
Electron Spin Relaxation of Photoexcited Porphyrin in Water—Glycerol Glass

 ,
,  , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
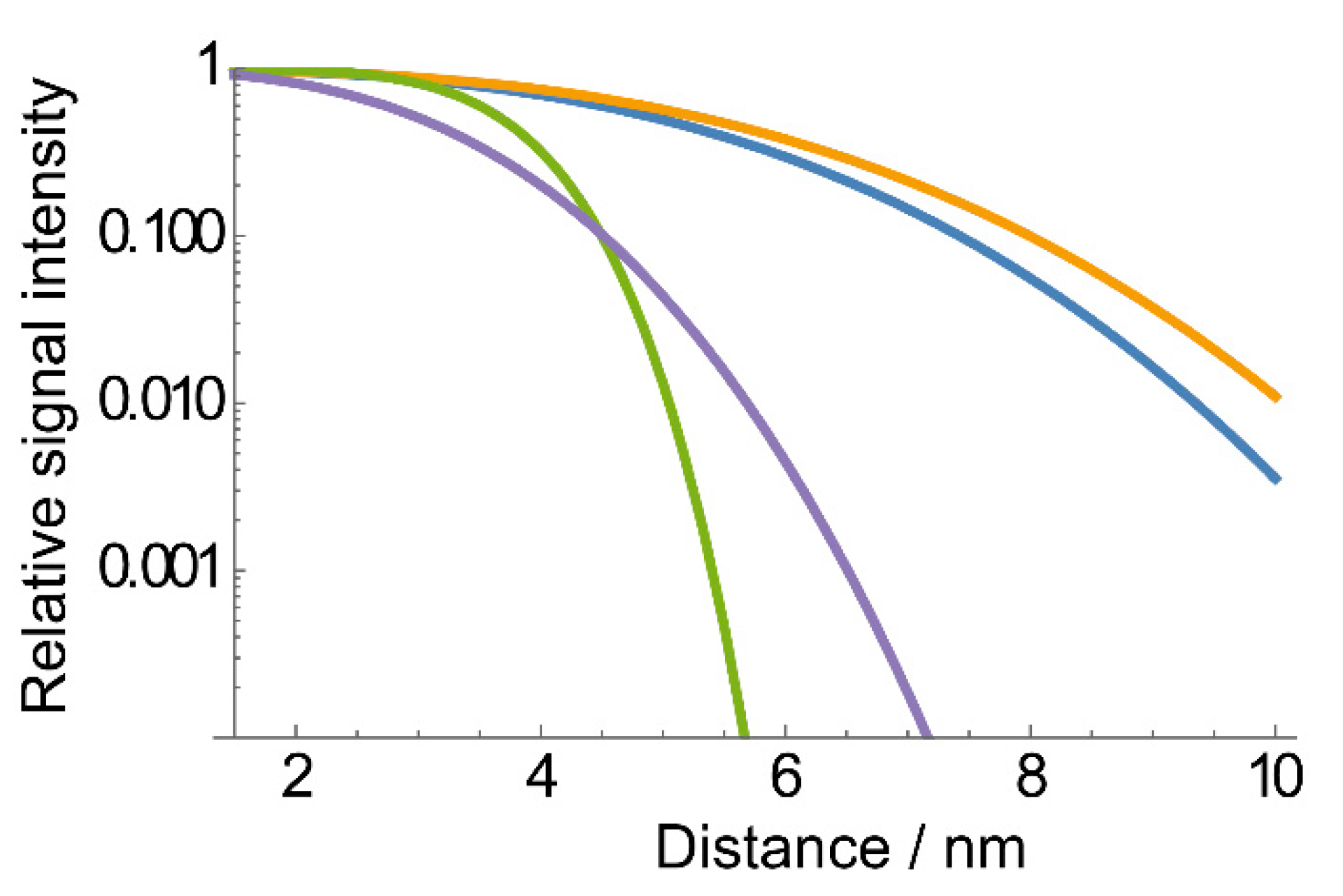{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
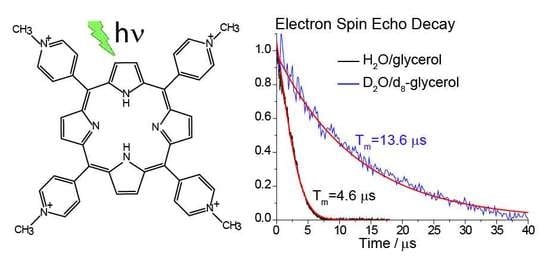
2.1. Tm in Photoexcited TMPyP4
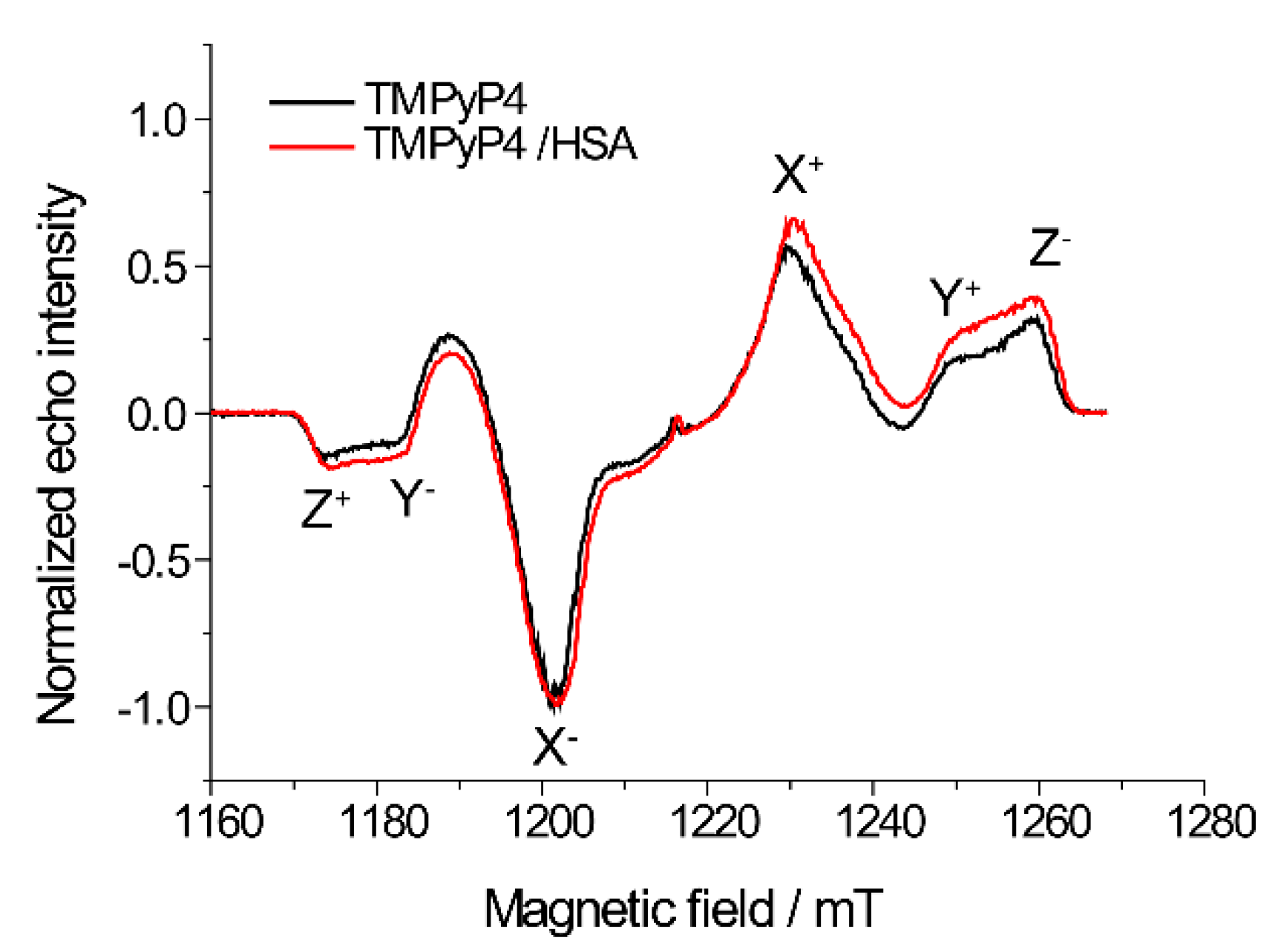
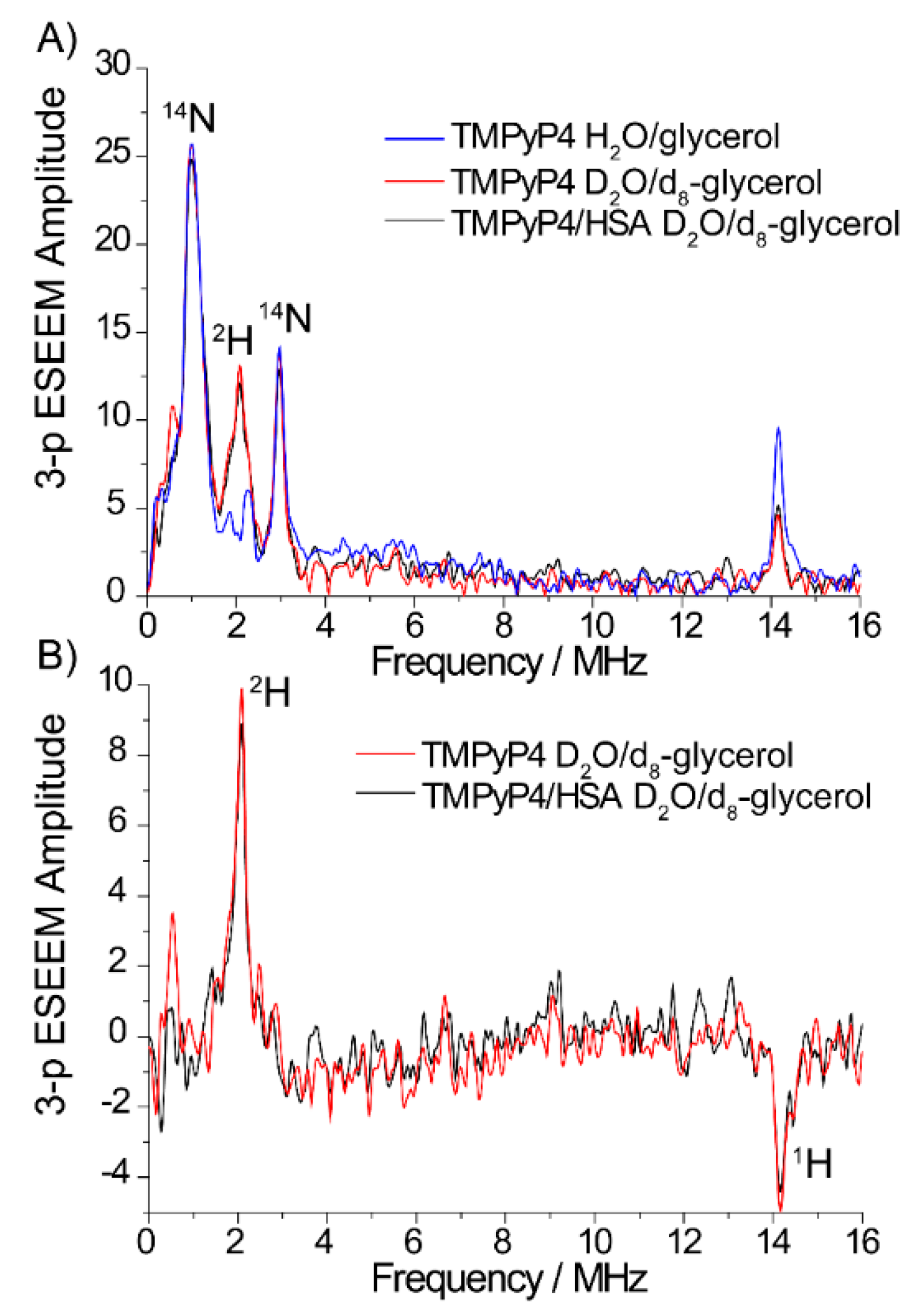
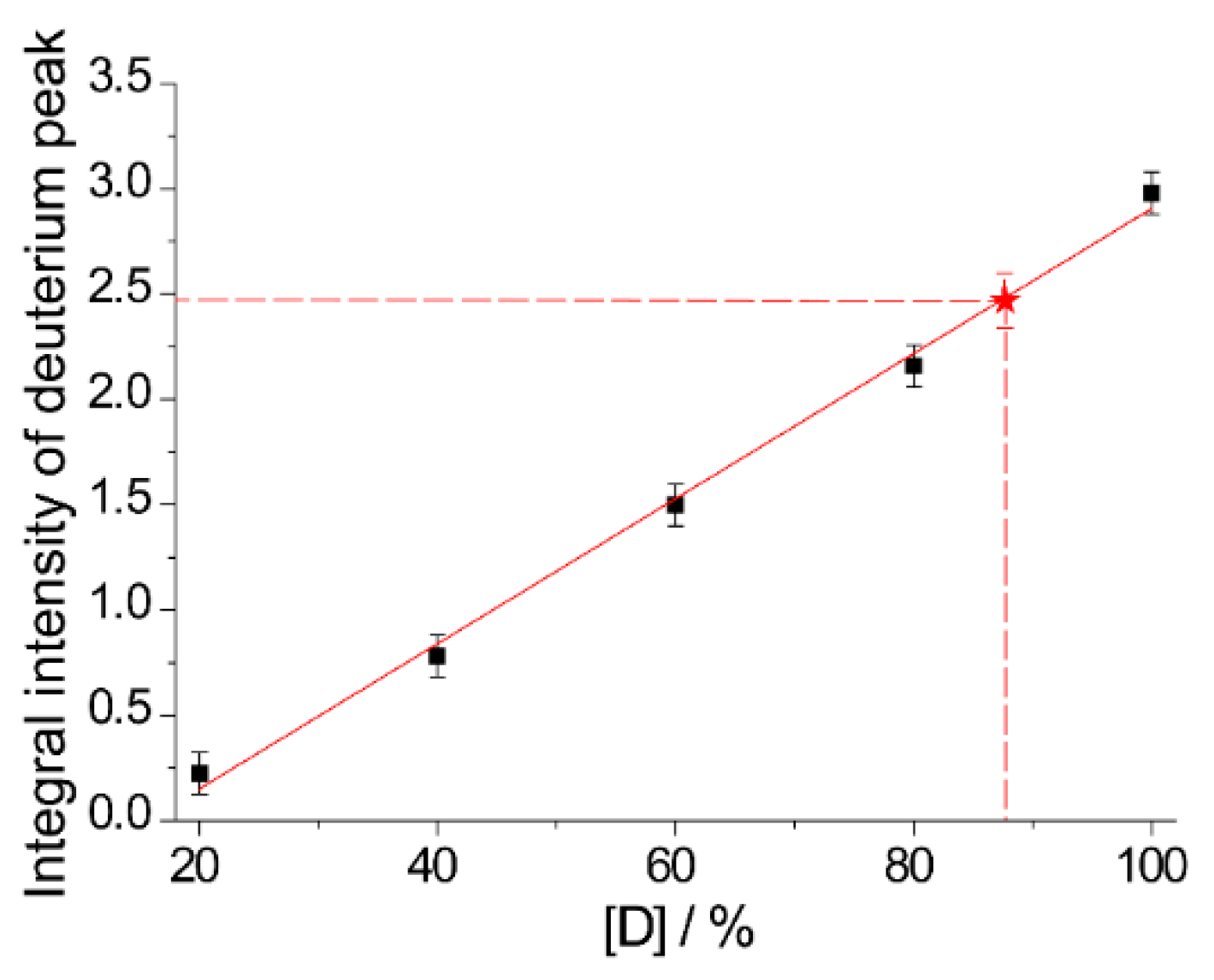
2.2. The Use of Tm to Determine Porphyrin Location in a Biopolymer
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duss, O.; Michel, E.; Yulikov, M.; Schubert, M.; Jeschke, G.; Allain, F.H.T. Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature 2014, 509, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef]
- Schiemann, O.; Prisner, T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q. Rev. Biophys. 2007, 40, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Malygin, A.A.; Krumkacheva, O.A.; Graifer, D.M.; Timofeev, I.O.; Ochkasova, A.S.; Meschaninova, M.I.; Venyaminova, A.G.; Fedin, M.V.; Bowman, M.; Karpova, G.G.; et al. Exploring the interactions of short RNAs with the human 40S ribosomal subunit near the mRNA entry site by EPR spectroscopy. Nucleic Acids Res. 2019, 47, 11850–11860. [Google Scholar] [CrossRef] [PubMed]
- Krumkacheva, O.A.; Shevelev, G.Y.; Lomzov, A.A.; Dyrkheeva, N.S.; Kuzhelev, A.A.; Koval, V.V.; Tormyshev, V.M.; Polienko, Y.F.; Fedin, M.V.; Pyshnyi, D.V.; et al. DNA complexes with human apurinic/apyrimidinic endonuclease 1: Structural insights revealed by pulsed dipolar EPR with orthogonal spin labeling. Nucleic Acids Res. 2019, 47, 7767–7780. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, Y.D.; Bowman, M.K.; Grishin, Y.A. Experimental Techniques BT—Pulsed Electron–Electron Double Resonance: Nanoscale Distance Measurement in the Biological, Materials and Chemical Sciences; Tsvetkov, Y.D., Bowman, M.K., Grishin, Y.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 37–65. ISBN 978-3-030-05372-7. [Google Scholar]
- Pannier, M.; Veit, S.; Godt, A.; Jeschke, G.; Spiess, H.W. Dead-time free measurement of dipole–dipole interactions between electron spins. J. Magn. Reson. 2000, 142, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Milov, A.; Salikohov, K.; Shirov, M. Application of ELDOR in Electron-Spin Echo for Paramagnetic Center Space Distribution in Solids. Fiz. Tverd. Tela 1981, 23, 975–982. [Google Scholar]
- Bagryanskaya, E.G.; Krumkacheva, O.A.; Fedin, M.V.; Marque, S.R.A. Development and Application of Spin Traps, Spin Probes, and Spin Labels. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2015; Volume 563, pp. 365–396. [Google Scholar]
- Weickert, S.; Cattani, J.; Drescher, M. Intrinsically disordered proteins (IDPs) studied by EPR and in-cell EPR. Electron. Paramagn. Reson. 2019, 26, 1–37. [Google Scholar]
- Jeschke, G. Dipolar spectroscopy-double-resonance methods. eMagRes 2016, 5, 1459–1476. [Google Scholar]
- Albertini, M.; Carbonera, D.; Gobbo, M.; Zurlo, E.; Di Valentin, M. Porphyrin Triplet State as a Potential Spin Label for Nanometer Distance Measurements by PELDOR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 6582–6585. [Google Scholar]
- Dal Farra, M.G.; Ciuti, S.; Gobbo, M.; Carbonera, D.; Di Valentin, M. Triplet-state spin labels for highly sensitive pulsed dipolar spectroscopy. Mol. Phys. 2019, 117, 2673–2687. [Google Scholar] [CrossRef]
- Carbonera, D.; Dal Farra, M.G.; Albertini, M.; Zurlo, E.; Polimeno, A.; Di Valentin, M.; Orian, L.; Gobbo, M. Light-Induced Porphyrin-Based Spectroscopic Ruler for Nanometer Distance Measurements. Chem. Eur. J. 2016, 22, 17204–17214. [Google Scholar]
- Hintze, C.; Bücker, D.; Domingo Köhler, S.; Jeschke, G.; Drescher, M. Laser-Induced Magnetic Dipole Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 2204–2209. [Google Scholar] [CrossRef]
- Krumkacheva, O.A.; Timofeev, I.O.; Politanskaya, L.V.; Polienko, Y.F.; Tretyakov, E.V.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Bagryanskaya, E.G.; et al. Triplet Fullerenes as Prospective Spin Labels for Nanoscale Distance Measurements by Pulsed Dipolar EPR Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 13271–13275. [Google Scholar] [CrossRef]
- Gonen, O.; Levanon, H. Energy transfer and fine structure axes determination in a hybrid porphyrin dimer oriented in a liquid crystal. Time resolved triplet EPR spectroscopy. J. Chem. Phys. 1986, 84, 4132–4141. [Google Scholar] [CrossRef]
- Levanon, H.; Vega, S. Analysis of the transient EPR signals in the photoexcited triplet state. Application to porphyrin molecules. J. Chem. Phys. 1974, 61, 2265–2274. [Google Scholar] [CrossRef]
- Wili, N.; Hintz, H.; Vanas, A.; Godt, A.; Jeschke, G. Distance measurement between trityl radicals by pulse dressed electron paramagnetic resonance with phase modulation. Magn. Reson. 2020, 1, 75–87. [Google Scholar] [CrossRef]
- Zecevic, A.; Eaton, G.R.; Eaton, S.S.; Lindgren, M. Dephasing of electron spin echoes for nitroxyl radicals in glassy solvents by non-methyl and methyl protons. Mol. Phys. 1998, 95, 1255–1263. [Google Scholar] [CrossRef]
- Mims, W.B. Electron spin echoes. In Electron Paramagnetic Resonance; Geschwind, S., Ed.; Plenum Press: New York, NY, USA, 1972; pp. 263–351. [Google Scholar]
- Salikhov, K.M.; Tsvetkov, Y.D. Electron spin-echo studies of spin-spin interactions in solids. In Time Domain Electron Spin Resonance; Kevan, L., Schwartz, R.N., Eds.; Wiley: New York, NY, USA, 1979; pp. 231–277. [Google Scholar]
- Brown, I.M. Electron spin echo studies of relaxation processes in molecular solids. In Time Domain Electron Spin Resonance; Kevan, L., Schwartz, R.N., Eds.; Wiley: New York, NY, USA, 1979; pp. 195–229. [Google Scholar]
- Dal Farra, M.G.; Richert, S.; Martin, C.; Larminie, C.; Gobbo, M.; Bergantino, E.; Timmel, C.R.; Bowen, A.M.; Di Valentin, M. Light-Induced Pulsed EPR Dipolar Spectroscopy on a Paradigmatic Hemeprotein. ChemPhysChem 2019, 20, 931–935. [Google Scholar] [CrossRef]
- Bieber, A.; Bücker, D.; Drescher, M. Light-induced dipolar spectroscopy—A quantitative comparison between LiDEER and LaserIMD. J. Magn. Reson. 2018, 296, 29–35. [Google Scholar] [CrossRef]
- Ward, R.; Bowman, A.; Sozudogru, E.; El-Mkami, H.; Owen-Hughes, T.; Norman, D.G. EPR distance measurements in deuterated proteins. J. Magn. Reson. 2010, 207, 164–167. [Google Scholar] [CrossRef]
- El Mkami, H.; Ward, R.; Bowman, A.; Owen-Hughes, T.; Norman, D.G. The spatial effect of protein deuteration on nitroxide spin-label relaxation: Implications for EPR distance measurement. J. Magn. Reson. 2014, 248, 36–41. [Google Scholar] [CrossRef]
- Schmidt, T.; Wälti, M.A.; Baber, J.L.; Hustedt, E.J.; Clore, G.M. Long Distance Measurements up to 160 Åin the GroEL Tetradecamer Using Q-Band DEER EPR Spectroscopy. Angew. Chem. Int. Ed. 2016, 55, 15905–15909. [Google Scholar] [CrossRef]
- Salikhov, K.M.; Semenov, A.G.; Tsvetkov, Y.D. Electron Spin Echo and Its Applications; Nauka: Novosibirsk, Russia, 1976. [Google Scholar]
- Halpern, H.J.; Quine, R.W.; Yong, L.; Barth, E.; Eaton, G.R.; Rinard, G.A.; Eaton, S.S.; Mailer, C.; Harbridge, J. Electron Spin Relaxation of Triarylmethyl Radicals in Fluid Solution. J. Magn. Reson. 2002, 152, 156–161. [Google Scholar]
- Eaton, S.S.; Eaton, G.R. Relaxation Times of Organic Radicals and Transition Metal Ions. In Distance Measurements in Biological Systems by EPR; Springer: Boston, MA, USA, 2002; pp. 29–154. [Google Scholar]
- Dzuba, S.A. Libration motion of guest spin probe molecules in organic glasses: CW EPR and electron spin echo study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2000, 56, 227–234. [Google Scholar] [CrossRef]
- Kirilina, E.P.; Dzuba, S.A.; Maryasov, A.G.; Tsvetkov, Y.D. Librational Dynamics of Nitroxide Molecules in a Molecular Glass Studied by Echo - Detected EPR. Appl. Magn. Reson. 2001, 21, 203–221. [Google Scholar] [CrossRef]
- Golysheva, E.A.; Shevelev, G.Y.; Dzuba, S.A. Dynamical transition in molecular glasses and proteins observed by spin relaxation of nitroxide spin probes and labels. J. Chem. Phys. 2017, 147, 064501. [Google Scholar] [CrossRef]
- Kay, C.W.M.; Elger, G.; Möbius, K. The photoexcited triplet state of free-base porphycene: A time-resolved EPR and electron spin echo investigation. Phys. Chem. Chem. Phys. 1999, 1, 3999–4002. [Google Scholar] [CrossRef]
- Angiolillo, P.J.; Vanderkooi, J.M. Electron paramagnetic resonance of the excited triplet state of metal-free and metal-substituted cytochrome c. Biophys. J. 1995, 68, 2505–2518. [Google Scholar] [CrossRef][Green Version]
- Dąbrowski, J.M.; Pucelik, B.; Regiel-Futyra, A.; Brindell, M.; Mazuryk, O.; Kyzioł, A.; Stochel, G.; Macyk, W.; Arnaut, L.G. Engineering of relevant photodynamic processes through structural modifications of metallotetrapyrrolic photosensitizers. Coord. Chem. Rev. 2016, 325, 67–101. [Google Scholar] [CrossRef]
- Lang, K.; Mosinger, J.; Wagnerová, D.M. Photophysical properties of porphyrinoid sensitizers non-covalently bound to host molecules; models for photodynamic therapy. Coord. Chem. Rev. 2004, 248, 321–350. [Google Scholar] [CrossRef]
- Zheng, X.H.; Nie, X.; Liu, H.Y.; Fang, Y.M.; Zhao, Y.; Xia, L.X. TMPyP4 promotes cancer cell migration at low doses, but induces cell death at high doses. Sci. Rep. 2016, 6, 26592. [Google Scholar] [CrossRef]
- Ouyang, D.; Inoue, S.; Okazaki, S.; Hirakawa, K. Tetrakis(N-methyl-p-pyridinio)porphyrin and its zinc complex can photosensitize damage of human serum albumin through electron transfer and singlet oxygen generation. J. Porphyr. Phthalocyanines 2016, 20, 813–821. [Google Scholar] [CrossRef]
- Groh, S.E.; Nagahisa, A.; Tan, S.L.; Orme-Johnson, W.H. Electron Spin Echo Modulation Demonstrates P-450scc Complexation. J. Am. Chem. Soc. 1983, 105, 7445–7446. [Google Scholar] [CrossRef]
- Mims, W.B.; Dams, J.L.; Peisach, J. The exchange of hydrogen ions and of water molecules near the active site of cytochrome c. J. Magn. Reson. 1990, 86, 273–292. [Google Scholar] [CrossRef]
- Erilov, D.A.; Bartucci, R.; Guzzi, R.; Shubin, A.A.; Maryasov, A.G.; Marsh, D.; Dzuba, S.A.; Sportelli, L. Water concentration profiles in membranes measured by ESEEM of spin-labeled lipids. J. Phys. Chem. B 2005, 109, 12003–12013. [Google Scholar] [CrossRef]
- Milov, A.D.; Samoilova, R.I.; Shubin, A.A.; Grishin, Y.A.; Dzuba, S.A. ESEEM measurements of local water concentration in D 2O- containing spin-labeled systems. Appl. Magn. Reson. 2008, 35, 73–94. [Google Scholar] [CrossRef]
- Syryamina, V.N.; De Zotti, M.; Peggion, C.; Formaggio, F.; Toniolo, C.; Raap, J.; Dzuba, S.A. A molecular view on the role of cholesterol upon membrane insertion, aggregation, and water accessibility of the antibiotic lipopeptide trichogin GA IV as revealed by EPR. J. Phys. Chem. B 2012, 116, 5653–5660. [Google Scholar] [CrossRef]
- Carmieli, R.; Papo, N.; Zimmermann, H.; Potapov, A.; Shai, Y.; Goldfarb, D. Utilizing ESEEM spectroscopy to locate the position of specific regions of membrane-active peptides within model membranes. Biophys. J. 2006, 90, 492–505. [Google Scholar] [CrossRef]
- Liu, L.; Mayo, D.J.; Sahu, I.D.; Zhou, A.; Zhang, R.; McCarrick, R.M.; Lorigan, G.A. Determining the Secondary Structure of Membrane Proteins and Peptides Via Electron Spin Echo Envelope Modulation (ESEEM) Spectroscopy. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2015; Volume 564, pp. 289–313. [Google Scholar]
- Cieslak, J.A.; Focia, P.J.; Gross, A. Electron spin-echo envelope modulation (ESEEM) reveals water and phosphate interactions with the KcsA potassium channel. Biochemistry 2010, 49, 1486–1494. [Google Scholar] [CrossRef][Green Version]
- Ionita, G.; Florent, M.; Goldfarb, D.; Chechik, V. Studying supramolecular assemblies by ESEEM spectroscopy: Inclusion complexes of cyclodextrins. J. Phys. Chem. B 2009, 113, 5781–5787. [Google Scholar] [CrossRef]
- Krumkacheva, O.A.; Fedin, M.V.; Polovyanenko, D.N.; Jicsinszky, L.; Marque, S.R.A.; Bagryanskaya, E.G. Structural equilibrium in new nitroxide-capped cyclodextrins: CW and pulse EPR study. J. Phys. Chem. B 2013, 117, 8223–8231. [Google Scholar] [CrossRef]
- Tait, C.E.; Neuhaus, P.; Anderson, H.L.; Timmel, C.R.; Carbonera, D.; Di Valentin, M. HYSCORE on Photoexcited Triplet States. Appl. Magn. Reson. 2015, 46, 389–409. [Google Scholar] [CrossRef]
- Kruk, N.N.; Dzhagarov, B.M.; Galievsky, V.A.; Chirvony, V.S.; Turpin, P.Y. Photophysics of the cationic 5,10,15,20-tetrakis(4-N-methylpyridyl) porphyrin bound to DNA, [poly(dA-dT)]2 and [poly(dG-dC)]2: Interaction with molecular oxygen studied by porphyrin triplet-triplet absorption and singlet oxygen luminescence. J. Photochem. Photobiol. B Biol. 1998, 42, 181–190. [Google Scholar] [CrossRef]
Sample Availability: Not available. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sannikova, N.; Timofeev, I.; Bagryanskaya, E.; Bowman, M.; Fedin, M.; Krumkacheva, O. Electron Spin Relaxation of Photoexcited Porphyrin in Water—Glycerol Glass. Molecules 2020, 25, 2677. https://doi.org/10.3390/molecules25112677
Sannikova N, Timofeev I, Bagryanskaya E, Bowman M, Fedin M, Krumkacheva O. Electron Spin Relaxation of Photoexcited Porphyrin in Water—Glycerol Glass. Molecules. 2020; 25(11):2677. https://doi.org/10.3390/molecules25112677
Chicago/Turabian StyleSannikova, Natalya, Ivan Timofeev, Elena Bagryanskaya, Michael Bowman, Matvey Fedin, and Olesya Krumkacheva. 2020. "Electron Spin Relaxation of Photoexcited Porphyrin in Water—Glycerol Glass" Molecules 25, no. 11: 2677. https://doi.org/10.3390/molecules25112677
APA StyleSannikova, N., Timofeev, I., Bagryanskaya, E., Bowman, M., Fedin, M., & Krumkacheva, O. (2020). Electron Spin Relaxation of Photoexcited Porphyrin in Water—Glycerol Glass. Molecules, 25(11), 2677. https://doi.org/10.3390/molecules25112677