Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs

 , ,
, ,  , and
, and
Abstract
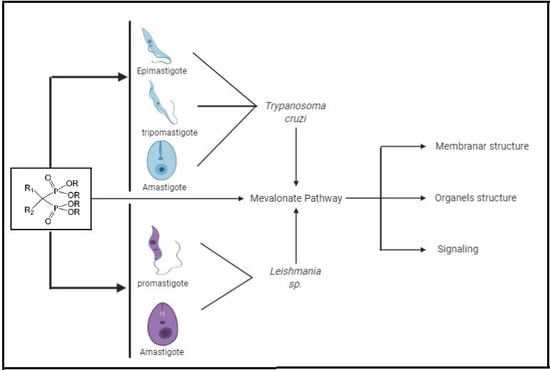
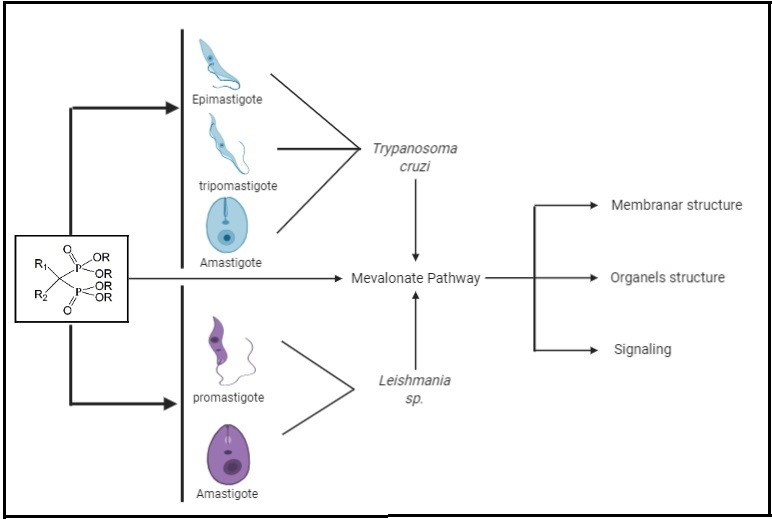{kind=link}
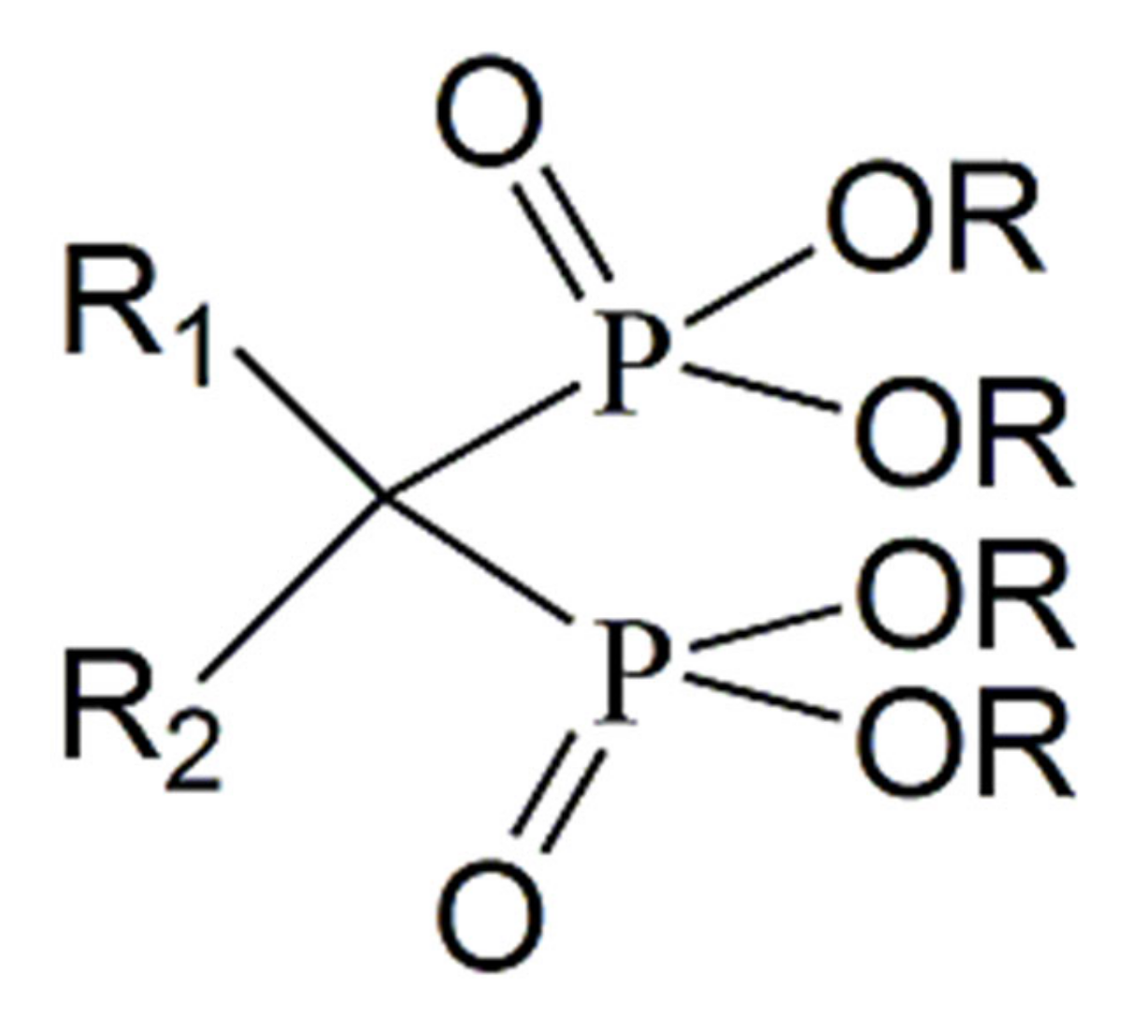{kind=link}
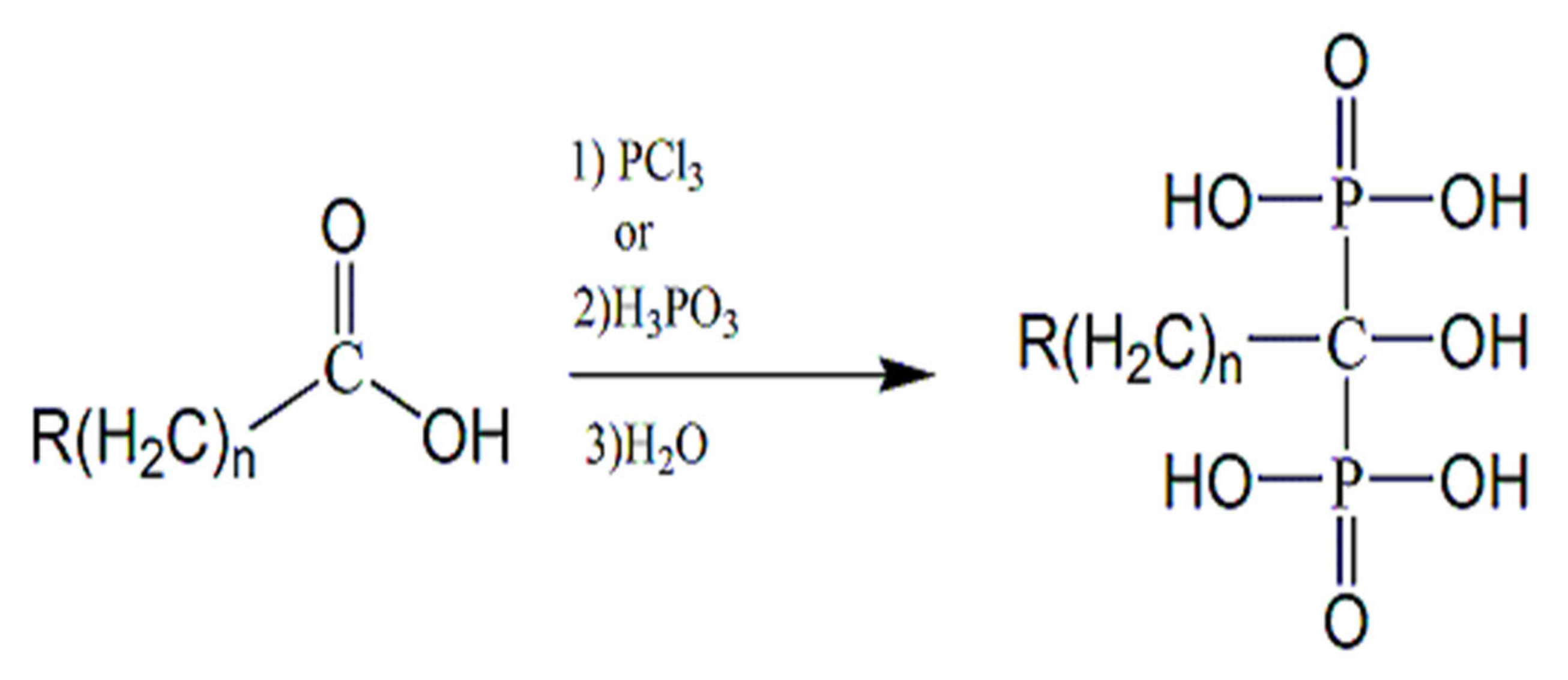{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. History of the Use of Bisphosphonates
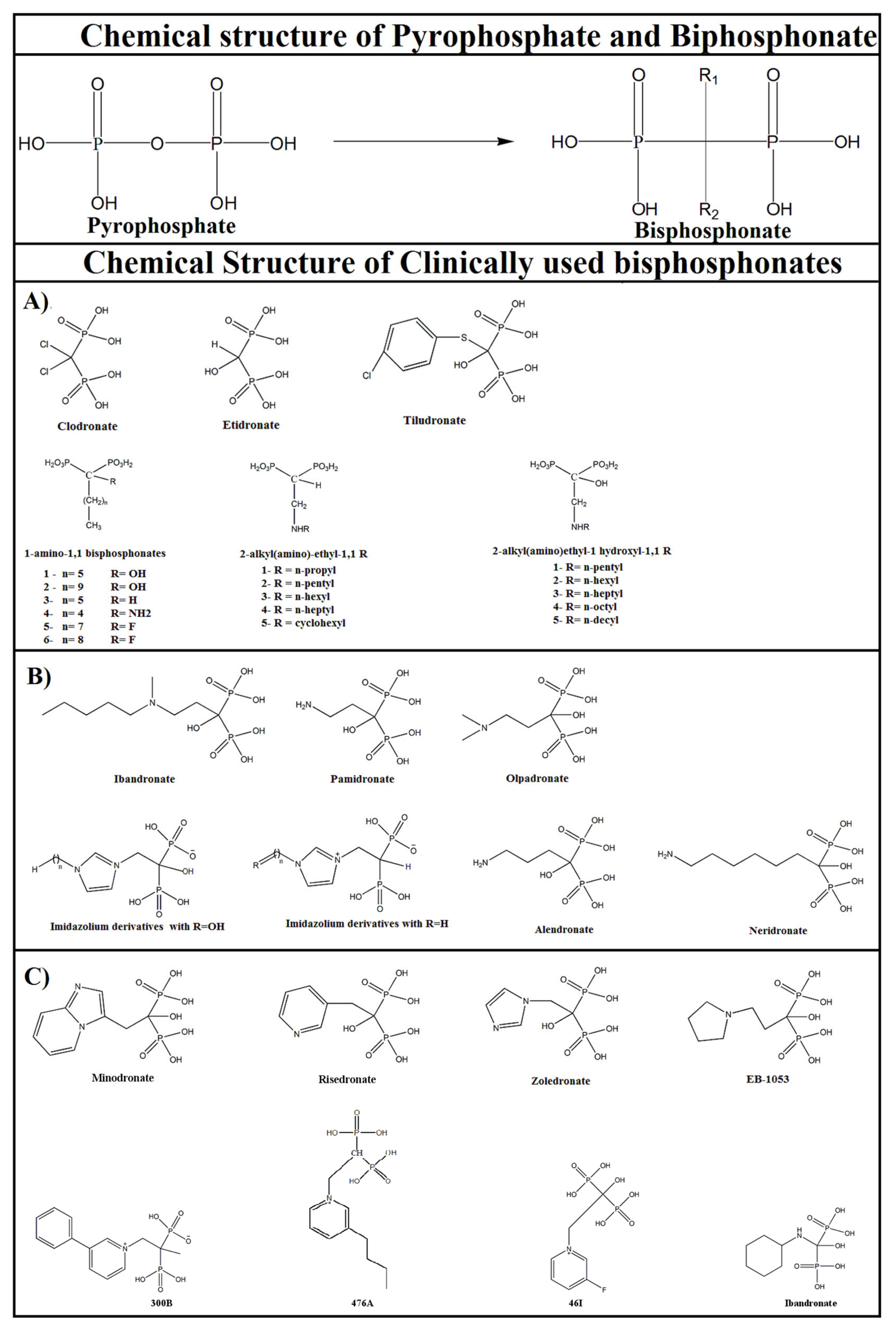
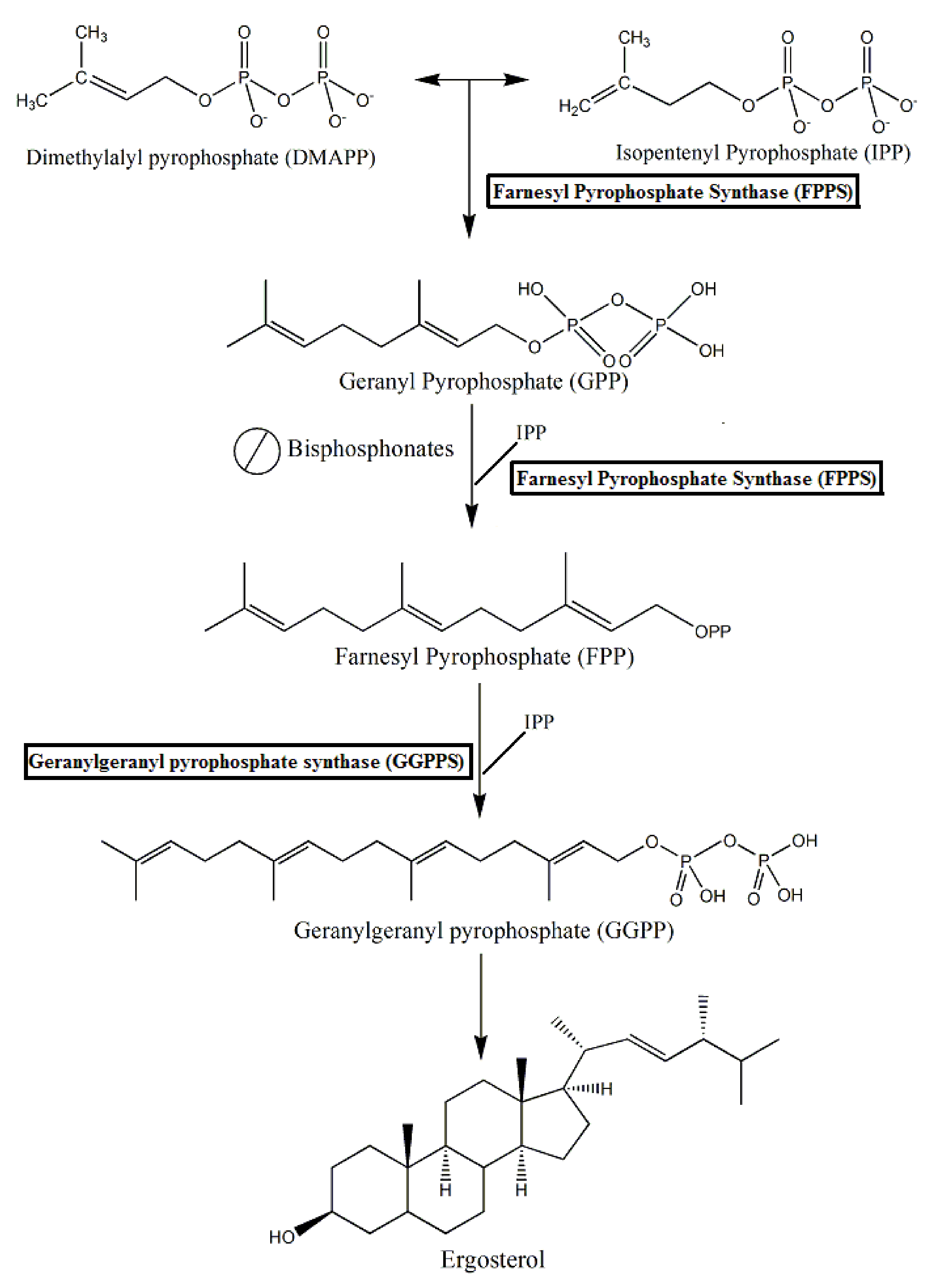
3. Chemical and Biological Characteristics of Bisphosphonate-Based Compounds
4. Bisphosphonate Compounds as Potential New Anti-Trypanosoma cruzi Drugs
5. Bisphosphonate Compounds as Potential New Anti-Leishmania spp. Drugs
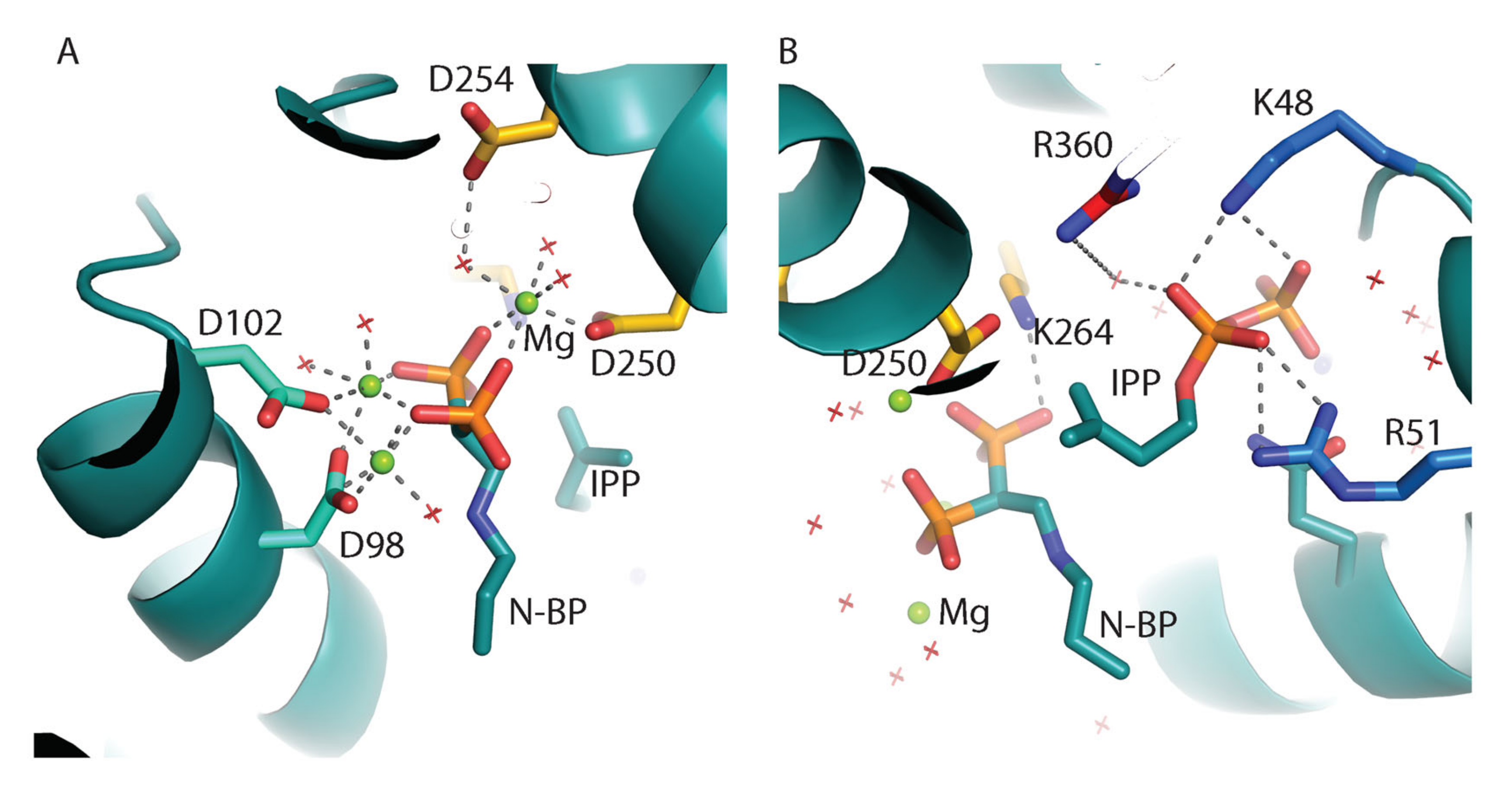
6. Overall Structure of LmFPPS and TcFPPS in Complex with Inhibitors
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Neglected Tropical Diseases. Available online: https://www.who.int/neglected_diseases/diseases/en/ (accessed on 27 May 2020).
- Andrade, C.H.; Kümmerle, A.E.; Guido, R.V.C. Medicinal chemistry perspectives for the 21st century: Challenges and opprotunities. Quim. Nova 2018, 41, 476–483. [Google Scholar]
- Weng, H.B.; Chen, H.X.; Wang, M.W. Innovation in neglected tropical disease drug discovery and development. Infect. Dis. Poverty 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rugani, J.N.; Quaresma, P.F.; Gontijo, C.F.; Soares, R.P.; Monte-Neto, R.L. Intraspecies susceptibility of Leishmania (Viannia) braziliensis to antileishmanial drugs: Antimony resistance in human isolates from atypical lesions. Biomed. Pharmacother. 2018, 108, 1170–1180. [Google Scholar] [CrossRef]
- Soeiro, M.D.N.C.; Souza, E.M.; Silva, C.F.; Batista, D.D.G.J.; Batista, M.M.; Pavão, B.P.; Britto, C. In vitro and in vivo studies of the antiparasitic activity of sterol 14α-demethylase (CYP51) inhibitor VNI against drug-resistant strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 2013, 57, 4151–4163. [Google Scholar] [CrossRef] [PubMed]
- Holanda, V.N.; Silva, W.V.; Nascimento, P.H.; Oliveira, R.N.; Lima, V.L.M.; Figueiredo, R.C.B.Q. Challengas and perspectives in the treatment of tegumentary leishmaniosis: Review. Rev. Interfaces Saúde Hum. Tecnol. 2018, 6, 140–157. [Google Scholar]
- Aripirala, S.; Gonzalez-Pacanowska, D.; Oldfield, E.; Kaiser, M.; Amzel, L.M.; Gabelli, S.B. Structural and thermodynamic basis of the inhibition of Leishmania major farnesyl diphosphate synthase by nitrogen-containing bisphosphonates. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 802–810. [Google Scholar] [CrossRef]
- Mukherjee, S.; Basu, S.; Zhang, K. Farnesyl pyrophosphate synthase is essential for the promastigote and amastigote stages in Leishmania major. Mol. Biochem. Parasitol. 2019, 230, 8–15. [Google Scholar] [CrossRef]
- Ortiz-Gómez, A.; Jiménez, C.; Estévez, A.M.; Carrero-Lérida, J.; Ruiz-Pérez, L.M.; González-Pacanowska, D. Farnesyl diphosphate synthase is a cytosolic enzyme in Leishmania major promastigotes and its overexpression confers resistance to risedronate. Eukaryot. Cell 2006, 5, 1057–1064. [Google Scholar] [CrossRef]
- Rodriguez, N.; Bailey, B.N.; Martin, M.B.; Oldfield, E.; Urbina, J.A.; Docampo, R. Radical cure of experimental cutaneous leishmaniasis by the bisphosphonate pamidronate. J. Infect. Dis. 2002, 186, 138–140. [Google Scholar] [CrossRef]
- Aripirala, S.; Szajnman, S.H.; Jakoncic, J.; Rodriguez, J.B.; Docampo, R.; Gabelli, S.B.; Amzel, L.M. Design, synthesis, calorimetry and crystallographic analysis of 2-alkylaminoethyl-1,1-bisphosphonates as inhibitors of Trypanosoma cruzi farnesyl diphosphate synthase. J. Med. Chem. 2012, 55, 6445–6454. [Google Scholar] [CrossRef]
- Galaka, T.; Falcone, B.N.; Li, C.; Szajnman, S.H.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-alkylaminomethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii. Bioorg. Med. Chem. 2019, 27, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Garzoni, L.R.; Caldera, A.; Meirelles, M.D.N.L.; Castro, S.L.; Docampo, R.; Meints, G.A.; Oldfield, E.; Urbina, J.A. Selective in vitro effects of the farnesyl pyrophosphate synthase inhibitor risedronate on Trypanosoma cruzi. Int. J. Antimicrob. Agents 2004, 23, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Cheong, Y.-K.; Ren, G.G.; Huang, M.; Detsch, R.; Boccaccini, A.R. Biological evaluations of novel 2,3,3-trisphosphonate in osteoclastic and osteoblastic activities. Gen. Med. Open 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kafarski, P. Physiologic activity of bisphosphonates—Recent advances. Open Pharm. Sci. J. 2016, 3, 56–78. [Google Scholar] [CrossRef]
- Kuzuyama, T. Mevalonate and nonmevalonate pathways for the biosynthesis of isoprene units. Biosci. Biotechnol. Biochem. 2002, 66, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Boucher, Y.; Kamekura, M.; Doolittle, W.F. Origins and evolution of isoprenoid lipid biosynthesis in archaea. Mol. Microbiol. 2004, 52, 515–527. [Google Scholar] [CrossRef]
- Vicent, D.; Maratos-Flier, E.; Kahn, C.R. The branch point enzyme of the mevalonate pathway for protein prenylation is overexpressed in the ob/ob mouse and induced by adipogenesis. Mol. Cell. Biol. 2000, 20, 2158–2166. [Google Scholar] [CrossRef]
- Cunillera, N.; Arró, M.; Delourme, D.; Karst, F.; Boronat, A.; Ferrer, A. Arabidopsis thaliana contains two differentially expressed farnesyl-diphosphate synthase genes. J. Biol. Chem. 1996, 271, 7774–7780. [Google Scholar] [CrossRef]
- Daudonnet, S.; Karst, F.; Tourte, Y. Expression of the farnesyldiphosphate synthase gene of Saccharomyces cerevisiae in tobacco. Mol. Breed. 1997, 3, 137–145. [Google Scholar] [CrossRef]
- Ohnuma, S.I.; Nakazawa, T.; Hemmi, H.; Hallberg, A.M.; Koyama, T.; Ogura, K.; Nishino, T. Conversion from farnesyl diphosphate synthase to geranylgeranyl diphosphate synthase by random chemical mutagenesis. J. Biol. Chem. 1996, 271, 10087–10095. [Google Scholar] [CrossRef]
- Montalvetti, A.; Bailey, B.N.; Martin, M.B.; Severin, G.W.; Oldfield, E.; Docampo, R. Bisphosphonates are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. J. Biol. Chem. 2001, 276, 33930–33937. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chan, J.M.W.; Lea, C.R.; Meints, G.A.; Lewis, J.C.; Tovian, Z.S.; Flessner, R.M.; Loftus, T.C.; Bruchhaus, I.; Kendrick, H.; et al. Effects of bisphosphonates on the growth of Entamoeba histolytica and Plasmodium species in vitro and in vivo. J. Med. Chem. 2004, 47, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Li, Z.H.; Miranda, K.; Oldfield, E.; Moreno, S.N.J. The farnesyl-diphosphate/geranylgeranyl-diphosphate synthase of Toxoplasma gondii is a bifunctional enzyme and a molecular target of bisphosphonates. J. Biol. Chem. 2007, 282, 30804–30816. [Google Scholar] [CrossRef] [PubMed]
- Yardley, V.; Khan, A.A.; Martin, M.B.; Slifer, T.R.; Araujo, F.G.; Moreno, S.N.J.; Docampo, R.; Croft, S.L.; Oldfield, E. In vivo activities of farnesyl pyrophosphate synthase inhibitors against Leishmania donovani and Toxoplasma gondii. Antimicrob. Agents Chemother. 2002, 46, 929–931. [Google Scholar] [CrossRef]
- Ling, Y.; Sahota, G.; Odeh, S.; Chan, J.M.W.; Araujo, F.G.; Moreno, S.N.J.; Oldfield, E. Bisphosphonate inhibitors of Toxoplasma gondii growth: In vitro, QSAR, and in vivo investigations. J. Med. Chem. 2005, 48, 3130–3140. [Google Scholar] [CrossRef]
- Martin, M.B.; Grimley, J.S.; Lewis, J.C.; Heath, H.T.; Bailey, B.N.; Kendrick, H.; Yardley, V.; Caldera, A.; Lira, R.; Urbina, J.A.; et al. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: A potential route to chemotherapy. J. Med. Chem. 2001, 44, 909–916. [Google Scholar] [CrossRef]
- Moreno, S.N.J.; Li, Z.H. Targeting the isoprenoid pathway of Toxoplasma gondii. Expert Opin. Ther. Targets 2008, 12, 253–263. [Google Scholar] [CrossRef]
- Rodan, G.A. Mechanisms of action of bisphosphonates. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 375–388. [Google Scholar] [CrossRef]
- Rosso, V.S.; Szajnman, S.H.; Malayil, L.; Galizzi, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of new 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2011, 19, 2211–2217. [Google Scholar] [CrossRef]
- Sarkar, S.; Strutz, S.E.; Frank, D.M.; Rivaldi, C.L.; Sissel, B.; Sánchez-Cordero, V. Chagas disease risk in Texas. PLoS Negl. Trop. Dis. 2010, 4, e836. [Google Scholar] [CrossRef]
- Szajnman, S.H.; García Liñares, G.E.; Li, Z.H.; Jiang, C.; Galizzi, M.; Bontempi, E.J.; Ferella, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorg. Med. Chem. 2008, 16, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Todolí, F.; Solano-Gallego, L.; Ojeda, A.; Quintana, J.; Lloret, A.; Roura, X.; Alberola, J.; Rodríguez-Cortés, A. Anti-Leishmania IgA in urine samples from dogs with clinical leishmaniasis. Vet. Parasitol. 2009, 159, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Leuret, F.; Lassaigne, J.L. Recherches Physiologiques et Chimiques pour Servir à L’histoire de la Digestion; Madame Huzard: Paris, France, 1825. [Google Scholar]
- Allen, D.W. Phosphines and Related P–C-Bonded Compounds. In Organophosphorus Chemistry; Allen, D.W., Tebby, J.C., Loakes, D., Eds.; Royal Society of Chemistry: London, UK, 2014; Volume 43. [Google Scholar]
- Fleisch, H.; Graham, R.; Russell, G.; Straumann, F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 1966, 212, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.; Bisaz, S. Isolation from urine of pyrophosphate, a calcification inhibitor. Am. J. Physiol. 1962, 203, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A.; Fleisch, H.A. Bisphosphonates: Mechanisms of action. J. Clin. Investig. 2009, 97, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Blomen, L.J.M.J. History of the bisphosphonates: Discovery and history of the non- medical uses of bisphosphonates. In Bisphosphonates on Bones; Elsevier: Amsterdam, The Netherlands, 1995; pp. 111–124. [Google Scholar]
- Galaka, T.; Casal, M.F.; Storey, M.; Li, C.; Chao, M.N.; Szajnman, S.H.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Antiparasitic activity of sulfur- and fluorine-containing bisphosphonates against trypanosomatids and apicomplexan parasites. Molecules 2017, 22, 82. [Google Scholar] [CrossRef]
- Ebetino, F.H.; Hogan, A.M.L.; Sun, S.; Tsoumpra, M.K.; Duan, X.; Triffitt, J.T.; Kwaasi, A.A.; Dunford, J.E.; Barnett, B.L.; Oppermann, U.; et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011, 49, 20–33. [Google Scholar] [CrossRef]
- Widler, L.; Jahnke, W.; Green, J.R. The chemistry of Bisphosphonates: From antiscaling agents to clinical therapeutics. Anti Cancer Agents Med. Chem. 2012, 12, 95–101. [Google Scholar] [CrossRef]
- Rasmusson, L.; Abtahi, J. Bisphosphonate associated osteonecrosis of the jaw: An update on pathophysiology, risk factors, and treatment. Int. J. Dent. 2014, 2014, 471035. [Google Scholar] [CrossRef]
- Forte, L.; Sarda, S.; Torricelli, P.; Combes, C.; Brouillet, F.; Marsan, O.; Salamanna, F.; Fini, M.; Boanini, E.; Bigi, A. Correction to “Multifunctionalization modulates hydroxyapatite surface interaction with bisphosphonate: Antiosteoporotic and antioxidative stress materials”. ACS Biomater. Sci. Eng. 2019, 5, 3429–3439. [Google Scholar] [CrossRef]
- Whitaker, M.; Guo, J.; Kehoe, T.; Benson, G. Bisphosphonates for osteoporosis—Where do we go from here? N. Eng. J. Med. 2012, 366, 2048–2051. [Google Scholar] [CrossRef] [PubMed]
- Ermer, M.A.; Kottmann, S.C.; Otten, J.E.; Wittmer, A.; Poxleitner, P.; Pelz, K. In vitro investigation of the antimicrobial effect of three bisphosphonates against different bacterial strains. J. Oral Maxillofac. Surg. 2018, 76, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Lipton, A. Anticancer properties of zoledronic acid. Cancer Investig. 2010, 28, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Ghosh, S.; Chan, J.M.W.; Meints, G.; Wang, H.; Raker, A.M.; Song, Y.; Colantino, A.; Burzynska, A.; Kafarski, P.; et al. Quantitative structure-activity relationships for γδ T cell activation by bisphosphonates. J. Med. Chem. 2004, 47, 375–384. [Google Scholar] [CrossRef]
- Martin, M.B.; Sanders, J.M.; Kendrick, H.; de Luca-Fradley, K.; Lewis, J.C.; Grimley, J.S.; Van Brussel, E.M.; Olsen, J.R.; Meints, G.A.; Burzynska, A.; et al. Activity of Bisphosphonates against Trypanosoma brucei rhodesiense. J. Med. Chem. 2002, 45, 2904–2914. [Google Scholar] [CrossRef]
- Yang, G.; Zhu, W.; Kim, K.; Byun, S.Y.; Choi, G.; Wang, K.; Cha, J.S.; Cho, H.S.; Oldfield, E.; No, J.H. In vitro and in vivo investigation of the inhibition of Trypanossoma brucei cell growth by lipophilic bisphosphonates. Antimicrob. Agents Chemother. 2015, 59, 7530–7539. [Google Scholar] [CrossRef]
- Lolli, M.L.; Lazzarato, L.; Di Stilo, A.; Fruttero, R.; Gasco, A. Michael addition of Grignard reagents to tetraethyl ethenylidenebisphosphonate. J. Organomet. Chem. 2002, 650, 77–83. [Google Scholar] [CrossRef]
- Degenhardt, C.R.; Burdsall, D.C. Synthesis of ethenylidenebis(phosphonic acid) and its tetraalkyl esters. J. Org. Chem. 1986, 51, 3488–3490. [Google Scholar] [CrossRef]
- Kieczykowski, G.R.; Jobson, R.B.; Melillo, D.G.; Reinhold, D.F.; Grenda, V.J.; Shinkai, I. Preparation or (4-Amino-l-hydroxybutylidene)bisphosphonic acid sodium Salt, MK-217 (Alendronate sodium). An improved procedure for the preparation of 1-Hydroxy-1,1-bisphosphonic acids. J. Org. Chem. 1995, 60, 8310–8312. [Google Scholar] [CrossRef]
- Nguyen, L.M.; Niesor, E.; Bentzen, C.L. Gem-diphosphonate and gem -phosphonate-phosphate compounds with specific high density lipoprotein inducing activity. Am. Chem. Soc. 1987, 30, 1426–1433. [Google Scholar] [CrossRef]
- Lai, C.; Xi, C.; Feng, Y. A facile approach for the synthesis of α-halogenated alkylidenediphosphonates by reaction of alkyllithium with chlorophosphate and halogen reagent. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 449–455. [Google Scholar] [CrossRef]
- Teulade, M.P.; Savignac, P.; Aboujaoude, E.E.; Liétge, S.; Collignon, N. Alkylidènediphosphonates et vinylphosphonates: Une démarche synthétiques sélective par voie carbanionique. J. Organomet. Chem. 1986, 304, 283–300. [Google Scholar] [CrossRef]
- Hutchinson, D.W.; Thornton, D.M. Michael addition reactions of ethenylidenebisphosphonates. J. Organomet. Chem. 1988, 346, 341–348. [Google Scholar] [CrossRef]
- Lehnert, W. Knoevenagel kondensationen mit TiCl4/base-IV. Umsetzungen von aldehyden und ketonen mit phosphonoessigester und methylendiphosphonsäureestern. Tetrahedron 1974, 30, 301–305. [Google Scholar] [CrossRef]
- Inoue, S.; Okauchi, T.; Minami, T. New synthesis of gem-bis(phosphono)ethylenes and their applications. Synthesis 2003, 13, 1971–1976. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Yoshida, Y.; Nakabayashi, N.; Shibuya, S. Simple and efficient method for preparation of conformationally constrained aminomethylene gem-diphosphonate derivatives via beckmann rearrangement. J. Org. Chem. 1994, 59, 7562–7564. [Google Scholar] [CrossRef]
- Wu, M.; Chen, R.; Huang, Y. Convenient synthesis of analogs of aminomethylene gem-diphosphonic acid from amines without catalyst. Synth. Commun. 2004, 34, 1393–1398. [Google Scholar] [CrossRef]
- Gouault-Bironneau, S.; Deprèle, S.; Sutor, A.; Montchamp, J.L. Radical reaction of sodium hypophosphite with terminal alkynes: Synthesis of 1,1-bis-H-phosphinates. Org. Lett. 2005, 7, 5909–5912. [Google Scholar] [CrossRef]
- Romanenko, V.; Kukhar, V. Progress in the development of pyrophosphate bioisosteres: Synthesis and biomedical potential of 1-Fluoro- and 1,1-Difluoromethylene-1,1-bisphosphonates. Curr. Org. Chem. 2016, 18, 1491–1512. [Google Scholar] [CrossRef]
- Puljula, E.; Turhanen, P.; Vepsäläinen, J.; Monteil, M.; Lecouvey, M.; Weisell, J. Structural requirements for bisphosphonate binding on hydroxyapatite: NMR study of bisphosphonate partial esters. ACS Med. Chem. Lett. 2015, 6, 397–401. [Google Scholar] [CrossRef]
- Qian, D.Q.; Shi, X.D.; Zeng, X.Z.; Cao, R.Z.; Liu, L.Z. Aminoalkylation of organophosphorus compounds with P-H bond by using Vilsmeier reagents. Tetrahedron Lett. 1997, 38, 6245–6246. [Google Scholar]
- Docampo, R.; Moreno, S.N. Bisphosphonates as chemotherapeutic agents against trypanosomatid and apicomplexan parasites. Curr. Drug Targets Infect. Disord. 2001, 1, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nagaoka, M.; Igarashi, K.; Hisashi, S. MPMBP, a novel bisphosphonate with an antioxidant side chain, stimulates bone formation through inhibition of NF-κB nuclear translocation. Folia Pharmacol. Jpn. 2019, 153, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Boanini, E. Calcium phosphates as delivery systems for bisphosphonates. J. Funct. Biomater. 2018, 9, 6. [Google Scholar] [CrossRef]
- Cromartie, T.H.; Fisher, K.J.; Grossman, J.N. The discovery of a novel site of action for herbicidal bisphosphonates. Pestic. Biochem. Physiol. 1999, 63, 114–126. [Google Scholar] [CrossRef]
- Giger, E.V.; Castagner, B.; Leroux, J.C. Biomedical applications of bisphosphonates. J. Control. Release 2013, 167, 175–188. [Google Scholar] [CrossRef]
- Russell, R.G.G. Bisphosphonates: Mode of Action and Pharmacology. Pediatrics 2007, 119, 150–162. [Google Scholar] [CrossRef]
- Russell, R.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef]
- Rodan, G.A.; Reszka, A.A. Bisphosphonate mechanism of action. Curr. Rheumatol. Rep. 2003, 2, 571–577. [Google Scholar]
- Frith, J.C.; Mönkkönen, J.; Blackburn, G.M.; Russell, R.G.G.; Rogers, M.J. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5′-(beta, gamma-dichloromethylene) triphosphate, by mammalian cells in vitro. J. Bone Miner. Res. 1997, 12, 1358–1367. [Google Scholar] [CrossRef]
- Ebetino, F.H.; Francis, M.D.; Rogers, M.J.; Russell, R.G.G. Mechanisms of action of etidronate and other bisphosphonates. Rev. Contemp. Pharmaco. 1998, 9, 233–243. [Google Scholar]
- Coleman, R.E. Risks and benefits of bisphosphonates. Br. J. Cancer 2008, 98, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Coxon, F.P.; Thompson, K.; Rogers, M.J. Recent advances in understanding the mechanism of action of bisphosphonates. Curr. Opin. Pharmacol. 2006, 6, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.B.; Arnold, W.; Heath, H.T.; Urbina, J.A.; Oldfield, E. Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem. Biophys. Res. Commun. 1999, 263, 754–758. [Google Scholar] [CrossRef]
- Reszka, A.A.; Rodan, G.A. Nitrogen-containing bisphosphonate mechanism of action. Curr. Rheumatol. Rep. 2003, 5, 65–74. [Google Scholar] [CrossRef]
- Reszka, A.A.; Rodan, G.A. Mechanism of action of bisphosphonates. Curr. Osteoporos. Rep. 2003, 1, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Cremers, S.; Papapoulos, S. Pharmacology of bisphosphonates. Bone 2011, 49, 42–49. [Google Scholar] [CrossRef]
- Zhang, L.; Ko, T.P.; Malwal, S.R.; Liu, W.; Zhou, S.; Yu, X.; Oldfield, E.; Guo, R.T.; Chen, C.C. Complex structures of MoeN5 with substrate analogues suggest sequential catalytic mechanism. Biochem. Biophys. Res. Commun. 2019, 511, 800–805. [Google Scholar] [CrossRef]
- Huang, C.H.; Gabelli, S.B.; Oldfield, E.; Mario Amzel, L. Binding of nitrogen-containing bisphosphonstes (N-BPs) to the Trypanosoma cruzi fernesyl diphosphate synthase homodimer. Proteins Struct. Funct. Bioinform. 2010, 78, 888–899. [Google Scholar] [CrossRef]
- Pool, F.; Currie, R.; Sweby, P.K.; Salazar, J.D.; Tindall, M.J. A mathematical model of the mevalonate cholesterol biosynthesis pathway. J. Theor. Biol. 2018, 443, 157–176. [Google Scholar] [CrossRef]
- Bishop, N.J.; Russell, G. Bisphosphonates. In Osteogenesis Imperfecta; Shapiro, J.R., Byers, P.H., Glorieux, F.H., Sponseller, P.D., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 495–500. ISBN 978-0-12-397165-4. [Google Scholar]
- Kumar, V.; Shahi, A.K. Nitrogen containing bisphosphonates associated osteonecrosis of the jaws: A review for past 10 year literature. Dent. Res. J. 2014, 11, 147. [Google Scholar]
- Sigman, L.; Sánchez, V.M.; Turjanski, A.G. Characterization of the farnesyl pyrophosphate synthase of Trypanosoma cruzi by homology modeling and molecular dynamics. J. Mol. Graph. Model. 2006, 25, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, D.; Singla, L.D. Pharmacokinetics and pharmacology to drugs used for control of emerging cryptosporidiosis and toxoplasmosis in livestock and humans. J. Entomol. Zool. Stud. 2019, 7, 1306–1313. [Google Scholar]
- Chagas, C. Nova tripanozomiaze humana: Estudos sobre a morfologia e o ciclo evolutive do Schizotrypanum cruzi n. gen. n. sp., agente etiológico de nova entidade mórbida do homem. Mem. Inst. Oswaldo Cruz 1909, 1, 159–218. [Google Scholar] [CrossRef]
- Requena-Méndez, A.; Aldasoro, E.; de Lazzari, E.; Sicuri, E.; Brown, M.; Moore, D.A.J.; Gascon, J.; Muñoz, J. Prevalence of Chagas Disease in Latin-American Migrants Living in Europe: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2015, 9, e0003540. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Salvador, F.; Sánchez-Montalvá, A.; Treviño, B.; Serre, N.; Avilés, A.S.; Almirante, B. Toxic profile of benznidazole in patients with chronic chagas disease: Risk factors and comparison of the product from two different manufacturers. Antimicrob. Agents Chemother. 2015, 59, 6125–6131. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas Disease. Seminar. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Rassi, A.; Marin-Neto, J.A.; Rassi, A. Chronic chagas cardiomyopathy: A review of the main pathogenic mechanisms and the efficacy of aetiological treatment following the benznidazole evaluation for interrupting trypanosomiasis (BENEFIT) trial. Mem. Inst. Oswaldo Cruz 2017, 112, 224–235. [Google Scholar] [CrossRef]
- World Health Organization. Chagas Disease (Also Known as American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 27 May 2020).
- World Health Organization. Research Priorities for Chagas Disease, Human African Trypanosomiasis and Leishmaniasis. In World Health Organization Technical Report Series; WHO: Geneva, Switzerland, 2012; pp. 1–100. [Google Scholar]
- Coura, J.R.; Borges-Pereira, J. Chagas disease: 100 years after its discovery. A systemic review. Acta Trop. 2010, 115, 5–13. [Google Scholar] [CrossRef]
- Hotez, P.J.; Woc-Colburn, L.; Bottazzi, M.E. Neglected tropical diseases in Central America and Panama: Review of their prevalence, populations at risk and impact on regional development. Int. J. Parasitol. 2014, 44, 597–603. [Google Scholar] [CrossRef]
- Pan American Health Organization. Chagas disease in the Americas: A Review of the Current Public Health Situation and a Vision for the Future. Available online: https://www.paho.org/hq/index.php?option=com_content&view=category&id=3591&Itemid=40370&lang=en (accessed on 27 May 2020).
- Edwards, M.S.; Stimpert, K.K.; Bialek, S.R.; Montgomery, S.P. Evaluation and management of congenital chagas disease in the United States. J. Pediatric Infect. Dis. Soc. 2019, 8, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Dumonteil, E.; Betancourt Cravioto, M.; Bottazzi, M.E.; Tapia-Conyer, R.; Meymandi, S.; Karunakara, U.; Ribeiro, I.; Cohen, R.M.; Pecoul, B. An unfolding tragedy of chagas disease in North America. PLoS Negl. Trop. Dis. 2013, 7, 2300–2304. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Montgomery, S.P. An estimate of the burden of chagas disease in the United States. Clin. Infect. Dis. 2009, 49, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Newsom-Davis, J.; Wray, D.; Shillito, P.; Harrison, J.; Betty, M.; Murray, N. Clinical and experimental-Observations in patients with congenital myasthenic syndromes. Ann. N. Y. Acad. Sci. 1993, 681, 451–460. [Google Scholar] [CrossRef]
- Quaresma, P.F.; De Brito, C.F.A.; Rugani, J.M.N.; Freire, J.D.M.; Baptista, R.D.P.; Moreno, E.C.; Gontijo, R.C.; Rego, F.D.; Diniz, J.E.; Melo, M.N.; et al. Distinct genetic profiles of Leishmania (Viannia) braziliensis associate with clinical variations in cutaneous-leishmaniasis patients from an endemic area in Brazil. Proc. Int. Astron. Union 2018, 145, 1161–1169. [Google Scholar] [CrossRef]
- Chatelain, E. Chagas disease research and development: Is there light at the end of the tunnel? Comput. Struct. Biotechnol. J. 2017, 15, 98–103. [Google Scholar] [CrossRef]
- Pérez-Ayala, A.; Pérez-Molina, J.A.; Norman, F.; Monge-Maillo, B.; Faro, M.V.; López-Vélez, R. Gastro-intestinal chagas disease in migrants to spain: Prevalence and methods for early diagnosis. Ann. Trop. Med. Parasitol. 2011, 105, 25–29. [Google Scholar] [CrossRef]
- Pinazo, M.J.; Espinosa, G.; Gállego, M.; López-Chejade, P.L.; Urbina, J.A.; Gascón, J. Case report: Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am. J. Trop. Med. Hyg. 2010, 82, 583–587. [Google Scholar] [CrossRef]
- Urbina, J.A. Specific chemotherapy of Chagas disease: Relevance, current limitations and new approaches. Acta Trop. 2010, 115, 55–68. [Google Scholar] [CrossRef]
- Bern, C.; Martin, D.L.; Gilman, R.H. Acute and Congenital Chagas Disease. Adv. Parasitol. 2011, 75, 19–47. [Google Scholar]
- Ferella, M.; Li, Z.H.; Andersson, B.; Docampo, R. Farnesyl diphosphate synthase localizes to the cytoplasm of Trypanosoma cruzi and T. brucei. Exp. Parasitol. 2008, 119, 308–312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cuevas, I.C.; Rohloff, P.; Sánchez, D.O.; Docampo, R. Characterization of farnesylated protein tyrosine phosphatase TcPRL-1 from Trypanosoma cruzi. Eukaryot. Cell 2005, 4, 1550–1561. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nepomuceno-Silva, J.L.; Yokoyama, K.; De Mello, L.D.B.; Mendonça, S.M.; Paixaøo, J.C.; Baron, R.; Faye, J.C.; Buckner, F.S.; Van Voorhis, W.C.; Gelb, M.H.; et al. TcRho1, a farnesylated Rho family homologue from Trypanosoma cruzi. Cloning, trans-splicing, and prenylation studies. J. Biol. Chem. 2001, 276, 29711–29718. [Google Scholar] [CrossRef] [PubMed]
- Docampo, R.; de Boiso, J.F.; Stoppani, A.O.M. Bba 47498 tricarboxylic acid cycle operation at the kinetoplast- mitochondrion complex of Trypanosoma cruzi. Biochim. Biophys. Acta Bioenerg. 1978, 502, 466–476. [Google Scholar] [CrossRef]
- Parod, A.J.; Quesada-Allue, L.A. Protein glycosylation in Trypanosoma cruzi. I. Characterization of dolichol-bound monosaccharides and oligosaccharides synthesized in vivo. J. Biol. Chem. 1982, 257, 7637–7640. [Google Scholar]
- Ferella, M.; Montalvetti, A.; Rohloff, P.; Miranda, K.; Fang, J.; Reina, S.; Kawamukai, M.; Búa, J.; Nilsson, D.; Pravia, C.; et al. A solanesyl-diphosphate synthase localizes in glycosomes of Trypanosoma cruzi. J. Biol. Chem. 2006, 281, 39339–39348. [Google Scholar] [CrossRef]
- Docampo, R.; Moreno, S.N.; Turrens, J.F.; Katzin, A.M.; Gonzalez-Cappa, S.M.; Stoppani, A.O. Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1981, 3, 169–180. [Google Scholar] [CrossRef]
- Bouzahzah, B.; Jelicks, L.A.; Morris, S.A.; Weiss, L.M.; Tanowitz, H.B. Risedronate in the treatment of Murine Chagas’ disease. Parasitol. Res. 2005, 96, 184–187. [Google Scholar] [CrossRef]
- Dunford, J.E.; Thompson, K.; Coxon, F.P.; Luckman, S.P.; Hahn, F.M.; Poulter, C.D.; Ebetino, F.H.; Rogers, M.J. Structure-Activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J. Pharmacol. Exp. Ther. 2001, 296, 235–242. [Google Scholar]
- Urbina, J.A.; Moreno, B.; Vierkotter, S.; Oldfield, E.; Payares, G.; Sanoja, C.; Bailey, B.N.; Yan, W.; Scott, D.A.; Moreno, S.N.J.; et al. Trypanosoma cruzi contains major pyrophosphate stores, and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J. Biol. Chem. 1999, 274, 33609–33615. [Google Scholar] [CrossRef]
- Brown, D.L.; Robbins, R. Developments in the therapeutic applications of Bisphosphonates. J. Clin. Pharmacol. 1999, 39, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Szajnman, S.H.; Bailey, B.N.; Docampo, R.; Rodriguez, J.B. Bisphosphonates derived from fatty acids are potent growth inhibitors of Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 2001, 11, 789–792. [Google Scholar] [CrossRef]
- Demoro, B.; Rostán, S.; Moncada, M.; Li, Z.H.; Docampo, R.; Olea Azar, C.; Maya, J.D.; Torres, J.; Gambino, D.; Otero, L. Ibandronate metal complexes: Solution behavior and antiparasitic activity. J. Biol. Inorg. Chem. 2018, 23, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Gabelli, S.B.; McLellan, J.S.; Montalvetti, A.; Oldfield, E.; Docampo, R.; Amzel, L.M. Structure and mechanism of the farnesyl diphosphate synthase from Trypanosoma cruzi: Implications for drug design. Proteins Struct. Funct. Genet. 2006, 62, 80–88. [Google Scholar] [CrossRef]
- Sanders, J.M.; Song, Y.; Chan, J.M.W.; Zhang, Y.; Jennings, S.; Kosztowski, T.; Odeh, S.; Flessner, R.; Schwerdtfeger, C.; Kotsikorou, E.; et al. Pyridinium-1-yl bisphosphonates are potent inhibitors of farnesyl diphosphate synthase and bone resorption. J. Med. Chem. 2005, 48, 2957–2963. [Google Scholar] [CrossRef]
- Demoro, B.; Caruso, F.; Rossi, M.; Benítez, D.; Gonzalez, M.; Cerecetto, H.; Otero, L. Risedronate metal complexes potentially active against Chagas disease. J. Inorg. Biochem. 2010, 104, 1252–1258. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Chatelain, E.; Kim, H.A.; Siqueira-Neto, J.L. Visceral leishmaniasis treatment: What do we have, what do we need and how to deliver it? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 11–19. [Google Scholar] [CrossRef]
- Lago, J.; Silva, J.A.; Borja, L.; Fraga, D.B.M.; Schriefer, A.; Arruda, S.; Lago, E.; Carvalho, E.M.; Bacellar, O. Clinical and histopathologic features of canine tegumentary leishmaniasis and the molecular characterization of Leishmania braziliensis in dogs. PLoS Negl. Trop. Dis. 2019, 13. [Google Scholar] [CrossRef]
- Lainson, R.; Shaw, J.J. Leishmaniasis of the New World: Taxonomic Problems. Br. Med. Bull. 1972, 28, 44–48. [Google Scholar] [CrossRef]
- World Health Organization. Leishmaniasis. Available online: https://www.who.int/leishmaniasis/en/ (accessed on 30 May 2020).
- Akhoundi, M.; Downing, T.; Votýpka, J.; Kuhls, K.; Lukeš, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef]
- Porcino, G.N.; Carvalho, K.S.S.; Braz, D.C.; Costa Silva, V.; Costa, C.H.N.; Santos, I.K.F.M. Evaluation of methods for detection of asymptomatic individuals infected with Leishmania infantum in the state of Piauí, Brazil. PLoS Negl. Trop. Dis. 2019, 13, e0007493. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Trinconi, C.T.; Miguel, D.C.; Silber, A.M.; Brown, C.; Mina, J.G.M.; Denny, P.W.; Heise, N.; Uliana, S.R.B. Tamoxifen inhibits the biosynthesis of inositolphosphorylceramide in Leishmania. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Weina, P.J.; Neafie, R.C.; Wortmann, G.; Polhemus, M.; Aronson, N.E. Old world leishmaniasis: An emerging infection among deployed US military and civilian workers. Clin. Infect. Dis. 2004, 39, 1674–1680. [Google Scholar] [CrossRef]
- Kreston, R. Discover Magazine Science for the Curious, 2011. Available online: https://www.discovermagazine.com/health/behind-enemy-lines-cutaneous-leishmaniasis-in-returning-us-troops-from-the-middle-east (accessed on 30 May 2020).
- Defense Base Act Compensation Blog—The Modern Day DBA Casualty. Available online: https://defensebaseactcomp.wordpress.com/2010/05/27/leishmaniasis-a-family-affair/ (accessed on 30 May 2020).
- Clarke, C.F.; Bradley, K.K.; Wright, J.H.; Glowicz, J. Case report: Emergence of autochthonous cutaneous leishmaniasis in northeastern Texas and southeastern Oklahoma. Am. J. Trop. Med. Hyg. 2013, 88, 157–161. [Google Scholar] [CrossRef]
- Beaumier, C.M.; Gomez-Rubio, A.M.; Hotez, P.J.; Weina, P.J. United States Military Tropical Medicine: Extraordinary Legacy, Uncertain Future. PLoS Negl. Trop. Dis. 2013, 7, 2448–2454. [Google Scholar] [CrossRef]
- Menezes, J.P.B.; Guedes, C.E.S.; Petersen, A.L.d.O.A.; Fraga, D.B.M.; Veras, P.S.T. Advances in development of new treatment for leishmaniasis. BioMed Res. Int. 2015, 2015, 15–18. [Google Scholar] [CrossRef]
- Wijnant, G.J.; Van Bocxlaer, K.; Yardley, V.; Harris, A.; Alavijeh, M.; Silva-Pedrosa, R.; Antunes, S.; Mauricio, I.; Murdan, S.; Croft, S.L. Comparative efficacy, toxicity and biodistribution of the liposomal amphotericin B formulations Fungisome® and AmBisome® in murine cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 223–228. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, A. Recent developments and future prospects in the treatment of visceral leishmaniasis. Ther. Adv. Infect. Dis. 2016, 3, 98–109. [Google Scholar] [CrossRef]
- Dos Santos Nogueira, F.; Avino, V.C.; Galvis-Ovallos, F.; Pereira-Chioccola, V.L.; Moreira, M.A.B.; Romariz, A.P.P.L.; Molla, L.M.; Menz, I. Use of miltefosine to treat canine visceral leishmaniasis caused by Leishmania infantum in Brazil. Parasites Vectors 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Resources for Health Professionals. Available online: https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html (accessed on 30 May 2020).
- Copeland, N.K.; Aronson, N.E. Leishmaniasis: Treatment updates and clinical practice guidelines review. Curr. Opin. Infect. Dis. 2015, 28, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Alcântara, L.M.; Ferreira, T.C.S.; Gadelha, F.R.; Miguel, D.C. Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Gadelha, A.P.R.; Brigagao, C.M.; da Silva, M.B.; Rodrigues, A.B.M.; Guimarães, A.C.R.; Paiva, F.; de Souza, W.; Henriques, C. Insights about the structure of farnesyl diphosphate synthase (FPPS) and the activity of bisphosphonates on the proliferation and ultrastructure of Leishmania and Giardia. Parasites Vectors 2020, 13, 168. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.L.; Guo, K.; Dunford, J.E.; Wu, X.; Knapp, S.; Ebetino, F.H.; Rogers, M.J.; Russell, R.G.G.; Oppermann, U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 7829–7834. [Google Scholar] [CrossRef]
- Huang, C.H.; Gabelli, S.B.; Docampo, R.; Oldfield, E.; Amzel, L.M. Structural basis for bisphosphonate inhibition of T-cruzi farnesyl diphosphate synthase. Biophys. J. 2007, 317–318. [Google Scholar]
- Tarshis, L.C.; Yan, M.; Poulter, C.D.; Sacchettini, J.C. Crystal structure of recombinant farnesyl diphosphate synthase at 2.6-.ANG. Resolution. Biochemistry 1994, 33, 10871–10877. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco Santos, J.C.; de Melo, J.A.; Maheshwari, S.; de Medeiros, W.M.T.Q.; de Freitas Oliveira, J.W.; Moreno, C.J.; Mario Amzel, L.; Gabelli, S.B.; Sousa Silva, M. Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs. Molecules 2020, 25, 2602. https://doi.org/10.3390/molecules25112602
Branco Santos JC, de Melo JA, Maheshwari S, de Medeiros WMTQ, de Freitas Oliveira JW, Moreno CJ, Mario Amzel L, Gabelli SB, Sousa Silva M. Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs. Molecules. 2020; 25(11):2602. https://doi.org/10.3390/molecules25112602
Chicago/Turabian StyleBranco Santos, Joice Castelo, Jonathas Alves de Melo, Sweta Maheshwari, Wendy Marina Toscano Queiroz de Medeiros, Johny Wysllas de Freitas Oliveira, Cláudia Jassica Moreno, L. Mario Amzel, Sandra B. Gabelli, and Marcelo Sousa Silva. 2020. "Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs" Molecules 25, no. 11: 2602. https://doi.org/10.3390/molecules25112602
APA StyleBranco Santos, J. C., de Melo, J. A., Maheshwari, S., de Medeiros, W. M. T. Q., de Freitas Oliveira, J. W., Moreno, C. J., Mario Amzel, L., Gabelli, S. B., & Sousa Silva, M. (2020). Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs. Molecules, 25(11), 2602. https://doi.org/10.3390/molecules25112602