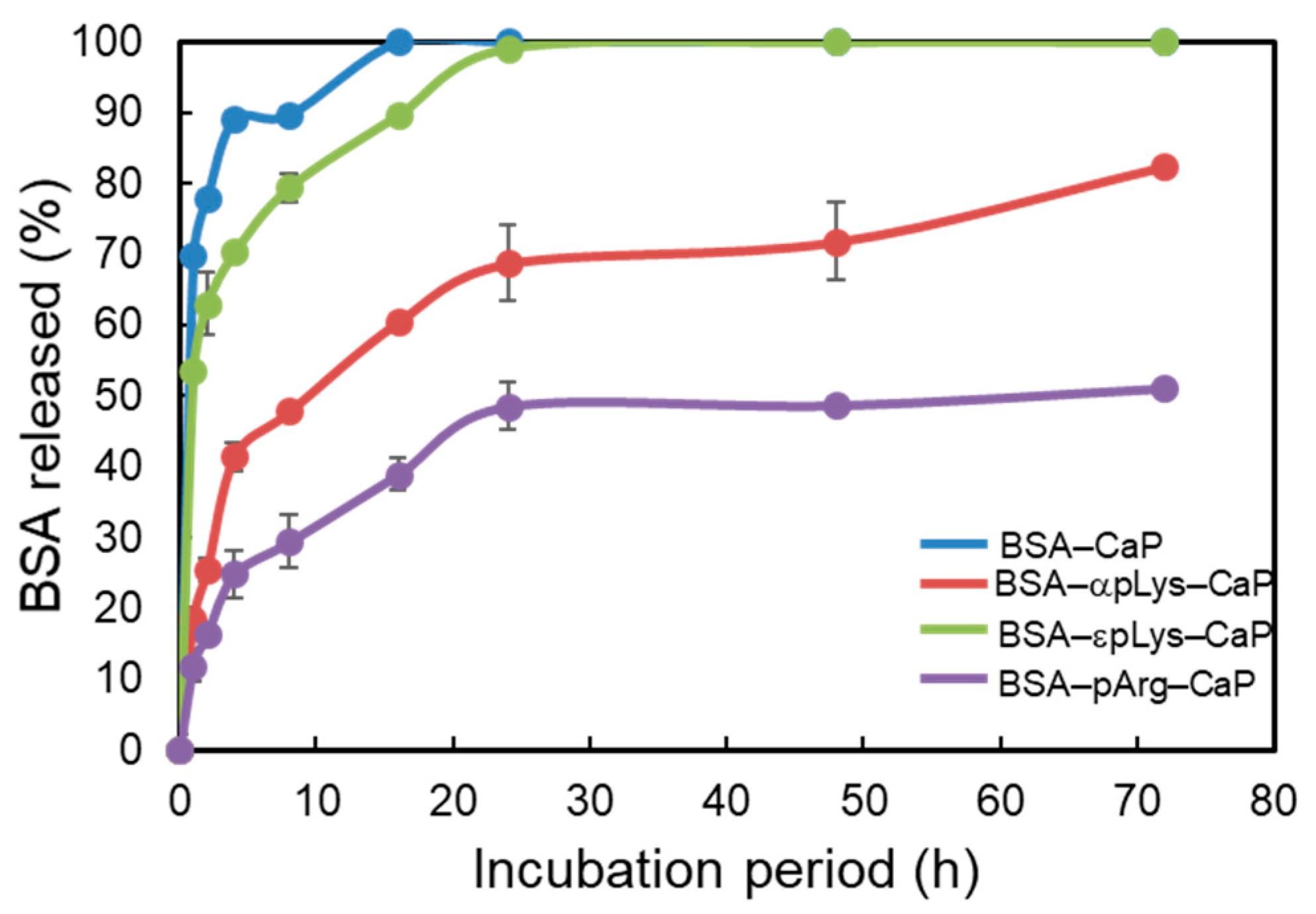
2.2. Controlled Release of Protein from the Protein–Peptide–CaP Composites
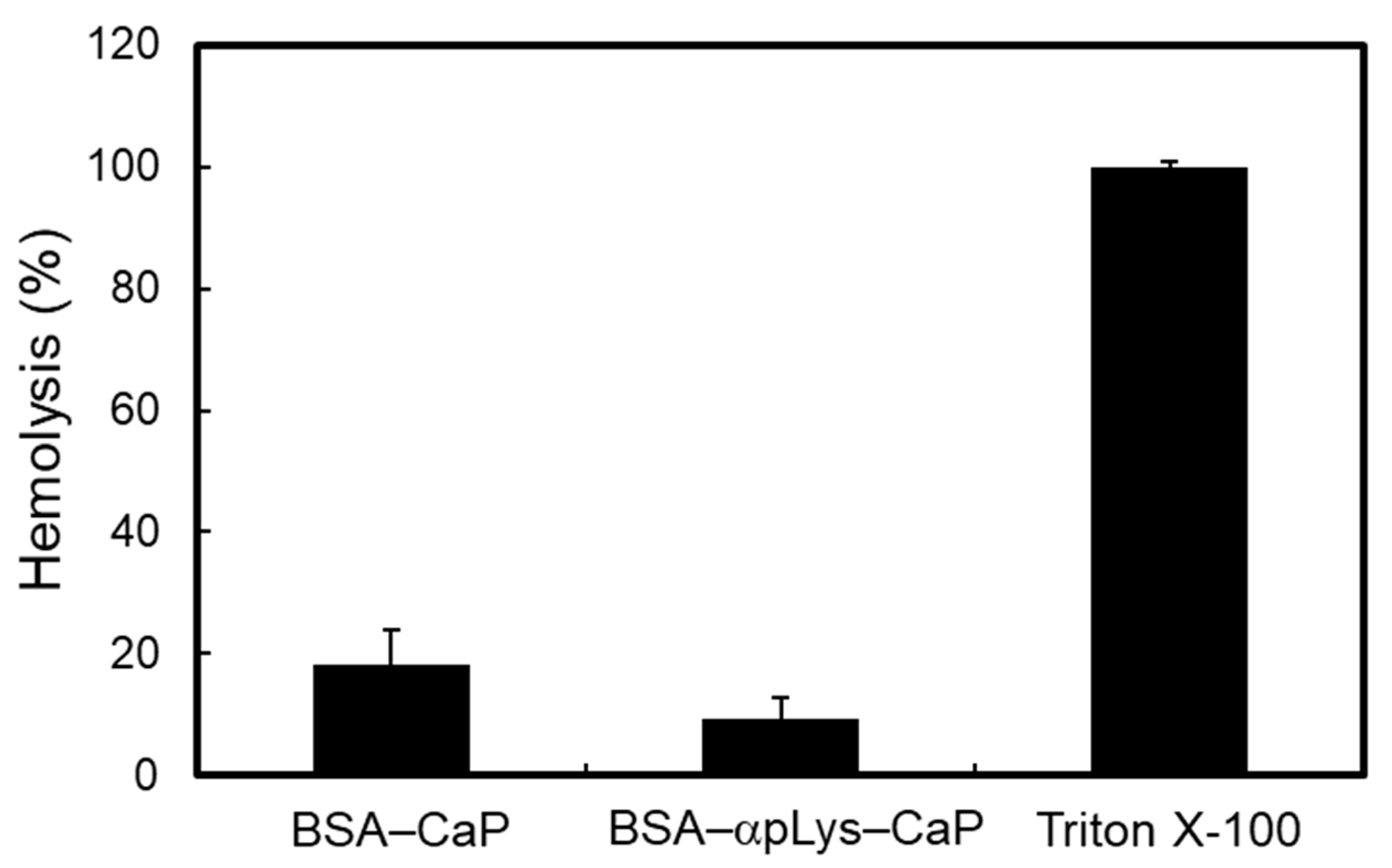
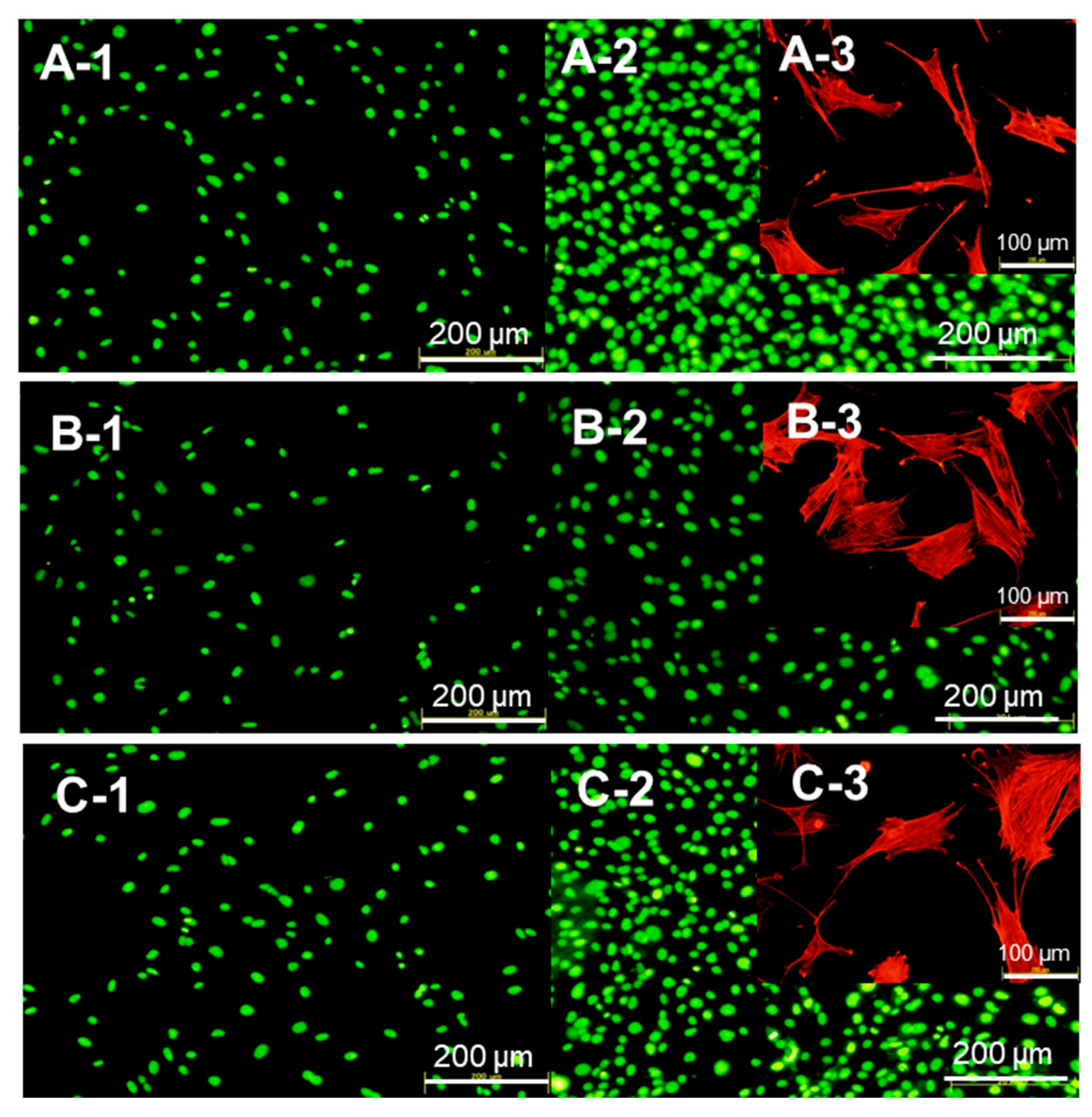
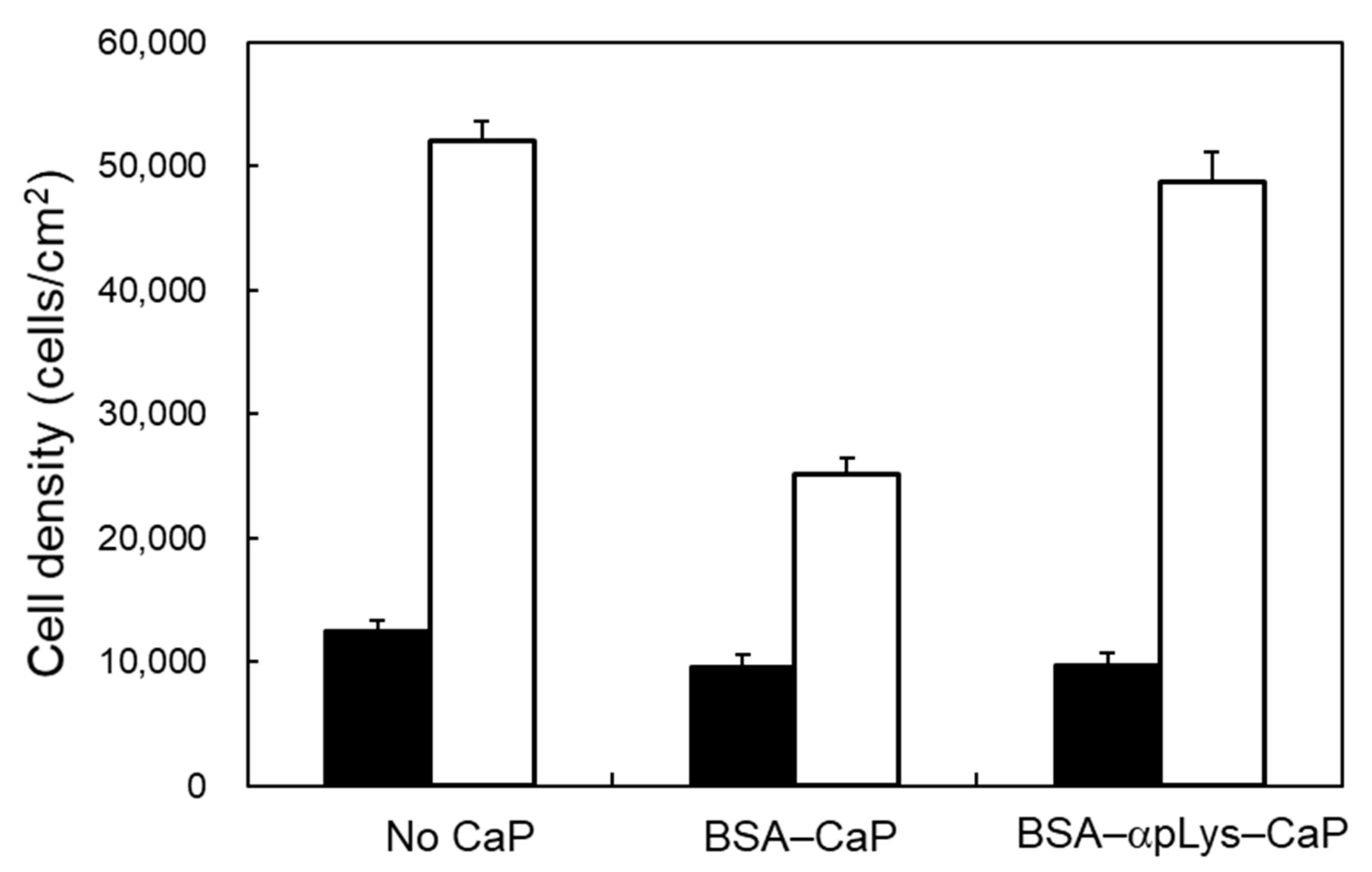
2.3. Biocompatibility of the Protein–Peptide–CaP Composites
3. Materials and Methods
3.1. Materials
3.2. Preparation of the Protein–Peptide–Calcium Phosphate (CaP) Composites
3.3. Characterization of the Protein–Peptide–Calcium Phosphate (CaP) Composites
3.4. Protein Release from the Protein–Peptide–CaP Composites
3.5. Hemocompatibility Assay
3.6. Cell Behavior After Being Cultured with the Protein–Peptide–CaP Composites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. 2012, 2, 2–11. [Google Scholar]
- Ratnaparkhi, M.P.; Gupta, J.P. Sustained release oral drug delivery system-An overview. Int. J. Pharm. Res. Rev. 2013, 2, 11–21. [Google Scholar]
- Zhang, L.; Li, Y.; Yu, J.C. Chemical modification of inorganic nanostructures for targeted and controlled drug delivery in cancer treatment. J. Mater. Chem. B 2014, 2, 452–470. [Google Scholar] [CrossRef]
- Wang, Z.; Denga, X.; Ding, J.; Zhou, W.; Zheng, X.; Tang, G. Mechanisms of drug release in pH-sensitive micelles for tumour targeted drug delivery system: A review. Int. J. Pharm. 2018, 535, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Asadi, N.; Davaran, S.; Panahi, Y.; Hasanzadeh, A.; Malakootikhah, J.; Moafi, H.F.; Akbarzadeh, A. Application of nanostructured drug delivery systems in immunotherapy of cancer: A review. Artif. Cells Nanomed. Biotechnol. 2017, 45, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gultepe, E.; Nagesha, D.; Sridhar, S.; Amiji, M. Nanoporous inorganic membranes or coatings for sustained drug delivery in implantable devices. Adv. Drug Deliv. Rev. 2010, 62, 305–315. [Google Scholar] [CrossRef]
- Lassalle, V.; Ferreira, M. PLGA based drug delivery systems (DDS) for the sustained release of insulin: Insight into the protein/polyester interactions and the insulin release behavior. J. Chem. Technol. Biotechnol. 2010, 85, 1588–1596. [Google Scholar] [CrossRef]
- Murai, K.; Higuchi, M.; Kinoshita, T.; Nagata, K.; Kato, K. Design of a nanocarrier with regulated drug release ability utilizing a reversible conformational transition of a peptide, responsive to slight changes in pH. Phys. Chem. Chem. Phys. 2013, 15, 11454–11460. [Google Scholar] [CrossRef]
- Lopes, J.R.; Santos, G.; Barata, P.; Oliveira, R.; Lopes, C.M. Physical and chemical stimuli-responsive drug delivery systems: Targeted delivery and main routes of administration. Curr. Pharm. Des. 2013, 19, 7169–7184. [Google Scholar] [CrossRef]
- Song, Y.; Li, Y.; Xu, Q.; Liu, Z. Mesoporous silica nanoparticles for stimuli-responsive controlled drug delivery: Advances, challenges, and outlook. Int. J. Nanomed. 2017, 12, 87–110. [Google Scholar] [CrossRef]
- Honda, M.; Asai, T.; Oku, N.; Arak, Y.; Tanaka, M.; Ebihara, N. Liposomes and nanotechnology in drug development: Focus on ocular targets. Int. J. Nanomed. 2013, 8, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Kalita, S.J.; Bhardwaj, A.; Bhatt, H.A. Nanocrystalline calcium phosphate ceramics in biomedical engineering. Mater. Sci. Eng. C 2007, 27, 441–449. [Google Scholar] [CrossRef]
- Samavedi, S.; Whittington, A.R.; Goldstein, A.S. Calcium phosphate ceramics in bone tissue engineering: A review of properties and their influence on cell behavior. Acta Biomater. 2013, 9, 8037–8045. [Google Scholar] [CrossRef]
- Ginebra, M.P.; Traykova, T.; Planell, J.A. Calcium phosphate cements as bone drug delivery systems: A review. J. Controlled Release 2006, 113, 102–110. [Google Scholar] [CrossRef]
- Ginebra, M.P.; Canal, C.; Espanol, M.; Pastorino, D.; Montufar, E.B. Calcium phosphate cements as drug delivery materials. Adv. Drug Deliv. Rev. 2012, 64, 1090–1110. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.P.; Liu, F.; Wilson, R.E.; Housley, P.R. Optimization of calcium phosphate transfection for bovine chromaffin cells: Relationship to calcium phosphate precipitate formation. Anal. Biochem. 1995, 226, 212–220. [Google Scholar] [CrossRef]
- Bisht, S.; Bhakta, G.r.; Mitra, S.; Maitr, A. pDNA loaded calcium phosphate nanoparticles: Highly efficient non-viral vector for gene delivery. Int. J. Pharm. 2005, 288, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Jana, N.R. Length-controlled synthesis of calcium phosphate nanorod and nanowire and application in intracellular protein delivery. ACS Appl. Mater. Interfaces 2016, 8, 8710–8720. [Google Scholar] [CrossRef]
- Paul, W.; Sharma, C.P. Development of porous spherical hydroxyapatite granules: Application towards protein delivery. J. Mater. Sci. Mater. Med. 1999, 10, 383–388. [Google Scholar] [CrossRef]
- Bose, S.; Tarafder, S.; Edgington, J.; Bandyopadhyay, A. Calcium phosphate ceramics in drug delivery. JOM 2011, 63, 93–98. [Google Scholar] [CrossRef]
- Parent, M.; Baradari, H.; Champion, E.; Damia, C.; Viana-Trecant, M. Design of calcium phosphate ceramics for drug delivery applications in bone diseases: A review of the parameters affecting the loading and release of the therapeutic substance. J. Controlled Release 2017, 252, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schnieders, J.; Gbureck, U.; Thull, R.; Kissel, T. Controlled release of gentamicin from calcium phosphate—poly(lactic acid-co-glycolic acid) composite bone cement. Biomaterials 2006, 27, 4239–4249. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.C.; Barriasa, C.C.; Barbosaa, M.A. Calcium phosphate-alginate microspheres as enzyme delivery matrices. Biomaterials 2004, 25, 4363–4373. [Google Scholar] [CrossRef] [PubMed]
- Inzana, J.A.; Olvera, D.; Fuller, S.M.; Kelly, J.P.; Graeve, O.A.; Schwarz, E.M.; Kates, S.L.; Awad, H.A. 3D printing of composite calcium phosphate and collagen scaffolds for bone regeneration. Biomaterials 2014, 35, 4026–4034. [Google Scholar] [CrossRef] [PubMed]
- Hadisi, Z.; Nourmohammadin, J.; Mohammadi, J. Composite of porous starch-silk fibroin nanofiber-calcium phosphate for bone regeneration. Ceram. Inter. 2015, 41, 10745–10754. [Google Scholar] [CrossRef]
- Nonoyama, T.; Ogasawara, H.; Tanaka, M.; Higuchi, M.; Kinoshita, T. Calcium phosphate biomineralization in peptide hydrogels for injectable bone-filling materials. Soft Matter. 2012, 8, 11531–11536. [Google Scholar] [CrossRef]
- Nonoyama, T.; Kinoshita, T.; Higuchi, M.; Nagata, K.; Tanaka, M.; Sato, K.; Kato, K. Multistep growth mechanism of calcium phosphate in the earliest stage of morphology-controlled biomineralization. Langmuir 2011, 27, 7077–7083. [Google Scholar] [CrossRef]
- Kojima, S.; Nakamura, H.; Lee, S.; Nagata, F.; Kato, K. Hydroxyapatite formation on self-assembling peptides with differing secondary structures and their selective adsorption for proteins. Int. J. Mol. Sci. 2019, 20, 4650. [Google Scholar] [CrossRef]
- Kojima, S.; Nagata, F.; Inagaki, M.; Kugimiya, S.; Kato, K. Enzyme immobilisation on poly-L-lysine-containing calcium phosphate particles for highly sensitive glucose detection. RSC Adv. 2019, 9, 10832–10841. [Google Scholar] [CrossRef]
- Kojima, S.; Nagata, F.; Inagaki, M.; Kugimiya, S.; Kato, K. Avidin-adsorbed peptide–calcium phosphate composites exhibiting high biotin-binding activity. New J. Chem. 2019, 43, 427–435. [Google Scholar] [CrossRef]
- Kojima, S.; Nagata, F.; Kugimiya, S.; Kato, K. Synthesis of peptide-containing calcium phosphate nanoparticles exhibiting highly selective adsorption of various proteins. Appl. Surf. Sci. 2018, 458, 438–445. [Google Scholar] [CrossRef]
- Shiraishi, N.; Anada, T.; Honda, Y.; Masuda, T.; Sasaki, K.; Suzuki, O. Preparation and characterization of porous alginate scaffolds containing various amounts of octacalcium phosphate (OCP) crystals. J. Mater. Sci. Mater. Med. 2010, 21, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Moriwaki, Y.; Takagi, T.; Moradian-Oldak, J. Effects of bovine amelogenins on the crystal morphology of octacalcium phosphate in a model system of tooth enamel formation. J. Cryst. Growth 2001, 222, 615–626. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Torchilin, V.P. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Pept. Sci. 2008, 90, 604–610. [Google Scholar] [CrossRef]
- Mayer, A.; Vadon, M.; Rinner, B.; Novak, A.; Wintersteiger, R.; Frohlich, E. The role of nanoparticle size in hemocompatibility. Toxicology 2009, 258, 139–147. [Google Scholar] [CrossRef]
- Orita, T.; Tomita, M.; Kato, K. Regulation of cellular responses to macroporous inorganic films prepared by the inverse-opal method. Colloids Surf. B. Biointerfaces 2011, 84, 187–197. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BSA–Peptide–CaP Composites | ζ-Potential a (mV) | Ca/P Molar Ratio b | Pore Volume c (cm3/g) | Specific Surface Area c (m2/g) |
|---|---|---|---|---|
| BSA–CaP | −27.5 | 1.37 | 1.10 | 174.9 |
| BSA–αpLys–CaP | −11.6 | 1.34 | 0.88 | 155.3 |
| BSA–εpLys–CaP | −4.5 | 1.33 | 0.88 | 178.3 |
| BSA–pArg–CaP | −11.2 | 1.32 | 0.90 | 147.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, K.; Lee, S.; Nagata, F. Preparation of Protein–Peptide–Calcium Phosphate Composites for Controlled Protein Release. Molecules 2020, 25, 2312. https://doi.org/10.3390/molecules25102312
Kato K, Lee S, Nagata F. Preparation of Protein–Peptide–Calcium Phosphate Composites for Controlled Protein Release. Molecules. 2020; 25(10):2312. https://doi.org/10.3390/molecules25102312
Chicago/Turabian StyleKato, Katsuya, Sungho Lee, and Fukue Nagata. 2020. "Preparation of Protein–Peptide–Calcium Phosphate Composites for Controlled Protein Release" Molecules 25, no. 10: 2312. https://doi.org/10.3390/molecules25102312
APA StyleKato, K., Lee, S., & Nagata, F. (2020). Preparation of Protein–Peptide–Calcium Phosphate Composites for Controlled Protein Release. Molecules, 25(10), 2312. https://doi.org/10.3390/molecules25102312