New Quinolone-Based Thiosemicarbazones Showing Activity Against Plasmodium falciparum and Mycobacterium tuberculosis

 , , , and
, , , and
Abstract
1. Introduction
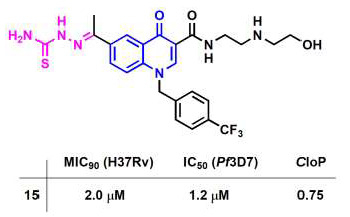
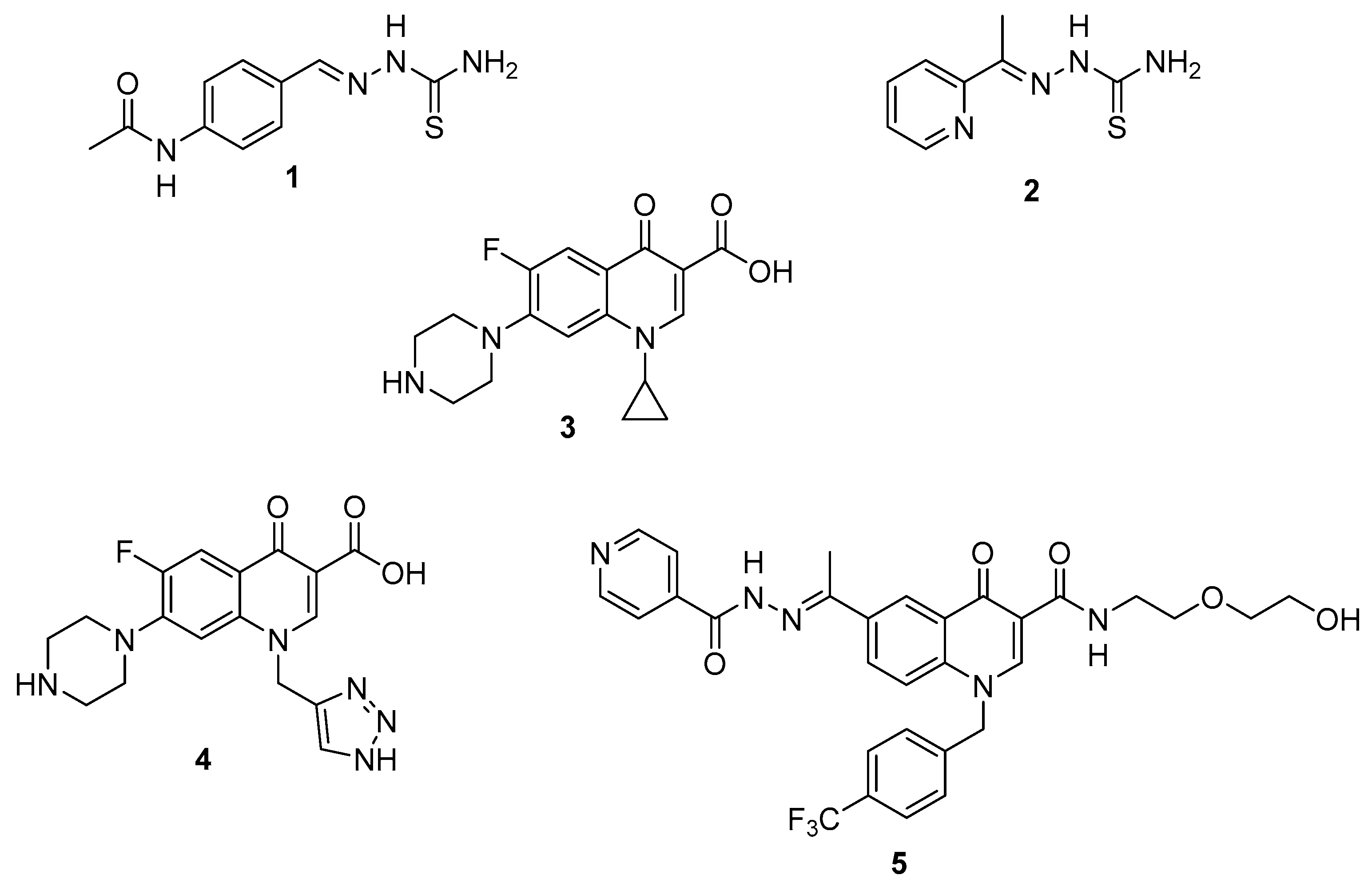
2. Results and Discussion
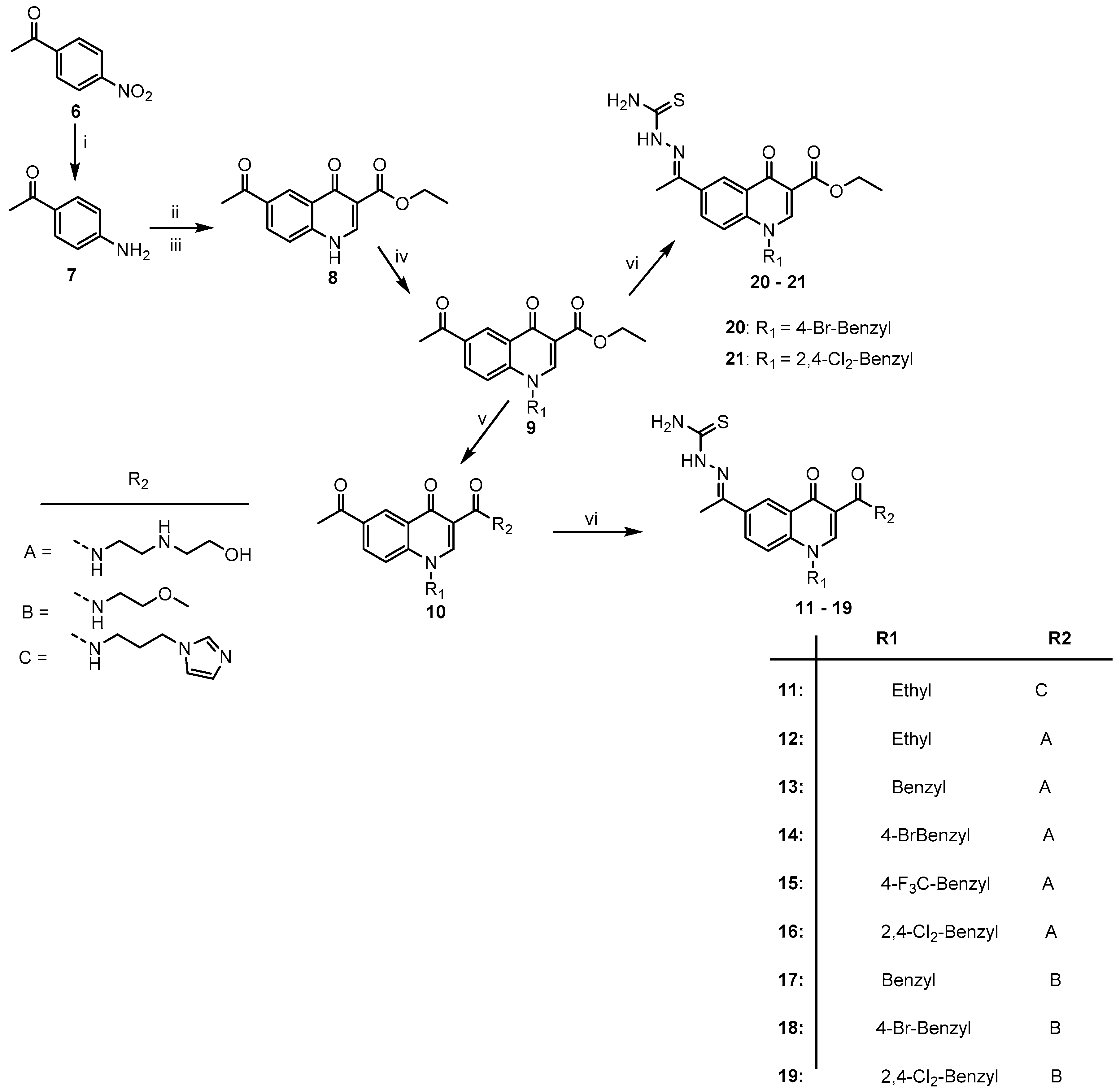
2.1. Chemistry
2.2. Pharmacology
3. Materials and Methods
3.1. General Information
3.2. General Synthetic Procedure for the Quinolone-Thiosemicarbazone Derivatives, 11–21
3.3. In Vitro Anti-Plasmodial Assay
3.4. In Vitro Cytotoxicity Assay
3.5. In Vitro Antimycobacterial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Delogu, G.; Sali, M.; Fadda, G. The biology of Mycobacterium tuberculosis infection. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013070. [Google Scholar] [CrossRef]
- Bañuls, A.L.; Sanou, A.; Anh, N.T.; Godreuil, S. Mycobacterium tuberculosis: Ecology and evolution of a human bacterium. J. Med. Microbiol. 2015, 64, 1261–1269. [Google Scholar] [CrossRef]
- Ahmad, S. Pathogenesis, immunolog, and diagnosis of latent Mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2011, 2011, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.-A.L. The end of the binary era: Revisiting the spectrum of tuberculosis. J. Immunol. 2018, 201, 2541–2548. [Google Scholar] [CrossRef]
- Ai, J.-W.; Ruan, Q.-L.; Liu, Q.-H.; Zhang, W.-H. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg. Microbes Infect. 2016, 5, e10. [Google Scholar] [CrossRef] [PubMed]
- Dargie, B.; Tesfaye, G.; Worku, A. Prevalence and associated factors of undernutrition among adult tuberculosis patients in some selected public health facilities of Addis Ababa, Ethiopia: A cross-sectional study. BMC Nutr. 2016, 2, 7. [Google Scholar] [CrossRef]
- Cudahy, P.; Shenoi, S. Diagnostics for pulmonary tuberculosis. Postgrad. Med. J. 2016, 92, 187–193. [Google Scholar] [CrossRef]
- Churchyard, G.; Kim, P.; Shah, N.; Rustomjee, R.; Gandhi, N.; Mathema, B.; Dowdy, D.; Kasmar, A.; Cardenas, V. What we know about tuberculosis transmission: An overview. J. Infect. Dis. 2017, 216, S629–S635. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2018; WHO: Geneva, Switzerland, 2018; ISBN 978-92-4-156564-6. [Google Scholar]
- Lohrasbi, V.; Talebi, M.; Bialvaei, A.; Fattorini, L.; Drancourt, M.; Heidary, M.; Darban-Sarokhalil, D. Trends in the discovery of new drugs for Mycobacterium tuberculosis therapy with a glance at resistance. Tuberculosis 2018, 109, 17–27. [Google Scholar] [CrossRef]
- Auld, S.C.; Shah, N.S.; Mathema, B.; Brown, T.S.; Ismail, N.; Omar, S.V.; Brust, J.C.; Nelson, K.; Allana, S.; Campbell, A.; et al. XDR tuberculosis in South Africa: Genomic evidence supporting transmission in communities. Eur. Respir. J. 2018, 52, 1800246. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2017; WHO: Geneva, Switzerland, 2017; pp. 32–43. [Google Scholar]
- WHO. World Malaria Report 2015; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Gomes, A.P.; Vitorino, R.R.; Costa, A.; de Mendonça, E.; Oliveira, M.; Siqueira-Batista, R. Severe Plasmodium falciparum malaria. Rev. Bras. Ter. Intensiva 2011, 23, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Olliaro, P. Mortality associated with severe Plasmodium falciparum malaria increases with age. Clin. Infect. Dis. 2008, 47, 158–160. [Google Scholar] [CrossRef]
- Snow, R.S. Global malaria eradication and the importance of Plasmodium falciparum epidemiology in Africa. BMC Med. 2015, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Salmanzadeh, S.; Foroutan-Rad, M.; Khademvatan, S.; Moogahi, S.; Bigdeli, S. Significant decline of malaria incidence in southwest of Iran (2001–2014). J. Trop. Med. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hay, S.I.; Guerra, C.A.; Tatem, A.J.; Atkinson, P.M.; Snow, R.W. Urbanization, malaria transmission and disease burden in Africa. Nat. Rev. Microbiol. 2005, 3, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.; Rizk, S.; Njimoh, D.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef]
- Mita, T.; Tanabe, K. Evolution of Plasmodium falciparum drug resistance: Implications for the development and containment of artemisinin resistance. Jpn. J. Infect. Dis. 2012, 65, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Pousibet-Puerto, J.; Salas-Coronas, J.; Sánchez-Crespo, A.; Molina-Arrebola, A.; Soriano-Pérez, M.; Giménez-López, M.; Vázquez-Villegas, J.; Cabezas-Fernández, M. Impact of using artemisinin-based combination therapy (ACT) in the treatment of uncomplicated malaria from Plasmodium falciparum in a non-endemic zone. Malar. J. 2016, 15, 339. [Google Scholar] [CrossRef]
- Pasvol, G. The treatment of complicated and severe malaria. Br. Med. Bull. 2005, 75–76, 29–47. [Google Scholar] [CrossRef]
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.; Rosenthal, P. Antimalarial drug resistance: Literature review and activities and findings of the ICEMR network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef]
- Marriner, G.A.; Nayyar, A.; Uh, E.; Wong, S.; Mukherjee, T.; Via, L.; Carroll, M.; Edwards, R.; Gruber, T.; Choi, I.; et al. The medicinal chemistry of tuberculosis chemotherapy. Top. Med. Chem. 2011, 7, 47–124. [Google Scholar]
- Li, X.-X.; Zhou, X.-N. Co-infection of tuberculosis and parasitic diseases in humans: A systematic review. Parasit. Vectors 2013, 6, 79. [Google Scholar] [CrossRef]
- Murphy, M.E.; Singh, K.P.; Laurenzi, M.; Brown, M.; Gillespie, S.H. Managing malaria in tuberculosis patients on fluoroquinolone-containing regimens: Assessing the risk of QT prolongation. Int. J. Tuberc. Lung Dis. 2012, 16, 144–149. [Google Scholar] [CrossRef]
- Grzegorzewicz, A.E.; Korduláková, J.; Jones, V.; Born, S.; Belardinelli, J.; Vaquié, A.; Gundi, V.; Madacki, J.; Slama, N.; Laval, F.; et al. A Common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by Isoxyl and Thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Mackey, Z.; Hansell, E.; Doyle, P.; Gut, J.; Caffrey, C.R.; Lehman, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and structure activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei and Trypanasoma cruzi. J. Med. Chem. 2004, 47, 3212–3219. [Google Scholar] [CrossRef]
- Akhtar, R.; Yousaf, M.; Naqvi, S.; Irfan, M.; Zahoor, A.; Hussain, A.; Chatha, S. Synthesis of ciprofloxacin-based compounds: A review. Syn. Commun. 2016, 46, 1849–1879. [Google Scholar] [CrossRef]
- Dixit, S.; Mishra, N.; Sharma, M.; Singh, S.; Agarwal, A.; Awasthi, S.; Bhasin, V. Synthesis and in vitro antiplasmodial activities of fluoroquinolone analogs. Eur. J. Med. Chem. 2012, 51, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Beteck, R.M.; Seldon, R.; Jordaan, A.; Warner, D.F.; Hoppe, H.C.; Laming, D.; Legoabe, L.J.; Khanye, S.D. Quinolone-isoniazid hybrids: Synthesis and preliminary in vitro cytotoxicity and anti-tuberculosis evaluation. Medchemcomm 2019, 10, 326–331. [Google Scholar] [CrossRef]
- Neelarapu, R.; Maignan, J.; Lichorowic, C.; Monastyrskyi, A.; Mutka, T.; LaCrue, A.; Blake, L.; Casandra, D.; Mashkouri, S.; Burrows, J.; et al. Design and synthesis of orally bioavailable piperazine substituted 4(1H)-quinolones with potent antimalarial activity: Structure-activity and structure-property relationship studies. J. Med. Chem. 2018, 61, 1450–1473. [Google Scholar] [CrossRef]
- Venkatachalam, T.K.; Pierens, G.K.; Reutens, D.C. Synthesis, NMR structural characterization and molecular modeling of substituted thiosemicarbazones and semicarbazones using DFT calculations to prove the syn/anti isomer formation. Magn. Reson. Chem. 2014, 52, 98–105. [Google Scholar] [CrossRef]
- Beteck, R.M.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Sysnthesis, in vitro cytotoxicity and trypanocidal evaluation of novel 1,3,6-substituted non-fluorquinolones. S. Afr. J. Chem. 2018, 71, 188–195. [Google Scholar] [CrossRef]
- Gumbo, M.; Beteck, R.M.; Mandizvo, T.; Seldon, R.; Warner, D.F.; Hoppe, H.C.; Isaacs, M.; Laming, D.; Tam, C.C.; Cheng, L.W.; et al. Cinnamoyl-oxaborole amides: Synthesis and their in vitro biological activity. Molecules 2018, 23, 2038. [Google Scholar] [CrossRef] [PubMed]
- Mbaba, M.; Mabhula, A.N.; Boelb, N.; Edkinsb, A.L.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Ferrocenyl and organic novobiocin derivatives: Synthesis and their in vitro biological activity. J. Inorg. Biochem. 2017, 172, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Oderinlo, O.O.; Tukulula, M.; Isaacs, M.; Hoppe, H.C.; Taylor, D.; Smith, V.J.; Khanye, S.D. New thiazolidine-2,4-dione derivatives combined with organometallic ferrocene: Synthesis, structure and antiparasitic activity. Appl. Organomet. Chem. 2018, 32, e4385. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Cox, J.G.; Spivey, V.L.; Loman, N.J.; Pallen, M.J.; Constantinindou, C.; Fernández, R.; Alemparte, C.; Remuñuinán, M.J.; Barros, D.; et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 2012, 7, e52951. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E., III. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. PNAS 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Compound | MIC90 (μM) | IC50 (μM) | ClogP a |
|---|---|---|---|
| H37Rv | Pf 3D7 | ||
| 11 | ˃125 | 3.7 | −0.02 |
| 12 | 73.4 | na | −1.17 |
| 13 | 2.7 | 4.1 | 0.18 |
| 14 | 10.3 | 4.7 | 0.95 |
| 15 | 2.0 | 1.2 | 0.75 |
| 16 | 4.8 | 3.1 | 1.38 |
| 17 | 44.2 | 24.6 | 1.07 |
| 18 | 15.0 | 9.9 | 1.84 |
| 19 | 10.2 | na | 2.27 |
| 20 | 102.5 | na | 2.55 |
| 21 | 31.6 | na | 3.52 |
| CQ | - | 0.012 | - |
| RF | 0.062 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beteck, R.M.; Seldon, R.; Jordaan, A.; Warner, D.F.; Hoppe, H.C.; Laming, D.; Khanye, S.D. New Quinolone-Based Thiosemicarbazones Showing Activity Against Plasmodium falciparum and Mycobacterium tuberculosis. Molecules 2019, 24, 1740. https://doi.org/10.3390/molecules24091740
Beteck RM, Seldon R, Jordaan A, Warner DF, Hoppe HC, Laming D, Khanye SD. New Quinolone-Based Thiosemicarbazones Showing Activity Against Plasmodium falciparum and Mycobacterium tuberculosis. Molecules. 2019; 24(9):1740. https://doi.org/10.3390/molecules24091740
Chicago/Turabian StyleBeteck, Richard M., Ronnett Seldon, Audrey Jordaan, Digby F. Warner, Heinrich C. Hoppe, Dustin Laming, and Setshaba D. Khanye. 2019. "New Quinolone-Based Thiosemicarbazones Showing Activity Against Plasmodium falciparum and Mycobacterium tuberculosis" Molecules 24, no. 9: 1740. https://doi.org/10.3390/molecules24091740
APA StyleBeteck, R. M., Seldon, R., Jordaan, A., Warner, D. F., Hoppe, H. C., Laming, D., & Khanye, S. D. (2019). New Quinolone-Based Thiosemicarbazones Showing Activity Against Plasmodium falciparum and Mycobacterium tuberculosis. Molecules, 24(9), 1740. https://doi.org/10.3390/molecules24091740