Ozone-Based Advanced Oxidation Processes for Primidone Removal in Water using Simulated Solar Radiation and TiO2 or WO3 as Photocatalyst



Abstract
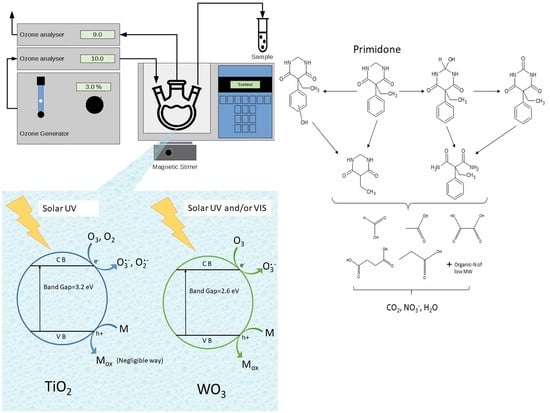
1. Introduction
2. Results
2.1. Degradation of PRM (5 mg L−1) in Ultrapure Water
2.1.1. Photolysis, Ozonation and Photolytic Ozonation of PRM
2.1.2. TiO2 P25 As Photocatalyst
2.1.3. WO3 As Photocatalyst
2.1.4. PRM Transformation Products
2.1.5. Ecotoxicity
2.2. Degradation of PRM in A Secondary Effluent
3. Materials and Methods
3.1. Materials
3.2. Experimental Procedure and Set-Up
3.3. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stuart, M.; Lapworth, D.; Crane, E.; Hart, A. Review of risk from potential emerging contaminants in UK groundwater. Sci. Total Environ. 2012, 416, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Baena-Nogueras, R.M.; González-Mazo, E.; Lara-Martín, P.A. Degradation kinetics of pharmaceuticals and personal care products in surface waters: Photolysis vs biodegradation. Sci. Total Environ. 2017, 590–591, 643–654. [Google Scholar] [CrossRef]
- Díaz-Garduño, B.; Pintado-Herrera, M.G.; Biel-Maeso, M.; Rueda-Márquez, J.J.; Lara-Martín, P.A.; Perales, J.A.; Manzano, M.A.; Garrido-Pérez, C.; Martín-Díaz, M.L. Environmental risk assessment of effluents as a whole emerging contaminant: Efficiency of alternative tertiary treatments for wastewater depuration. Water Res. 2017, 119, 136–149. [Google Scholar] [CrossRef]
- Vazquez-Roig, P.; Andreu, V.; Blasco, C.; Picó, Y. Risk assessment on the presence of pharmaceuticals in sediments, soils and waters of the Pego-Oliva Marshlands (Valencia, eastern Spain). Sci. Total Environ. 2012, 440, 24–32. [Google Scholar] [CrossRef]
- Bottoni, P.; Caroli, S. Presence of residues and metabolites of pharmaceuticals in environmental compartments, food commodities and workplaces: A review spanning the three-year period 2014–2016. Microchem. J. 2016, 136, 2–24. [Google Scholar] [CrossRef]
- Glaze, W.H.; Kang, J.-W.; Chapin, D.H. The Chemistry of Water Treatment Processes Involving Ozone, Hydrogen Peroxide and Ultraviolet Radiation. Ozone Sci. Eng. 1987, 9, 335–352. [Google Scholar] [CrossRef]
- Neamţu, M.; Grandjean, D.; Sienkiewicz, A.; Le Faucheur, S.; Slaveykova, V.; Colmenares, J.J.V.; Pulgarín, C.; De Alencastro, L.F. Degradation of eight relevant micropollutants in different water matrices by neutral photo-Fenton process under UV254 and simulated solar light irradiation—A comparative study. Appl. Catal. B Environ. 2014, 158–159, 30–37. [Google Scholar] [CrossRef]
- Noguera-Oviedo, K.; Aga, D.S. Lessons learned from more than two decades of research on emerging contaminants in the environment. J. Hazard. Mater. 2016, 316, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Márquez, G.; Rodríguez, E.M.; Beltrán, F.J.; Álvarez, P.M. Solar photocatalytic ozonation of a mixture of pharmaceutical compounds in water. Chemosphere 2014, 113, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mehrjouei, M.; Müller, S.; Möller, D. A review on photocatalytic ozonation used for the treatment of water and wastewater. Chem. Eng. J. 2015, 263, 209–219. [Google Scholar] [CrossRef]
- Yang, L.; Hu, C.; Nie, Y.; Qu, J. Catalytic Ozonation of Selected Pharmaceuticals over Mesoporous Alumina-Supported Manganese Oxide. Environ. Sci. Technol. 2009, 43, 2525–2529. [Google Scholar] [CrossRef]
- Aguinaco, A.; Beltrán, F.J.; García-Araya, J.F.; Oropesa, A. Photocatalytic ozonation to remove the pharmaceutical diclofenac from water: Influence of variables. Chem. Eng. J. 2012, 189–190, 275–282. [Google Scholar] [CrossRef]
- Rodríguez, E.M.; Márquez, G.; León, E.A.; Álvarez, P.M.; Amat, A.M.; Beltrán, F.J. Mechanism considerations for photocatalytic oxidation, ozonation and photocatalytic ozonation of some pharmaceutical compounds in water. J. Environ. Manag. 2013, 127, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Solís, R.R.; Rivas, F.J.; Martínez-Piernas, A.; Agüera, A. Ozonation, photocatalysis and photocatalytic ozonation of diuron: Intermediates identification. Chem. Eng. J. 2016, 292, 72–81. [Google Scholar] [CrossRef]
- Regulska, E.; Karpińska, J. Photocatalytic degradation of olanzapine in aqueous and river waters suspension of titanium dioxide. Appl. Catal B Environ. 2012, 117–118, 96–104. [Google Scholar] [CrossRef]
- Regulska, E.; Karpińska, J. Investigation of novel material for effective photodegradation of bezafibrate in aqueous samples. Environ. Sci Pollut. Res. 2014, 21, 5242–5248. [Google Scholar] [CrossRef]
- Malato, S.; Fernández-Ibáñez, P.; Maldonado, M.I.; Blanco, J.; Gernjak, W. Decontamination and disinfection of water by solar photocatalysis: Recent overview and trends. Catal. Today 2009, 147, 1–59. [Google Scholar] [CrossRef]
- Scaife, D.E. Oxide semiconductors in photoelectrochemical conversion of solar energy. Sol. Energy 1980, 25, 41–54. [Google Scholar] [CrossRef]
- Nishimoto, S.; Mano, T.; Kameshima, Y.; Miyake, M. Photocatalytic water treatment over WO3 under visible light irradiation combined with ozonation. Chem. Phys. Lett. 2010, 500, 86–89. [Google Scholar] [CrossRef]
- Mena, E.; Rey, A.; Contreras, S.; Beltrán, F.J. Visible light photocatalytic ozonation of DEET in the presence of different forms of WO3. Catal. Today 2014, 252, 100–106. [Google Scholar] [CrossRef]
- Mena, E.; Rey, A.; Rodríguez, E.M.; Beltrán, F.J. Reaction mechanism and kinetics of DEET visible light assisted photocatalytic ozonation with WO3 catalyst. Appl. Catal. B Environ. 2017, 202, 460–472. [Google Scholar] [CrossRef]
- Mano, T.; Nishimoto, S.; Kameshima, Y.; Miyake, M. Investigation of photocatalytic ozonation treatment of water over WO3 under visible light irradiation. J. Ceram. Soc. Japan 2011, 119, 822–827. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, J.; Cao, H.; Guo, Z.; Rabeah, J.; Brückner, A.; Xie, Y. The role of ozone and influence of band structure in WO3 photocatalysis and ozone integrated process for pharmaceutical wastewater treatment. J. Hazard. Mater. 2018, 360, 481–489. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Some Drugs and Herbal Products; WHO Press, Ed.; International Agency for Research on Cancer: Lyon, France, 2013; Volume 108, ISBN 9789283201748. [Google Scholar]
- Lafont, O.; Cavé, C.; Ménager, S.; Miocque, M. New chemical aspects of primidone metabolism. Eur. J. Med. Chem. 1990, 25, 61–66. [Google Scholar] [CrossRef]
- Bourgin, M.; Beck, B.; Boehler, M.; Borowska, E.; Fleiner, J.; Salhi, E.; Teichler, R.; von Gunten, U.; Siegrist, H.; McArdell, C.S. Evaluation of a full-scale wastewater treatment plant upgraded with ozonation and biological post-treatments: Abatement of micropollutants, formation of transformation products and oxidation by-products. Water Res. 2018, 129, 486–498. [Google Scholar] [CrossRef]
- Aminot, Y.; Fuster, L.; Pardon, P.; Le Menach, K.; Budzinski, H. Suspended solids moderate the degradation and sorption of waste water-derived pharmaceuticals in estuarine waters. Sci. Total Environ. 2018, 612, 39–48. [Google Scholar] [CrossRef]
- Heberer, T. Occurrence, fate, and removal of pharmaceutical residues in the aquatic environment: A review of recent research data. Toxicol. Lett. 2002, 131, 5–17. [Google Scholar] [CrossRef]
- Wert, E.C.; Rosario-Ortiz, F.L.; Snyder, S.A. Effect of ozone exposure on the oxidation of trace organic contaminants in wastewater. Water Res. 2009, 43, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Gerrity, D.; Stanford, B.D.; Trenholm, R.A.; Snyder, S.A. An evaluation of a pilot-scale nonthermal plasma advanced oxidation process for trace organic compound degradation. Water Res. 2010, 44, 493–504. [Google Scholar] [CrossRef]
- Dong, M.M.; Trenholm, R.; Rosario-Ortiz, F.L. Photochemical degradation of atenolol, carbamazepine, meprobamate, phenytoin and primidone in wastewater effluents. J. Hazard. Mater. 2015, 282, 216–223. [Google Scholar] [CrossRef]
- Guo, Y.C.; Krasner, S.W. Occurrence of Primidone, Carbamazepine, Caffeine, and Precursors for N-Nitrosodimethylamine in Drinking Water Sources Impacted by Wastewater1. JAWRA J. Am. Water Resour. Assoc. 2009, 45, 58–67. [Google Scholar] [CrossRef]
- Morasch, B. Occurrence and dynamics of micropollutants in a karst aquifer. Environ. Pollut. 2013, 173, 133–137. [Google Scholar] [CrossRef]
- Ternes, T.A.; Meisenheimer, M.; McDowell, D.; Sacher, F.; Brauch, H.-J.; Haist-Gulde, B.; Preuss, G.; Wilme, U.; Zulei-Seibert, N. Removal of Pharmaceuticals during Drinking Water Treatment. Environ. Sci. Technol. 2002, 36, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Real, F.J.; Javier Benitez, F.; Acero, J.L.; Sagasti, J.J.P.; Casas, F. Kinetics of the chemical oxidation of the pharmaceuticals primidone, ketoprofen, and diatrizoate in ultrapure and natural waters. Ind. Eng. Chem. Res. 2009, 48, 3380–3388. [Google Scholar] [CrossRef]
- Liu, N.; Wang, T.; Zheng, M.; Lei, J.; Tang, L.; Hu, G.; Xu, G.; Wu, M. Radiation induced degradation of antiepileptic drug primidone in aqueous solution. Chem. Eng. J. 2015, 270, 66–72. [Google Scholar] [CrossRef]
- Sijak, S.; Liu, N.; Zheng, M.; Xu, G.; Tang, L.; Yao, J.; Wu, M. Degradation of Anticonvulsant Drug Primidone in Aqueous Solution by UV Photooxidation Processes. Environ. Eng. Sci. 2015, 32, 436–444. [Google Scholar] [CrossRef]
- Checa, M.; Figueredo, M.; Aguinaco, A.; Beltrán, F.J. Graphene oxide/titania Photocatalytic ozonation of Primidone in a visible LED photoreactor. J. Hazard. Mater. 2019, 369, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, F.J. Ozone Reaction Kinetics for Water and Wastewater Systems; Lewis publishers: Boca Raton, FL, USA, 2004; Volume 1, ISBN 1-56670-629-7. [Google Scholar]
- Charpentier, J.C. Mass-transfer rates in gas-liquid absorbers and reactors. Adv. Chem. Eng. 1981, 11, 1–133. [Google Scholar]
- David Yao, C.C.; Haag, W.R. Rate constants for direct reactions of ozone with several drinking water contaminants. Water Res. 1991, 25, 761–773. [Google Scholar] [CrossRef]
- Alam, M.S.; Rao, B.S.M.; Janata, E. OH reactions with aliphatic alcohols: Evaluation of kinetics by direct optical absorption measurement. A pulse radiolysis study. Radiat. Phys. Chem. 2003, 67, 723–728. [Google Scholar] [CrossRef]
- Beltrán, F.J.; Pocostales, P.; Alvarez, P.; Oropesa, A. Diclofenac removal from water with ozone and activated carbon. J. Hazard. Mater. 2009, 163, 768–776. [Google Scholar] [CrossRef]
- Fathinia, M.; Khataee, A. Photocatalytic ozonation of phenazopyridine using TiO2 nanoparticles coated on ceramic plates: Mechanistic studies, degradation intermediates and ecotoxicological assessments. Appl. Catal. A Gen. 2015, 491, 136–154. [Google Scholar] [CrossRef]
- Bauer, D.; Dïottone, L.; Hynes, A.J.; February, A. O 1 D quantum yields from O photolysis in the near UV region 3 between 305 and 375 nm. PCCP 2000, 1421–1424. [Google Scholar] [CrossRef]
- Matsumi, Y.; Comes, F.J.; Hancock, G.; Hofzumahaus, A.; Hynes, A.J.; Kawasaki, M.; Ravishankara, A.R. Quantum yields for production of O(1D) in the ultraviolet photolysis of ozone: Recommendation based on evaluation of laboratory data. J. Geophys. Res. Atmos. 2002, 107, ACH 1-1–ACH 1-12. [Google Scholar] [CrossRef]
- Ohtani, B.; Prieto-Mahaney, O.O.; Li, D.; Abe, R. What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test. J. Photochem. Photobiol. A Chem. 2010, 216, 179–182. [Google Scholar] [CrossRef]
- Turchi, S.C.; Ollis, D.F. Photocatalytic degradation of organic water contaminants: Mechanisms involving hydroxyl radical attack. J. Catal. 1990, 122, 178–192. [Google Scholar] [CrossRef]
- Rodríguez, E.M.; Márquez, G.; Tena, M.; Álvarez, P.M.; Beltrán, F.J. Determination of main species involved in the first steps of TiO2 photocatalytic degradation of organics with the use of scavengers: The case of ofloxacin. Appl. Catal. B Environ. 2015, 178, 44–53. [Google Scholar] [CrossRef]
- Andreozzi, R.; Insola, A.; Caprio, V.; D’Amore, M. The kinetics of Mn(II)-catalysed ozonation of oxalic acid in aqueous solution. Water Res. 1992, 26, 917–921. [Google Scholar] [CrossRef]
- Hayon, E.; McGarvey, J.J. Flash photolysis in the vacuum ultraviolet region of SO42−, CO32− and OH− ions in aqueous solutions. J. Phys. Chem. 1967, 71, 1472–1477. [Google Scholar] [CrossRef]
- LaVerne, J.A.; Pimblott, S.M. Scavenger and time dependences of radicals and molecular products in the electron radiolysis of water: Examination of experiments and models. J. Phys. Chem. 1991, 95, 3196–3206. [Google Scholar] [CrossRef]
- Cao, H.; Lin, X.; Zhan, H.; Zhang, H.; Lin, J. Photocatalytic degradation kinetics and mechanism of phenobarbital in TiO2 aqueous solution. Chemosphere 2013, 90, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.M.; Fernández, G.; Klamerth, N.; Maldonado, M.I.; Álvarez, P.M.; Malato, S. Efficiency of different solar advanced oxidation processes on the oxidation of bisphenol A in water. Appl. Catal. B Environ. 2010, 95, 228–237. [Google Scholar] [CrossRef]
- Autin, O.; Hart, J.; Jarvis, P.; MacAdam, J.; Parsons, S.A.; Jefferson, B. The impact of background organic matter and alkalinity on the degradation of the pesticide metaldehyde by two advanced oxidation processes: UV/H2O2 and UV/TiO2. Water Res. 2013, 47, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, B.; Jarvis, P.; Bhagianathan, G.K.; Smith, H.; Autin, O.; Goslan, E.H.; MacAdam, J.; Carra, I. Effect of elevated UV dose and alkalinity on metaldehyde removal and THM formation with UV/TiO2 and UV/H2O2. Chem. Eng. J. 2016, 288, 359–367. [Google Scholar] [CrossRef]
- Li, L.; Sillanpää, M.; Risto, M. Influences of water properties on the aggregation and deposition of engineered titanium dioxide nanoparticles in natural waters. Environ. Pollut. 2016, 219, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Wicaksana, Y.; Liu, S.; Scott, J.; Amal, R. Tungsten trioxide as a visible light photocatalyst for volatile organic carbon removal. Molecules 2014, 19, 17747–17762. [Google Scholar] [CrossRef]
- Bader, H.; Hoigné, J. Determination of ozone in water by the indigo method. Water Res. 1981, 15, 449–456. [Google Scholar] [CrossRef]
- Masschelein, W.; Denis, M.; Ledent, R. Spectrophotometric determination of residual hydrogen peroxide. Water Sew. Work. 1977, 69–72. [Google Scholar]
- APHA/AWWA/WEF. Standard Methods for the Examination of Water and Wastewater. Stand. Methods 2012, 541. [Google Scholar]
- Hatchard, C.G.; Parker, C.A. A New Sensitive Chemical Actinometer. II. Potassium Ferrioxalate as a Standard Chemical Actinometer. Proc. R. Soc. A Math. Phys. Eng. Sci. 1956, 235, 518–536. [Google Scholar]
- OECD. Daphnia acute Immobilisation Test and Reproduction Test. OECD Guidel. Test. Chem. 1984, 202, 1–16. [Google Scholar]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Units |
|---|---|---|
| pH | 8.2 | - |
| Electrical conductivity | 533 | µS cm−1 |
| COD | 47 | mg L−1 O2 |
| BOD5 | 11 | mg L−1 O2 |
| TOC | 14.16 | mg L−1 |
| DOC | 13.8 | mg L−1 |
| IC | 28.8 | mg L−1 |
| Alkalinity (1) | 240 | mg L−1 CaCO3 |
| A254nm | 0.222 | - |
| SUVA254 | 1.61 | L (mg DOC m) −1 |
| F− | 0.43 | mg L−1 |
| Cl− | 78.30 | mg L−1 |
| NO3− | 22.6 | mg L−1 |
| NO2− | 0.18 | mg L−1 |
| SO4= | 52.5 | mg L−1 |
| PO43− | 2.6 | mg L−1 |
| System | Ultrapure Water kObs, min−1 (R2) | Secondary Effluent kObs, min−1 (R2) | Ratio kObs(SE)/kObs |
|---|---|---|---|
| O3 | 0.321 (0.98) | 0.122 (0.99) | 0.38 |
| UV-Vis/O3 | 0.429 (0.98) | 0.140 (0.99) | 0.33 |
| UV-Vis/TiO2 | 0.093 (0.99) | 0.011 (0.99) | 0.12 |
| UV-Vis/TiO2/O3 | 0.380 (0.94) | 0.254 (0.98) | 0.67 |
| Vis/WO3/O3 | 0.224 (0.99) | 0.073 (0.98) | 0.32 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueredo, M.A.; Rodríguez, E.M.; Checa, M.; Beltran, F.J. Ozone-Based Advanced Oxidation Processes for Primidone Removal in Water using Simulated Solar Radiation and TiO2 or WO3 as Photocatalyst. Molecules 2019, 24, 1728. https://doi.org/10.3390/molecules24091728
Figueredo MA, Rodríguez EM, Checa M, Beltran FJ. Ozone-Based Advanced Oxidation Processes for Primidone Removal in Water using Simulated Solar Radiation and TiO2 or WO3 as Photocatalyst. Molecules. 2019; 24(9):1728. https://doi.org/10.3390/molecules24091728
Chicago/Turabian StyleFigueredo, Manuel A., Eva M. Rodríguez, Manuel Checa, and Fernando J. Beltran. 2019. "Ozone-Based Advanced Oxidation Processes for Primidone Removal in Water using Simulated Solar Radiation and TiO2 or WO3 as Photocatalyst" Molecules 24, no. 9: 1728. https://doi.org/10.3390/molecules24091728
APA StyleFigueredo, M. A., Rodríguez, E. M., Checa, M., & Beltran, F. J. (2019). Ozone-Based Advanced Oxidation Processes for Primidone Removal in Water using Simulated Solar Radiation and TiO2 or WO3 as Photocatalyst. Molecules, 24(9), 1728. https://doi.org/10.3390/molecules24091728