
An Efficient Greener Approach for N-acylation of Amines in Water Using Benzotriazole Chemistry

 , ,
, ,  , ,
, ,
Abstract
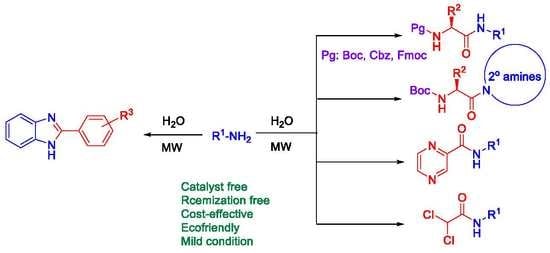
1. Introduction
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. General Methods for N-acylation
4.2. General Methods for Preparation of 2-Substituted Benzimidazoles
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Savanur, H.M.; Malunavar, S.S.; Prabhala, P.; Sutar, S.M.; Kalkhambkar, R.G.; Laali, K.K. Synthesis of diverse libraries of carboxamides via chemoselective N-acylation of amines by carboxylic acids employing Brønsted acidic IL [BMIM(SO3H)][OTf]. Tetrahedron Lett. 2019, 60, 151159. [Google Scholar] [CrossRef]
- Humphrey, J.M.; Chamberlin, A.R. Chemical Synthesis of Natural Product Peptides: Coupling Methods for the Incorporation of Noncoded Amino Acids into Peptides. Chem. Rev. 1997, 97, 2243–2266. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and antiviral sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Holzer, W.; Olofsson, B. Increasing the Reactivity of Amides towards Organometallic Reagents: An Overview. Adv. Synth. Catal. 2014, 356, 3697–3736. [Google Scholar] [CrossRef]
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Material Science; Wiley- Interscience: New York, NY, USA, 2000. [Google Scholar]
- Panda, S.S.; Girgis, A.S.; Mishra, B.B.; Elagawany, M.; Devarapalli, V.; Littlefield, W.F.; Samir, A.; Fayad, W.; Fawzy, N.G.; Srour, A.M.; et al. Synthesis, computational studies, antimycobacterial and antibacterial properties of pyrazinoic acid–isoniazid hybrid conjugates. RSC Adv. 2019, 9, 20450–20462. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. β-Lactams: Versatile building blocks for the stereoselective synthesis of non-β-Lactam products. Chem. Rev. 2007, 107, 4437–4492. [Google Scholar] [CrossRef] [PubMed]
- Roughley, S.D.; Jordan, A.M. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
- Ishihara, K. Dehydrative condensation catalyses. Tetrahedron 2009, 65, 1085–1109. [Google Scholar] [CrossRef]
- Lanigan, R.M.; Sheppard, T.D. Recent developments in amide synthesis: Direct amidation of carboxylic acids and transamidation reactions. Eur. J. Org. Chem. 2013, 2013, 7453–7465. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef]
- Sabatini, M.T.; Boulton, L.T.; Sneddon, H.F.; Sheppard, T.D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10–17. [Google Scholar] [CrossRef]
- Wang, X. Challenges and outlook for catalytic direct amidation reactions. Nat. Catal. 2019, 2, 98–102. [Google Scholar] [CrossRef]
- Rathi, J.O.; Shankarling, G.S. Concentrated solar radiation aided energy efficient and chemoselective protocol for N-acylation and N-formylation reactions in aqueous medium. Sol. Energy 2019, 189, 471–479. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.S.; Laffan, D.; Thomson, C.; Williams, M.T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Brenner, S. An active amide group in the molecule of drugs that induce pemphigus: A casual or causal relationship? Dermatology 1994, 189, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Rajput, P.; Sharma, A. Synthesis and biological importance of amide analogues. J. Pharmacol. Med. Chem. 2018, 2, 22–31. [Google Scholar]
- Katritzky, A.R.; He, H.-Y.; Suzuki, K. N-Acylbenzotriazoles: Neutral Acylating Reagents for the Preparation of Primary, Secondary, and Tertiary Amides. J. Org. Chem. 2000, 65, 8210–8213. [Google Scholar] [CrossRef]
- Mali, S.M.; Bhaisare, R.D.; Gopi, H.N. Thioacids Mediated Selective and Mild N-Acylation of Amines. J. Org. Chem. 2013, 78, 5550–5555. [Google Scholar] [CrossRef]
- Panda, S.S.; Dennis Hall, C.; Scriven, E.; Katritzky, A.R. Aminoacyl benzotriazolides: Versatile reagents for the preparation of peptides, their mimetics and conjugates. Aldrichimica Acta. 2013, 46, 43–55. [Google Scholar]
- Allen, C.L.; Williams, J.M.J. Metal-catalyzed approaches to amide bond formation. Chem. Soc. Rev. 2011, 40, 3405–3415. [Google Scholar] [CrossRef] [PubMed]
- Cossy, J.; Palegrosdemange, C. Convenient synthesis of amides from carboxylic acids and primary amines. Tetrahedron Lett. 1989, 30, 2771–2774. [Google Scholar] [CrossRef]
- Spivey, A.C.; Arseniyadis, S. Nucleophilic catalysis by 4-(dialkylamino)pyridines revisited - The search for optimal reactivity and selectivity. Chem. Int. Ed. 2004, 43, 5436–5441. [Google Scholar] [CrossRef]
- Taylor, J.E.; Jones, M.D.; Williams, J.M.J.; Bull, S.D. N-Acyl DBN tetraphenylborate salts as N-acylating agents. J. Org. Chem. 2012, 77, 2808–2818. [Google Scholar] [CrossRef] [PubMed]
- Owston, N.A.; Parker, A.J.; Williams, J.M.J. Highly efficient ruthenium-catalyzed oxime to amide rearrangement. Org. Lett. 2007, 9, 3599–3601. [Google Scholar] [CrossRef]
- Ramalingan, C.; Park, Y.-T. Mercury-Catalyzed Rearrangement of Ketoximes into Amides and Lactams in Acetonitrile. J. Org. Chem. 2007, 72, 4536–4538. [Google Scholar] [CrossRef]
- Yoo, W.J.; Li, C.J. Highly Efficient Oxidative Amidation of Aldehydes with Amine Hydrochloride Salts. J. Am. Chem. Soc. 2006, 128, 13064–13065. [Google Scholar] [CrossRef]
- Ekoue-Kovi, K.; Wolf, C. One-pot oxidative esterification and amidation of aldehydes. Chem. Eur. J. 2008, 14, 6302–6315. [Google Scholar] [CrossRef]
- Yang, X.; Birman, V.B. Acyl Transfer Catalysis with 1,2,4-Triazole Anion. Org. Lett. 2009, 11, 1499–1502. [Google Scholar] [CrossRef]
- Miller, S.J. In Search of Peptide-Based Catalysts for Asymmetric Organic Synthesis. Acc. Chem. Res. 2004, 37, 601–610. [Google Scholar] [CrossRef]
- Ishihara, K.; Kosugi, Y.; Akakura, M. Rational design of an L-histidine-derived minimal artificial acylase for the kinetic resolution of racemic alcohols. J. Am. Chem. Soc. 2004, 126, 12212–12213. [Google Scholar] [CrossRef] [PubMed]
- Seliem, I.A.; Panda, S.S.; Girgis, A.S.; Nagy, Y.I.; George, R.F.; Fayad, W.; Fawzy, N.G.; Ibrahim, T.S.; Al-Mahmoudy, A.M.M.; Sakhuja, R.; et al. Design, synthesis, antimicrobial, and DNA gyrase inhibitory properties of fluoroquinolone–dichloroacetic acid hybrids. Chem. Biol. Drug Des. 2020, 95, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Girgis, A.S.; Mishra, B.B.; Elagawany, M.; Devarapalli, V.; Littlefield, W.F.; Samir, A.; Fawzy, N.G.; Srour, A.M. Novel pyrazinoic acid-isoniazid conjugates with amino acid linker: Microwave assisted synthesis, anti-infective properties, and molecular modeling studies. RSC Adv. 2019, 9, 20450–20462. [Google Scholar]
- Panda, S.S.; Asiri, A.M.; Elagawany, M.; Buchanan, D.D.; Torkian, B.; Bathala, K.; Thomas, S.J.; Capito, J.E.; Arshad, M.N.; Al-Romaizan, A.N. Efficient synthesis of pyrazinoic acid hybrid conjugates. SynOpen 2017, 1, 50–58. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rachwal, S. Synthesis of Heterocycles Mediated by Benzotriazole. 1. Monocyclic Systems. Chem. Rev. 2010, 110, 1564–1610. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rachwal, S. Synthesis of Heterocycles Mediated by Benzotriazole. 2. Bicyclic Systems. Chem. Rev. 2011, 111, 7063–7120. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Yao, G.; Lan, X.; Zhao, X. The conversion of secondary into tertiary amides using benzotriazole methodology. J. Org. Chem. 1993, 58, 2086–2093. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Strah, S.; Belyakov, S.A. The preparation of functionalized amines and amides using benzotriazole derivatives and organozinc reagents. Tetrahedron 1998, 54, 7167–7178. [Google Scholar] [CrossRef]
- Katritzky, A.R.; El Khatib, M.; Bol’shakov, O.; Khelashvili, L.; Steel, P.J. Benzotriazol-1-yl-sulfonyl Azide for Diazotransfer and Preparation of Azidoacylbenzotriazoles. J. Org. Chem. 2010, 75, 6532–6539. [Google Scholar] [CrossRef]
- Küçükbay, F.Z.; Küçükbay, H.; Tanc, M.; Supuran, C.T. Synthesis and carbonic anhydrase inhibitory properties of amino acid–coumarin/quinolinone conjugates incorporating glycine, alanine and phenylalanine moieties. J. Enzyme Inhib. Med. Chem. 2016, 31, 1198–1202. [Google Scholar] [CrossRef]
- Laconde, G.; Amblard, M.; Martinez, J. Unexpected Reactivity of N-Acyl-Benzotriazoles with Aromatic Amines in Acidic Medium (ABAA Reaction). Eur. J. Org. Chem. 2019, 2019, 85–90. [Google Scholar] [CrossRef]
- Denny, W.A.; Rewcastle, G.W.; Bauley, B.C. Potential antitumor agents. 59. Structure-activity relationships for 2-phenylbenzimidazole-4-carboxamides, a new class of minimal DNA-intercalating agents which may not act via topoisomerase II. J. Med. Chem. 1990, 33, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Seyhan, E.; Sultan, N.; Nilgun, A.; Noyanalpan, N. Synthesis and antimicrobial activity of 5-substituted 1-dialkylaminomethyl-2-arylbenzimidazole derivatives. Arzneim.-Forsch. 1997, 47, 410–412. [Google Scholar]
- Gungor, T.; Fouquet, A.; Eulon, J.-M.; Provost, D.; Cazes, M.; Cloarec, A. Cardiotonic agents. Synthesis and cardiovascular properties of novel 2-arylbenzimidazoles and azabenzimidazoles. J. Med. Chem. 1992, 35, 4455–4463. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Neugebauer, A.; Klein, C.D. Metal-mediated inhibition of Escherichia coli methionine aminopeptidase: Structure-activity relationships and development of a novel scoring function for metal-ligand interactions. J. Med. Chem. 2006, 49, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Preston, P.N. Benzimidazoles and congeneric tricyclic compounds. In The Chemistry of Heterocyclic Compounds; Weissberger, A., Taylor, E.C., Eds.; Wiley: New York, NY, USA, 1981; Volume 40, pp. 6–60. [Google Scholar]
- Grimmett, M.R. Imidazoles and their benzo derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon: Oxford, England, 1984; Volume 5, pp. 457–487. [Google Scholar]
- Benincori, T.; Sannicolo, F. New benzimidazole synthesis. J. Heterocycl. Chem. 1988, 25, 1029–1033. [Google Scholar] [CrossRef]
- Tumelty, D.; Cao, K.; Holmes, C.P. Traceless solid-phase synthesis of substituted benzimidazoles via a base-cleavable linker. Org. Lett. 2001, 3, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, K.; Khodaei, M.M.; Naali, F. Mild and highly efficient method for the synthesis of 2-arylbenzimidazoles and 2-arylbenzothiazoles. J. Org. Chem. 2008, 73, 6835–6837. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Ibrahim, M.A.; Oliferenko, A.A.; Asiri, A.M.; Katritzky, A.R. Catalyst-free facile synthesis of 2-substituted benzothiazoles. Green Chem. 2013, 15, 2709–2712. [Google Scholar] [CrossRef]
- Panda, S.S.; Jain, S.C. A greener protocol for the highly efficient synthesis of spiro-indoles via Schiff’s bases. Monatsh. Chem. 2012, 143, 1187–1194. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Panda, S.S.; Alamry, K.A.; Katritzky, A.R. Green, Catalyst-Free Synthesis of Mesalazine Conjugates. Synthesis 2013, 45, 3255–3258. [Google Scholar]
- Meegalla, S.K.; Wall, M.J.; Chen, J.; Wilson, K.J.; Ballentine, S.K.; Desjarlais, R.L.; Schubert, C.; Crysler, C.S.; Chen, Y.; Molloy, C.J.; et al. Structure-based optimization of a potent class of arylamide FMS inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3632–3637. [Google Scholar] [CrossRef]
- Talanian, R.V.; Quinlan, C.; Trautz, S.; Hackett, M.C.; Mankovich, J.A.; Banach, D.; Ghayur, T.; Brady, K.D.; Wong, W.W. Substrate specificities of caspase family proteas. J. Biol. Chem. 1997, 272, 9677–9682. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Tsuda, Y.; Li, T.; Motoyama, T.; Takahashi, M.; Shimizu, Y.; Yokoi, T.; Sasaki, Y.; Ambo, A.; Kita, A.; et al. Development of Potent Bifunctional Endomorphin-2 Analogues with Mixed μ-/δ-Opioid Agonist and δ-Opioid Antagonist Properties. J. Med. Chem. 2004, 47, 3591–3599. [Google Scholar] [CrossRef] [PubMed]
- Pelagatti, P.; Carcelli, M.; Calbiani, F.; Cassi, C.; Elviri, L.; Pelizzi, C.; Rizzotti, U.; Rogolino, D. Transfer Hydrogenation of Acetophenone Catalyzed by Half-Sandwich Ruthenium(II) Complexes Containing Amino Amide Ligands. Detection of the Catalytic Intermediates by Electrospray Ionization Mass Spectrometry. Organometallics 2005, 24, 5836–5844. [Google Scholar] [CrossRef]
- Geurts, M.; Poupaert, J.H.; Scriba, G.K.E.; Lambert, D.M. N-(Benzyloxycarbonyl)Glycine Esters and Amides as New Anticonvulsants. J. Med. Chem. 1998, 41, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Dong, K.; Li, M. Visible Light-Induced Amide Bond Formation. Org. Lett. 2020, 22, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Elmore, D.T.; Toseland, P.A. Degradative Studies on Peptides and Proteins. IV. Formation of Salts of 2-Acylaminothiazol-5-Ones by Acid-Catalyzed Degradation of N-Acylthiocarbamoylpeptides and Their Behavior towards Nucleophilic Reagents. J. Chem. Soc. 1957, 2460–2466. [Google Scholar] [CrossRef]
- Tortorella, V.; Bettoni, G. Optical Rotatory Dispersion Curves of N-(N-Oxido-2-Pyridyl)Amino Compounds. IV. Amino Amides. Gazz. Chim. Ital. 1968, 98, 324–330. [Google Scholar]
- Nuijens, T.; Cusan, C.; Kruijtzer, J.A.W.; Rijkers, D.T.S.; Liskamp, R.M.J.; Quaedflieg, P.J.L.M. Enzymatic Synthesis of C-Terminal Arylamides of Amino Acids and Peptides. J. Org. Chem. 2009, 74, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, K.; Kubo, K.; Igarashi, T.; Sakurai, T. Electron Transfer-Initiated Asymmetric Photocyclization of Chiral Auxiliary-Substituted N-Acyl-α-Dehydro(1-Naphthyl)Alaninamides to the Corresponding 3,4-Dihydrobenzo[f]Quinolinone Derivatives. Tetrahedron 2005, 61, 11211–11224. [Google Scholar] [CrossRef]
- Abernethy, J.L.; Howell, F.G.; Ledesma, A.; Doose, D.; Everett, R. Behavior of Amides of N-Acylamino Acids toward Aniline and Substituted Anilines under Papain Catalysis. Tetrahedron 1975, 31, 2659–2662. [Google Scholar] [CrossRef]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Hogan, F.; Bai, R.; Chapuis, J.-C.; et al. Antineoplastic agents 365. Dolastatin 10 SAR probes. Anticancer. Drug Des. 1998, 13, 243–277. [Google Scholar] [PubMed]
- Lampariello, L.R.; Peruzzi, D.; Sega, A.; Taddei, M. Some Observations on the Preparation of Amides and Their Reduction to Amines in Ionic Liquids. Lett. Org. Chem. 2005, 2, 265–270. [Google Scholar] [CrossRef]
- Elzein, E.; Ibrahim, P.; Palle, V.; Rehder, K.; Zablocki, J.A. 1-akan-2-ol substituted piperazine and piperidine compounds. PCT Int Appl. Patent WO2005061470A1, 7 July 2005. [Google Scholar]
- Pfleiderer, G.; Celliers, P.G.; Stanulovic, M.; Wachsmuth, E.D.; Determann, H.; Braunitzer, G. Properties and Analytical Application of the Kidney-Particle Aminopeptidase. Biochem. Z. 1964, 340, 552–564. [Google Scholar]
- Antoine, M.; Marchand, P.; Le Baut, G.; Czech, M.; Baasner, S.; Gunther, E. Side Chain Modifications of (Indol-3-Yl)Glyoxamides as Antitumor Agents. J. Enzyme Inhib. Med. Chem. 2008, 23, 686–695. [Google Scholar] [CrossRef][Green Version]
- Orsy, G.; Fülöp, F.; Mándity, I.M. N-Acetylation of Amines in Continuous-Flow with Acetonitrile—No Need for Hazardous and Toxic Carboxylic Acid Derivatives. Molecules 2020, 25, 1985. [Google Scholar] [CrossRef]
- Dolezal, M.; Cmedlova, P.; Palek, L.; Vinsova, J.; Kunes, J.; Buchta, V.; Jampilek, J.; Kralova, K. Synthesis and Antimycobacterial Evaluation of Substituted Pyrazinecarboxamides. Eur. J. Med. Chem. 2008, 43, 1105–1113. [Google Scholar] [CrossRef]
- Robba, M. Certain Derivatives of Diazines. I. Synthesis of Nitriles of Diazines. Ann. Chim. 1960, 5, 351–379. [Google Scholar]
- Yang, Y.; Shang, P.; Cheng, C.; Wang, D.; Yang, P.; Zhang, F.; Li, T.; Lu, A.; Zhao, Y. Novel N-Phenyl Dichloroacetamide Derivatives as Anticancer Reagents: Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2010, 45, 4300–4306. [Google Scholar] [CrossRef]
- Panda, S.S.; Jain, S.C. Synthesis of 2-Arylbenzimidazoles in Water. Synth. Commun. 2011, 41, 729–735. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| Entry | Reaction Temp. (°C) | Reaction Time | Yield a (%) |
| 1 | 20 (Room temp.) | 1 h | 74 |
| 2 | 20 (Room temp.) | 2 h | 86 |
| 3 | 50 (Conv.) | 30 min | 77 |
| 4 | 50 (Conv.) | 1 h | 82 |
| 5 | 70 (MW) | 15 min | 96 |
| Entry | R-NH2 | Product | Yield (%) | Mp (°C) |
|---|---|---|---|---|
| 1 |  |  | 94 | 137–139 |
| 2 |  |  | 93 | 123–125 |
| 3 |  |  | 95 | 145–146 |
| 4 |  |  | 91 | 165–167 |
| 5 |  |  | 95 | 118–120 |
| 6 |  |  | 88 | 152–154 |
| 7 |  |  | 96 | 158–160 |
| 8 |  |  | 92 | 152–154 |
| 9 |  |  | 97 | 183–185 |
| 10 |  |  | 79 | 185–187 |
| 11 |  |  | 94 | 193–195 |
| 12 |  |  | 75 | 172–174 |
| 13 |  |  | 80 | 164–166 |
| 14 |  |  | 85 | 160–162 |
| 15 |  |  | 98 | 185–187 |
| 16 |  |  | 94 | 138–140 |
| 17 |  |  | 77 | 191–193 |
| 18 |  |  | 84 | 174–176 |
| 19 |  |  | 90 | 192–194 |
| 20 |  |  | 90 | 142–143 |
| 21 |  |  | 98 | 152–154 |
| 22 |  |  | 95 | 160–162 |
| 23 |  |  | 90 | 182–184 |
| 24 |  |  | 73 | 163–165 |
| 25 |  |  | 82 | 150–152 |
| 26 |  |  | 90 | 197–199 |
| 27 |  |  | 98 | 210–212 |
| 28 |  |  | 70 | 162–164 |
| 29 |  |  | 70 | 95–97 |
| 30 |  |  | 90 | 180–182 |
| 31 |  |  | 89 | 152–154 |
| 32 |  |  | 97 | 155–157 |
| 33 |  |  | 71 | 138–140 |
| 34 |  |  | 81 | 147–149 |
| 35 |  |  | 98 | 155–157 |
| 36 |  |  | 85 | 157–159 |
| 37 |  |  | 80 | 130–132 |
| 38 |  |  | 80 | 143–145 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, T.S.; Seliem, I.A.; Panda, S.S.; Al-Mahmoudy, A.M.M.; Abdel-Samii, Z.K.M.; Alhakamy, N.A.; Asfour, H.Z.; Elagawany, M. An Efficient Greener Approach for N-acylation of Amines in Water Using Benzotriazole Chemistry. Molecules 2020, 25, 2501. https://doi.org/10.3390/molecules25112501
Ibrahim TS, Seliem IA, Panda SS, Al-Mahmoudy AMM, Abdel-Samii ZKM, Alhakamy NA, Asfour HZ, Elagawany M. An Efficient Greener Approach for N-acylation of Amines in Water Using Benzotriazole Chemistry. Molecules. 2020; 25(11):2501. https://doi.org/10.3390/molecules25112501
Chicago/Turabian StyleIbrahim, Tarek S., Israa A. Seliem, Siva S. Panda, Amany M. M. Al-Mahmoudy, Zakaria K. M. Abdel-Samii, Nabil A. Alhakamy, Hani Z. Asfour, and Mohamed Elagawany. 2020. "An Efficient Greener Approach for N-acylation of Amines in Water Using Benzotriazole Chemistry" Molecules 25, no. 11: 2501. https://doi.org/10.3390/molecules25112501
APA StyleIbrahim, T. S., Seliem, I. A., Panda, S. S., Al-Mahmoudy, A. M. M., Abdel-Samii, Z. K. M., Alhakamy, N. A., Asfour, H. Z., & Elagawany, M. (2020). An Efficient Greener Approach for N-acylation of Amines in Water Using Benzotriazole Chemistry. Molecules, 25(11), 2501. https://doi.org/10.3390/molecules25112501