Advanced Isoconversional Kinetic Analysis for the Elucidation of Complex Reaction Mechanisms: A New Method for the Identification of Rate-Limiting Steps


Abstract
:
1. Introduction
2. Theoretical Part
3. Data Simulation
4. Results
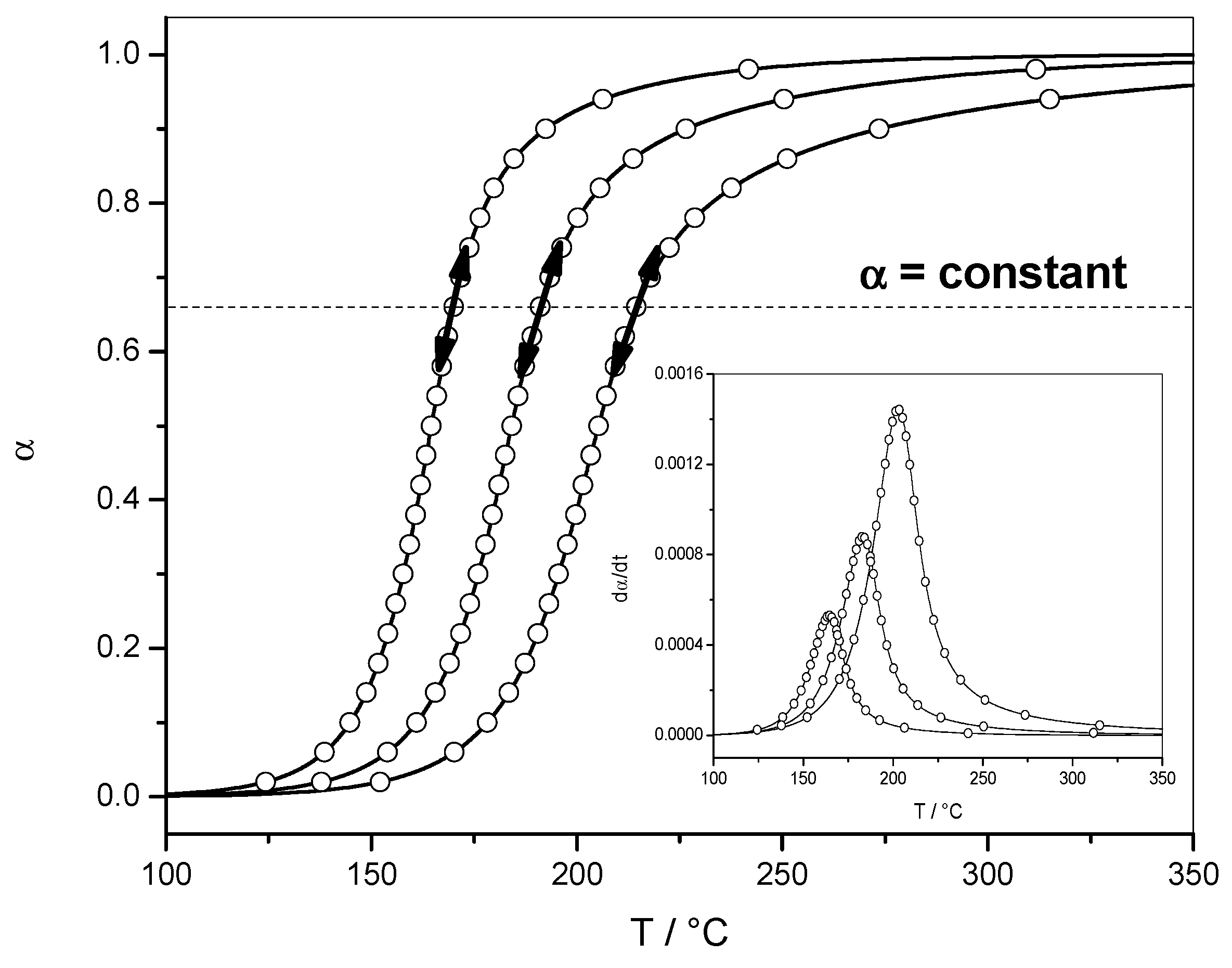
4.1. Autocatalytic Reaction with Diffusion-Controlled Part (Data Set 1)
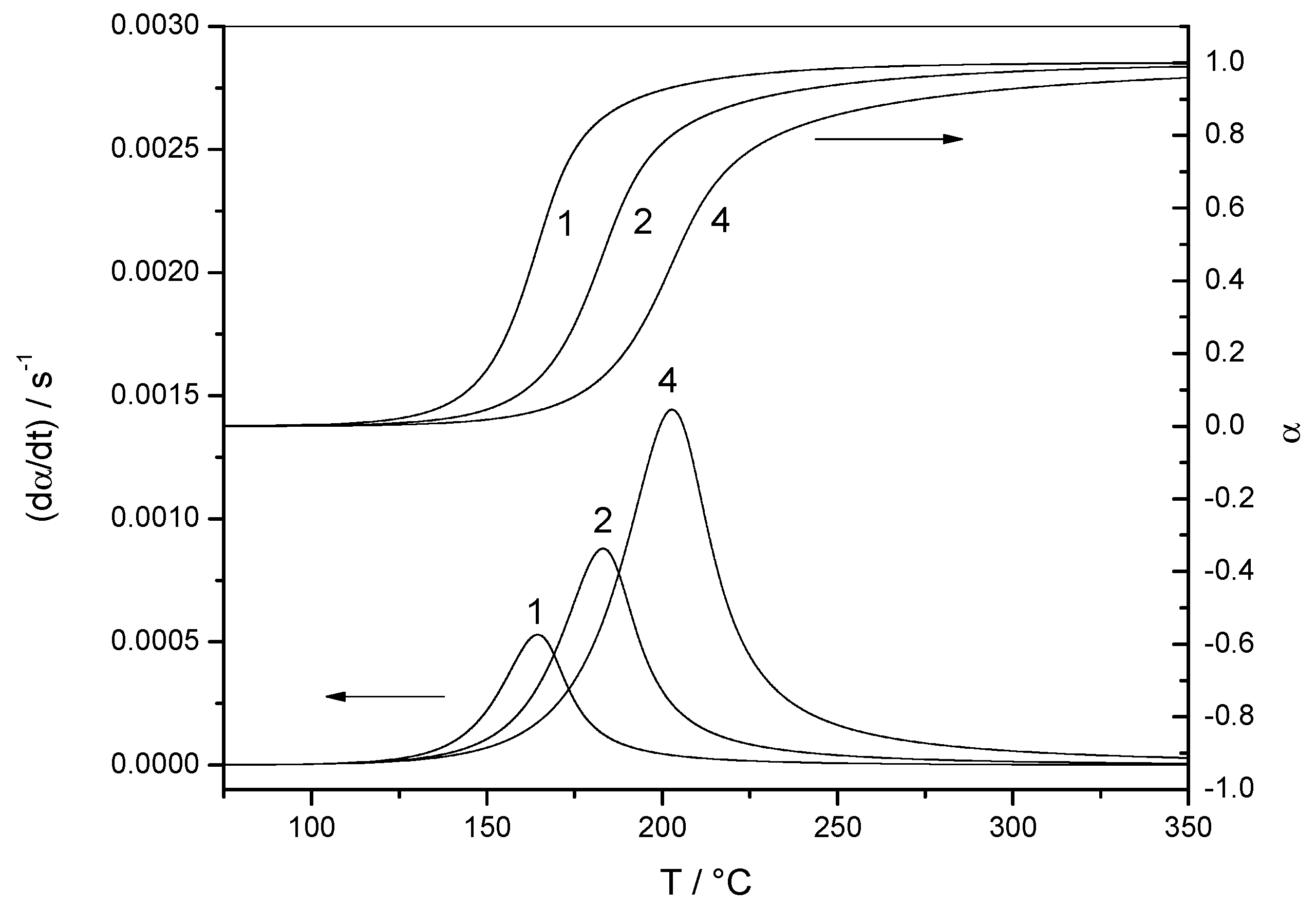
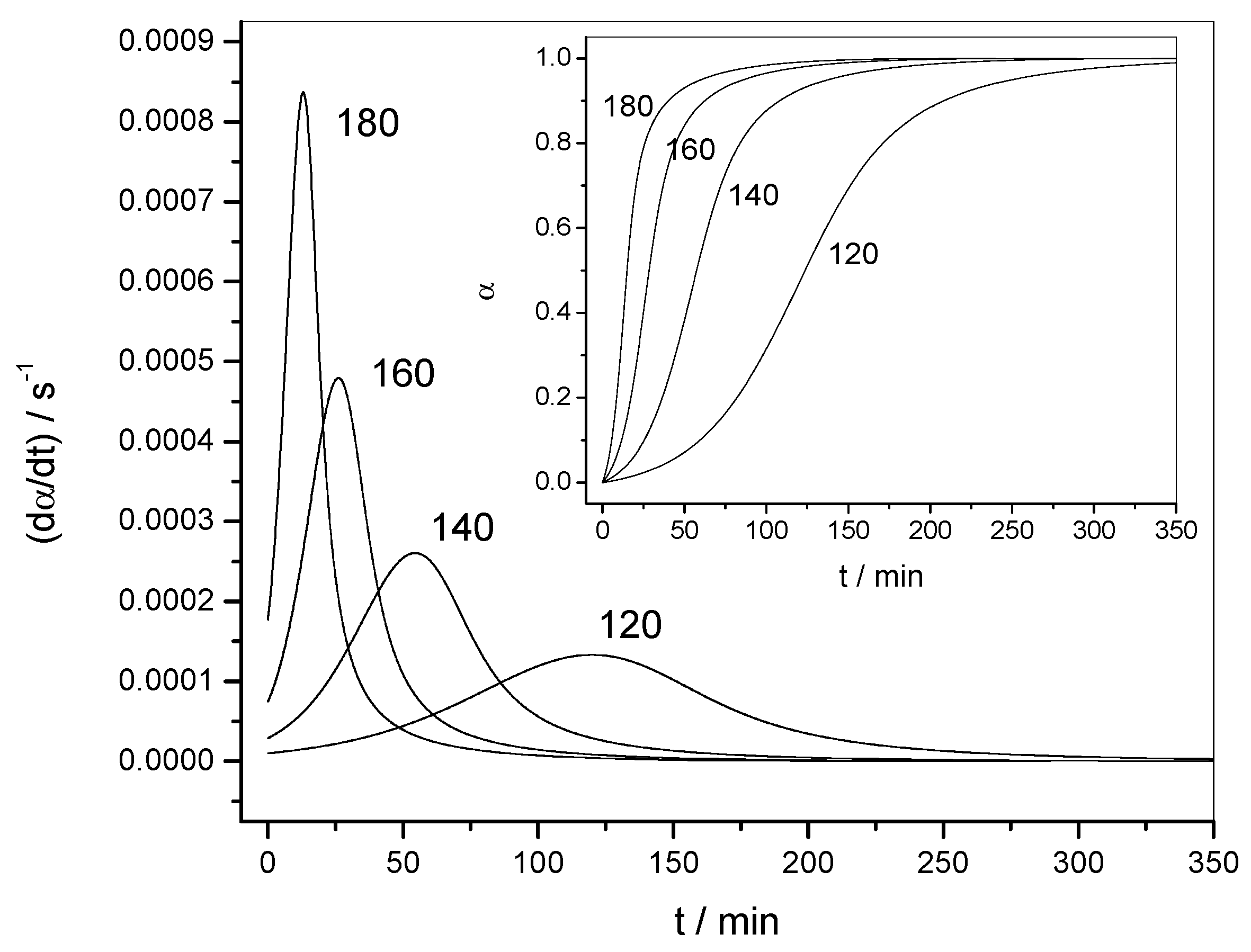
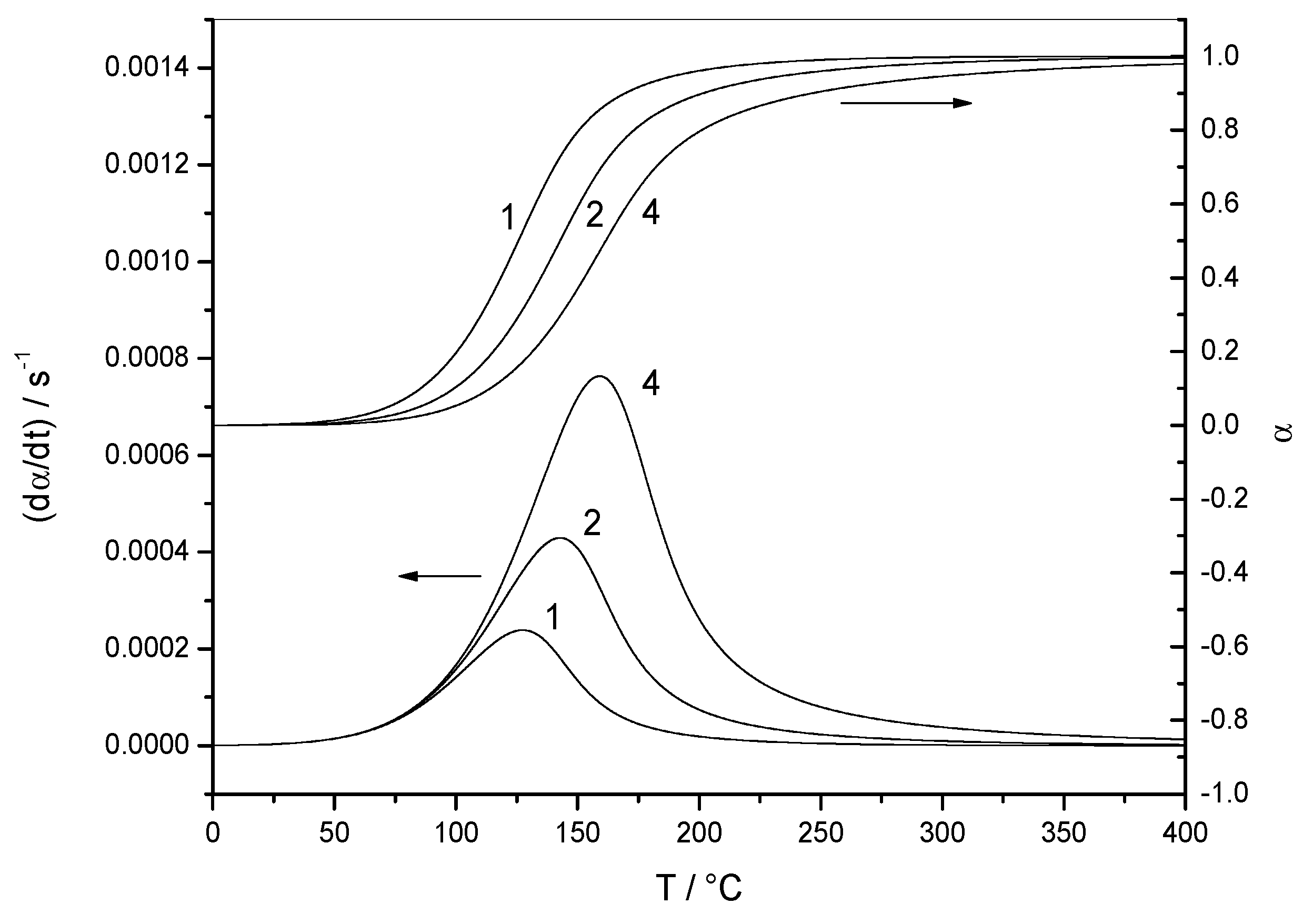
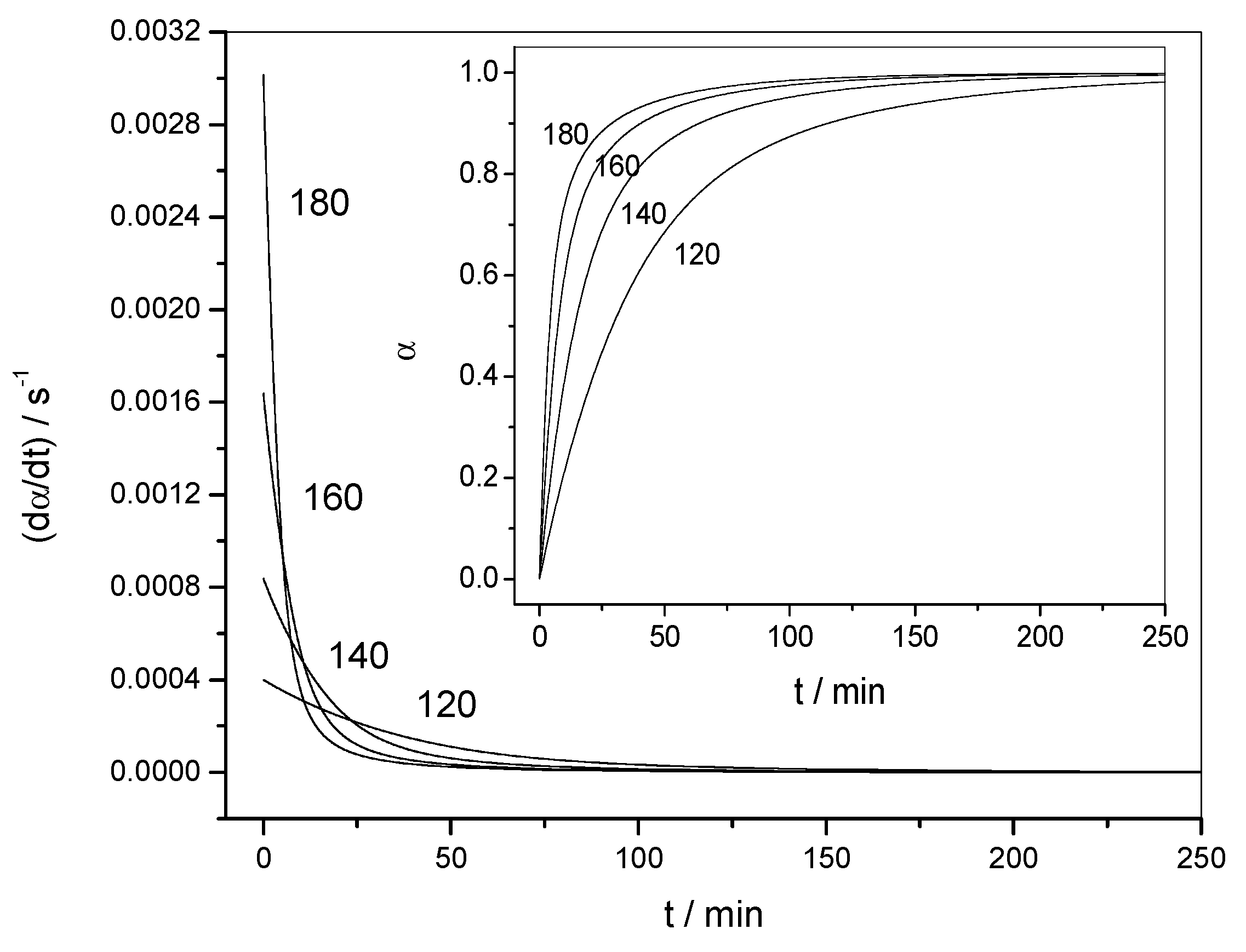
4.1.1. Reaction Rate and Extent of Conversion for Non-Isothermal and Isothermal Conditions
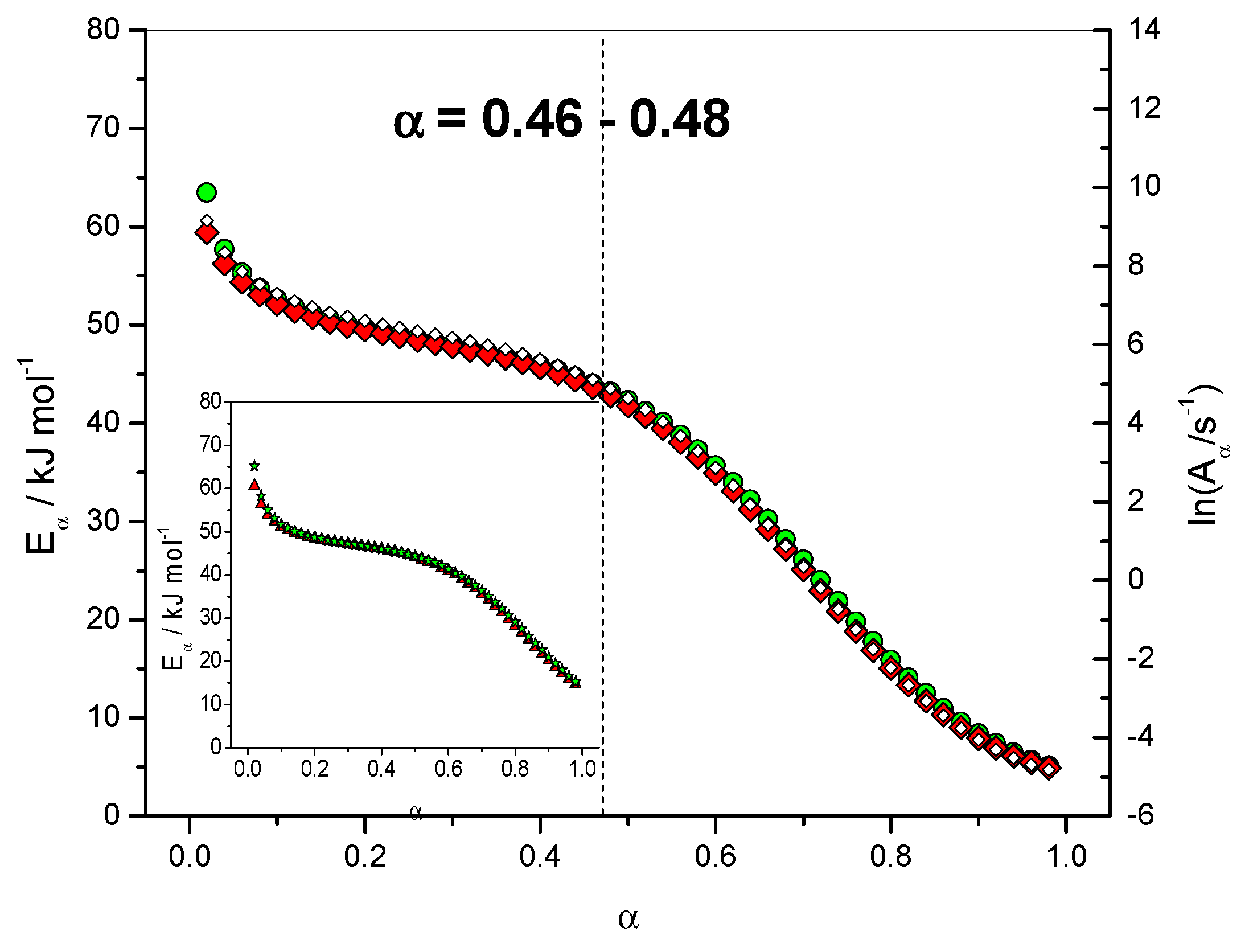
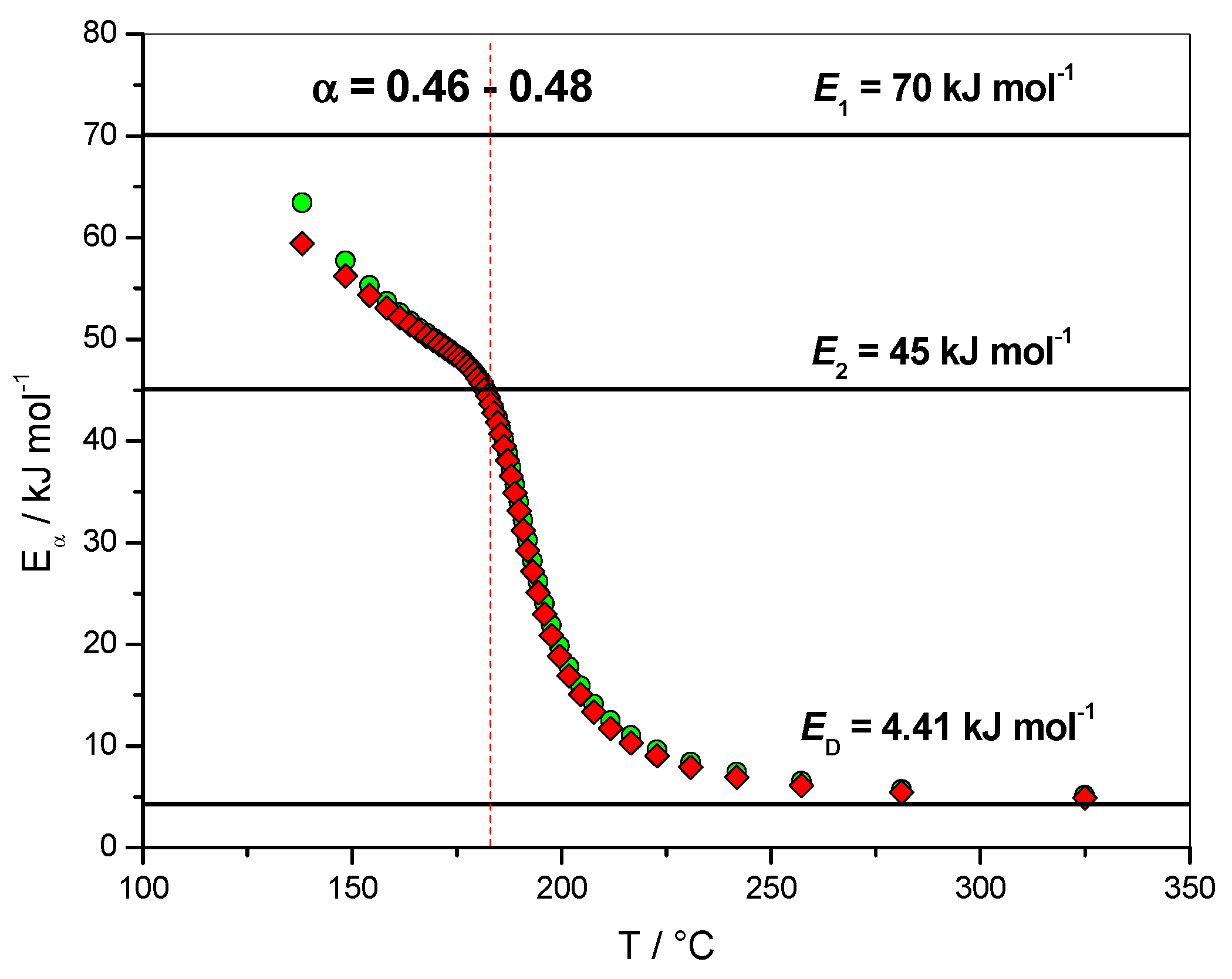
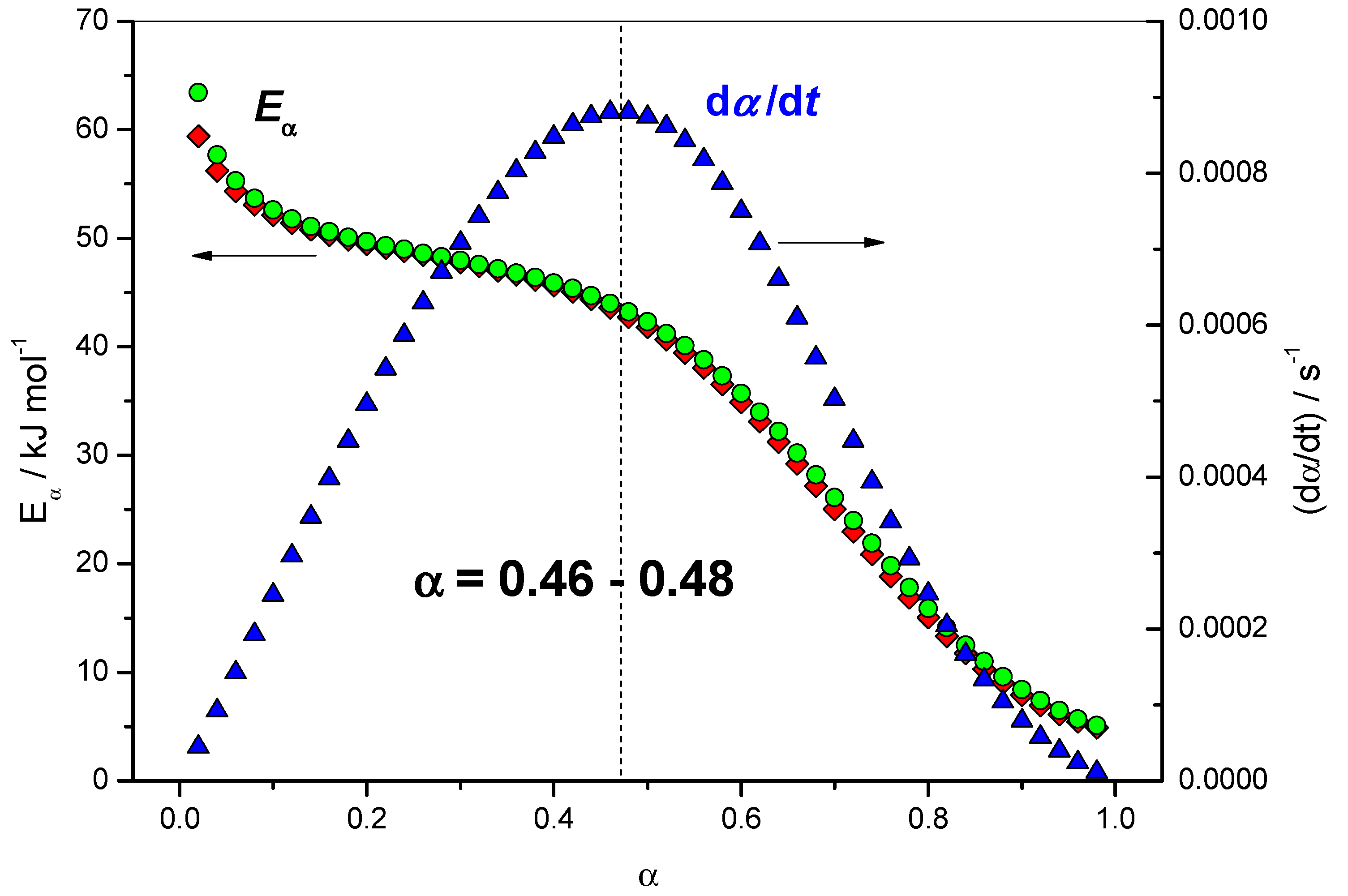
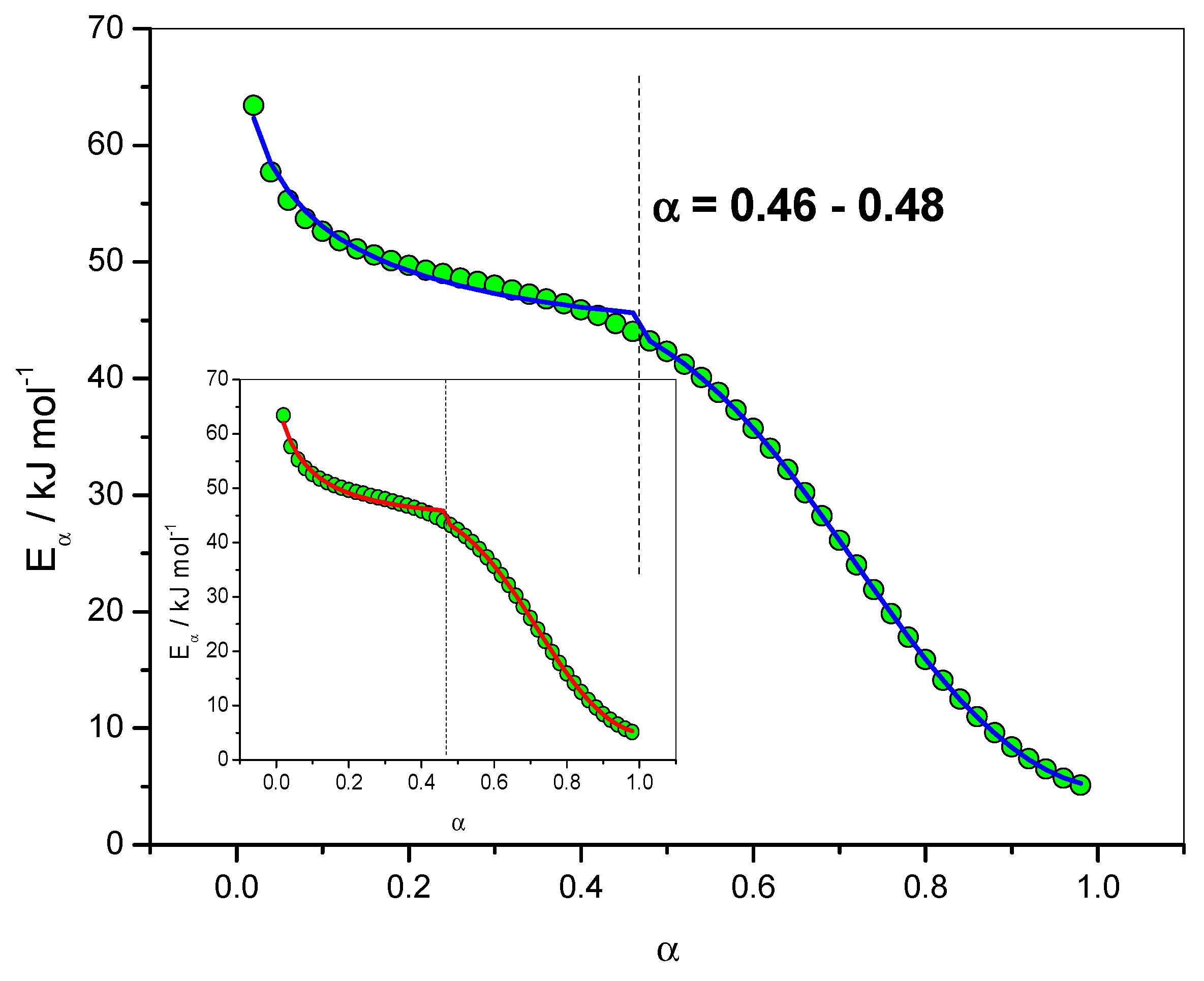
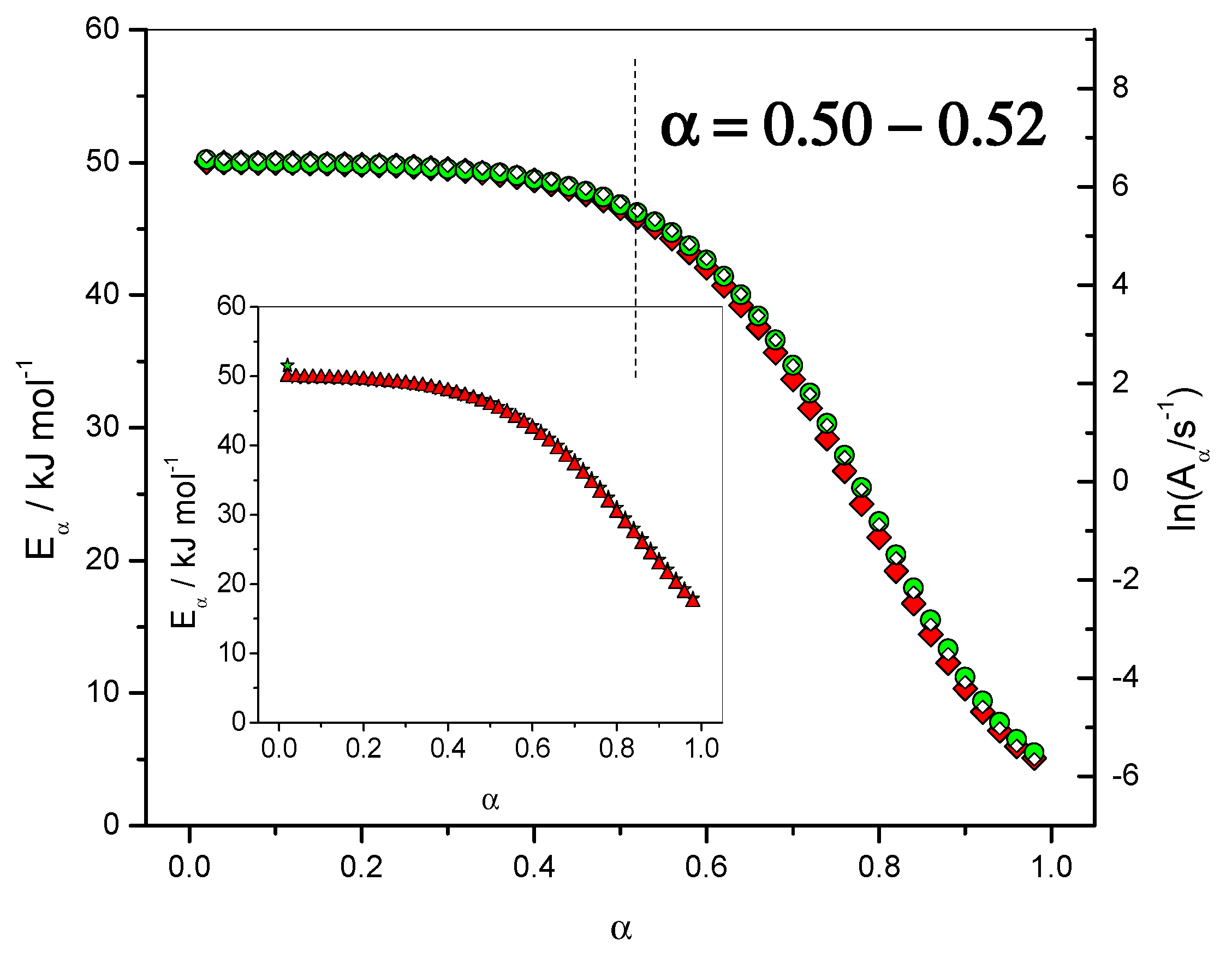
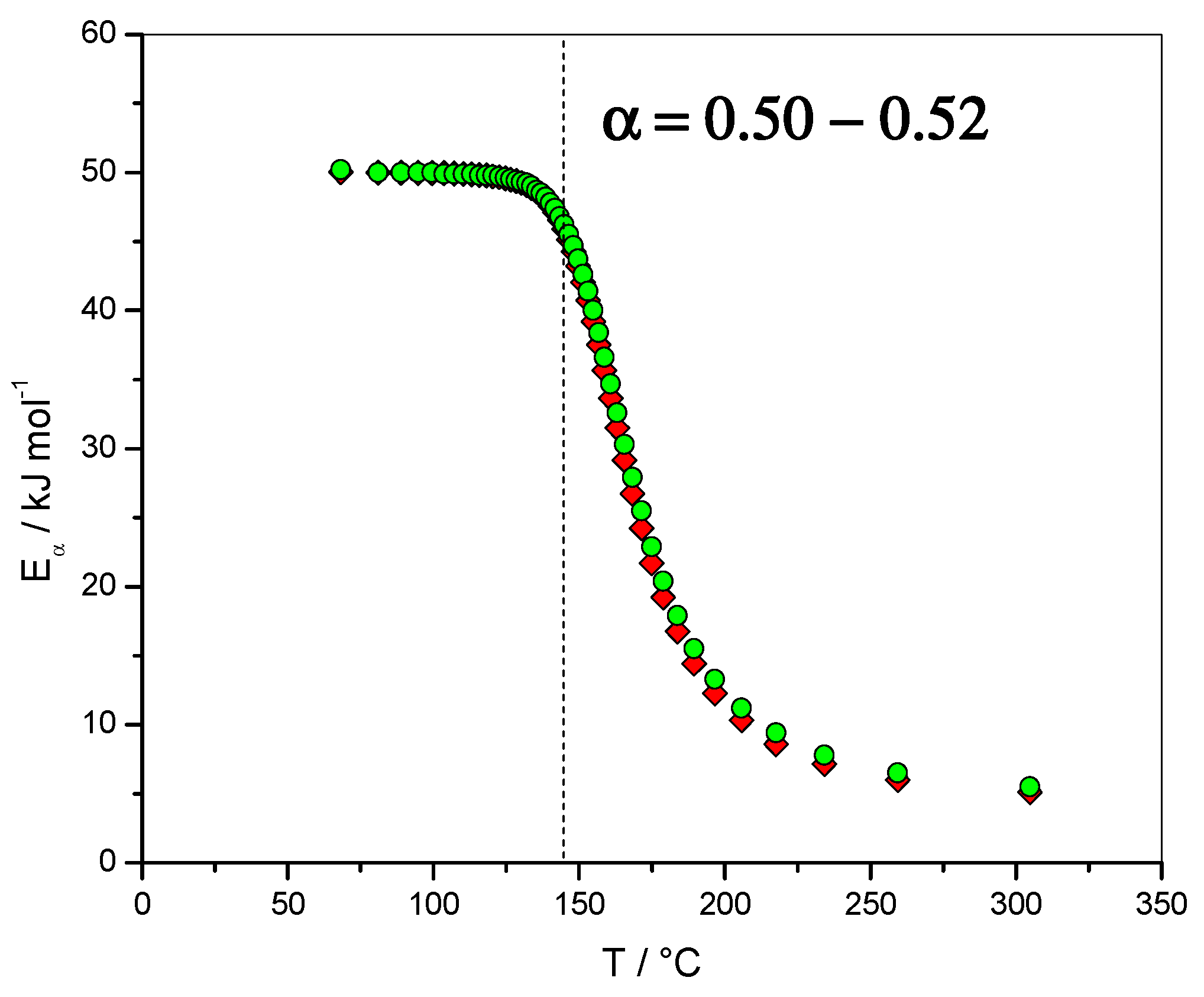
4.1.2. Dependence of the Effective Activation Energy and of the Pre-Exponential Factor
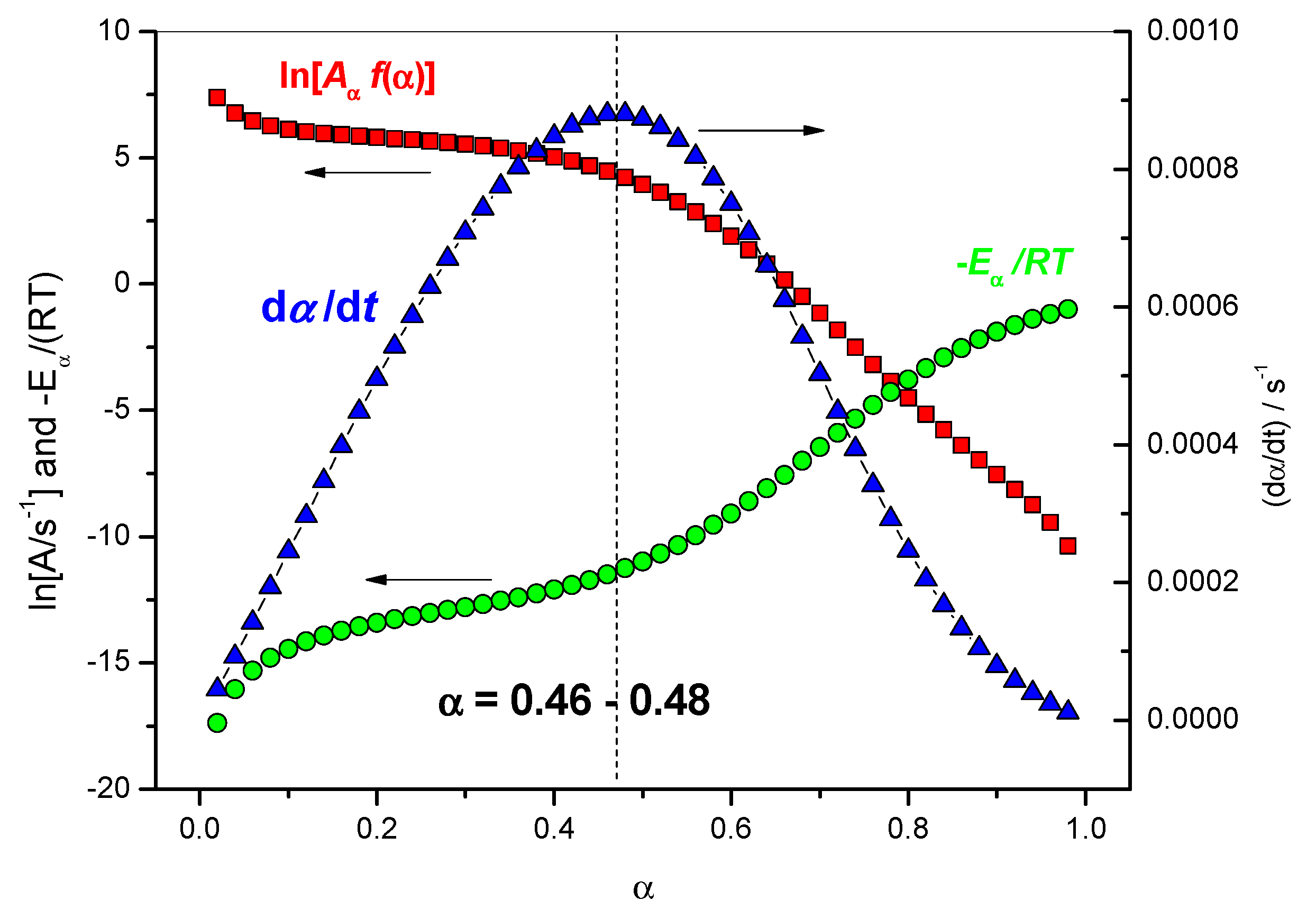
4.1.3. Variation of the Reaction Rate with the Extent of Conversion
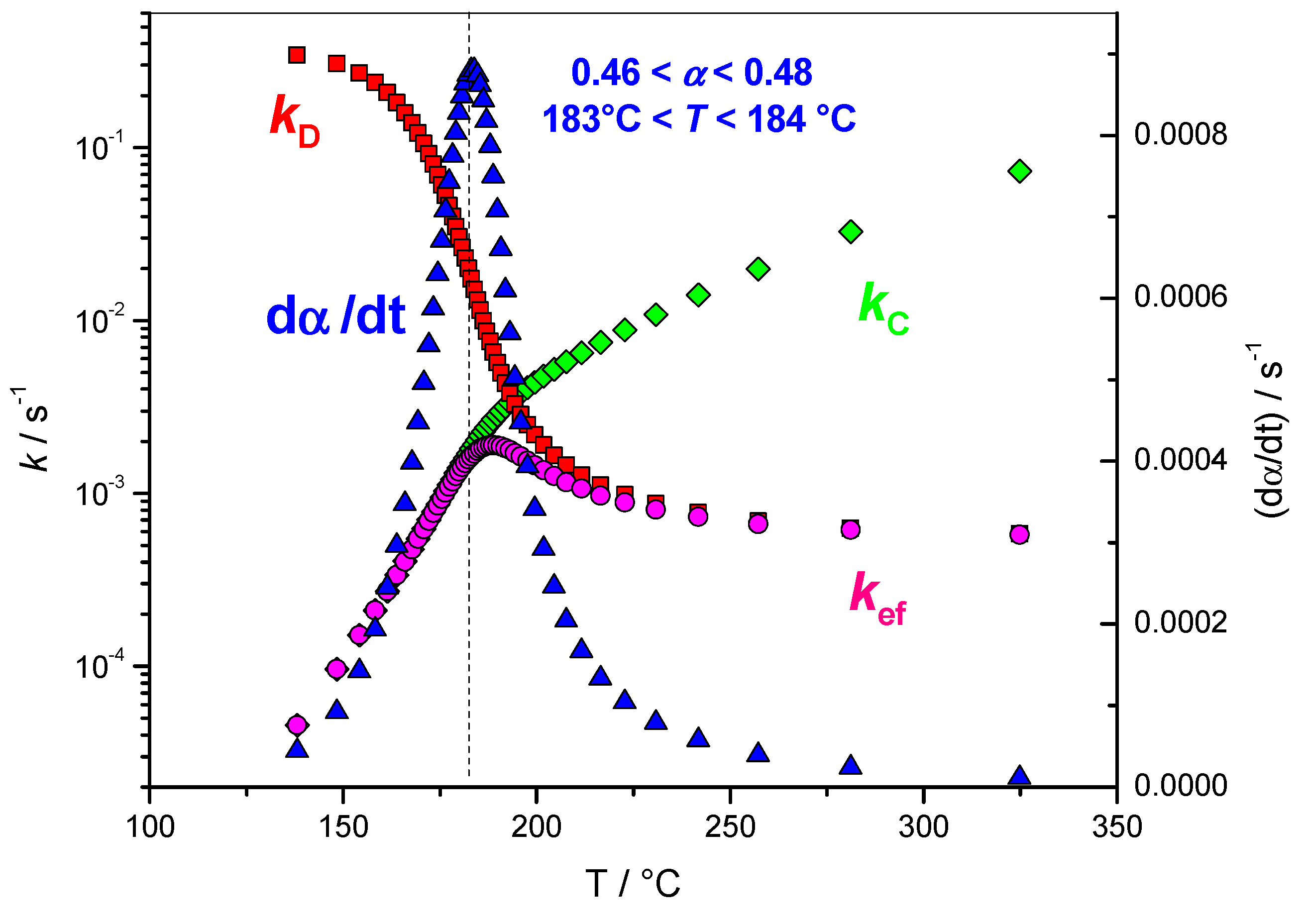
4.1.4. Variation of the Rate Coefficients with Extent of Conversion
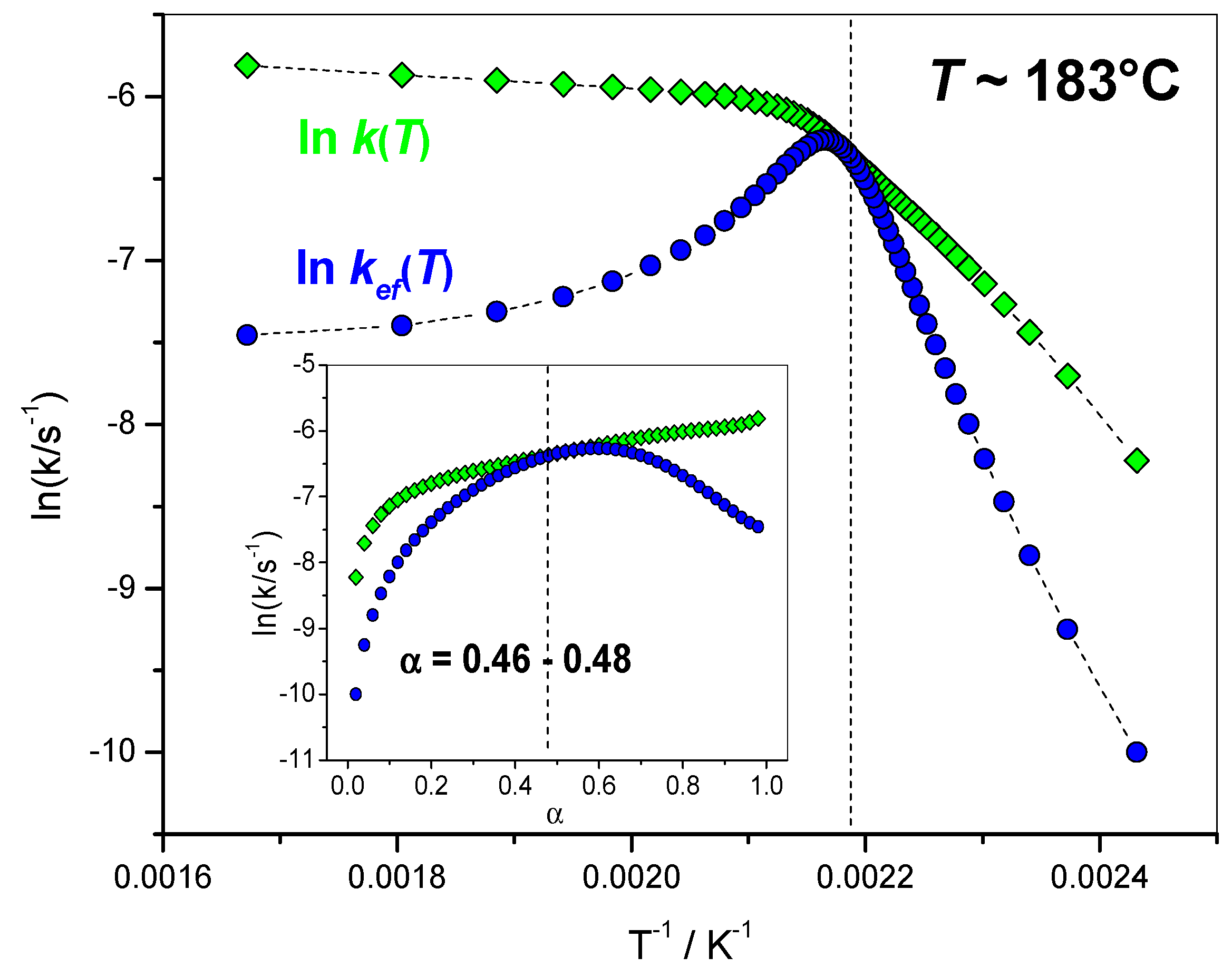
4.1.5. Variation of the Overall Rate Coefficient k(T) and of the Effective Rate Coefficient kef(T) with Reciprocal Temperature
4.1.6. Fit of the Eα Dependence with the Sourour and Kamal and Diffusion Models
4.2. First-Order Reaction with Diffusion-Controlled Part (Data Set 2)
5. Conclusions
Funding
Conflicts of Interest
References
- Pascault, J.P.; Sautereau, H.; Verdu, J.; Williams, R.J.J. Thermosetting polymers; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Hale, A.; Macosko, C.W.; Bair, H.E. Glass Transition Temperature as a Function of Conversion in Thermosetting Polymers. Macromolecules 1991, 24, 2610–2621. [Google Scholar] [CrossRef]
- Pascault, J.P.; Williams, R.J.J. Glass Transition Temperature Versus Conversion Relationships for Thermosetting Polymers. J. Polym. Sci. Part B Polym. Phys. 1990, 28, 85–95. [Google Scholar] [CrossRef]
- Alzina, C.; Sbirrazzuoli, N.; Mija, A. Hybrid Nanocomposites: Advanced Nonlinear Method for Calculating Key Kinetic Parameters of Complex Cure Kinetics. J. Phys. Chem. B 2010, 114, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Vyazovkin, S.; Sbirrazzuoli, N. Isoconversional kinetic analysis of thermally stimulated processes in polymers. Macromol. Rapid Comm. 2006, 27, 1515–1532. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Vyazovkin, S. Isoconversional Kinetics of Thermally Stimulated Processes; Springer: Berlin, Germany, 2015. [Google Scholar]
- Vyazovkin, S. Evaluation of activation energy of thermally stimulated solid-state reactions under arbitrary variation of temperature. J. Comput. Chem. 1997, 18, 393–402. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Vincent, L.; Vyazovkin, S. Comparison of several computational procedures for evaluating the kinetics of thermally stimulated condensed phase reactions. Chemometr. Intell. Lab 2000, 54, 53–60. [Google Scholar] [CrossRef]
- Vyazovkin, S. Modification of the integral isoconversional method to account for variation in the activation energy. J. Comput. Chem. 2001, 22, 178–183. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N. Determination of pre-exponential factors and of the mathematical functions f(α) or G(α) that describe the reaction mechanism in a model-free way. Thermochim. Acta 2013, 564, 59–69. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N. Is the Friedman method applicable to transformations with temperature dependent reaction heat? Macromol. Chem. Phys. 2007, 208, 1592–1597. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Vincent, L.; Vyazovkin, S. Electronic solution to the problem of a kinetic standard for DSC measurements. Chemometr. Intell. Lab 2000, 52, 23–32. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Brunel, D.; Elegant, L. Different kinetic equations analysis. J. Therm. Anal. 1992, 38, 1509–1524. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Girault, Y.; Elegant, L. Simulations for evaluation of kinetic methods in differential scanning calorimetry. Part 3—peak maximum evolution methods and isoconversional methods. Thermochim. Acta 1997, 293, 25–37. [Google Scholar] [CrossRef]
- Falco, G.; Guigo, N.; Vincent, L.; Sbirrazzuoli, N. FA polymerization disruption by protic polar solvent. Polymers 2018, 10, 529. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J. Polym. Sci. Part C 1964, 6, 183–195. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Sbirrazzuoli, N. Mechanism and kinetics of epoxy-amine cure studied by differential scanning calorimetry. Macromolecules 1996, 29, 1867–1873. [Google Scholar] [CrossRef]
- Ryan, M.E.; Dutta, A. Kinetics of epoxy cure: A rapid technique for kinetic parameter estimation. Polymer 1979, 20, 203–206. [Google Scholar] [CrossRef]
- Sourour, S.; Kamal, M.R. Differential scanning calorimetry of epoxy cure: Isothermal cure kinetics. Thermochim. Acta 1976, 14, 41–59. [Google Scholar] [CrossRef]
- Yamini, G.; Shakeri, A.; Vafayan, M.; Zohuriaan-Mehr, M.J.; Kabiri, K.; Zolghadr, M. Cure kinetics of modified lignosulfonate/epoxy blends. Thermochim. Acta 2019, 675, 18–28. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tα/°C | Eα (FR)/kJ·mol−1 | α | ln (Aα/s−1) | [Aα f(α)]/s−1 | exp(−Eα/(RTα) | (dα/dt)/s−1 |
|---|---|---|---|---|---|---|
| 181.56 | 45.05 | 0.42 | 5.47 | 130.25 | 6.632 × 10−6 | 8.638 × 10−4 |
| 182.34 | 44.39 | 0.44 | 5.30 | 108.30 | 8.076 × 10−6 | 8.746 × 10−4 |
| 183.11 | 43.63 | 0.46 | 5.11 | 87.43 | 1.007 × 10−5 | 8.802 × 10−4 |
| 183.89 | 42.76 | 0.48 | 4.89 | 68.30 | 1.289 × 10−5 | 8.801 × 10−4 |
| 184.66 | 41.78 | 0.50 | 4.63 | 51.45 | 1.699 × 10−5 | 8.740 × 10−4 |
| 185.45 | 40.68 | 0.52 | 4.35 | 37.26 | 2.313 × 10−5 | 8.618 × 10−4 |
| 2 < α < 46% | A1/A2 | E1/kJ·mol−1 | E2/kJ·mol−1 | m | MSSDa |
| Autocatalytic | 5918.03 | 79.8 | 38.2 | 0.97 | 0.4435 |
| 48 < α < 98% | A/D0 | E2/kJ·mol−1 | ED/kJ·mol−1 | K | MSSDa |
| Diffusion | 362.67 | 48.9 | 4.9 | −7.84 | 0.0023 |
| A1/s−1 | A2/s−1 | E1/kJ·mol−1 | E2/kJ·mol−1 | m | MSSDa | |
|---|---|---|---|---|---|---|
| 2 < α < 46% | 20756.17 | 498.52 | 67.6 | 42.6 | 1.2 | 0.6011 |
| 2 < α < 24% | 20258.31 | 510.84 | 76.1 | 46.7 | 1.3 | 0.0345 |
| A2/s−1 | D0/s−1 | E2/kJ·mol−1 | ED/kJ·mol−1 | K | MSSDa | |
| 48 < α < 98% | 498.9 | 1.43 | 48.8 | 4.9 | −7.87 | 0.0024 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbirrazzuoli, N. Advanced Isoconversional Kinetic Analysis for the Elucidation of Complex Reaction Mechanisms: A New Method for the Identification of Rate-Limiting Steps. Molecules 2019, 24, 1683. https://doi.org/10.3390/molecules24091683
Sbirrazzuoli N. Advanced Isoconversional Kinetic Analysis for the Elucidation of Complex Reaction Mechanisms: A New Method for the Identification of Rate-Limiting Steps. Molecules. 2019; 24(9):1683. https://doi.org/10.3390/molecules24091683
Chicago/Turabian StyleSbirrazzuoli, Nicolas. 2019. "Advanced Isoconversional Kinetic Analysis for the Elucidation of Complex Reaction Mechanisms: A New Method for the Identification of Rate-Limiting Steps" Molecules 24, no. 9: 1683. https://doi.org/10.3390/molecules24091683
APA StyleSbirrazzuoli, N. (2019). Advanced Isoconversional Kinetic Analysis for the Elucidation of Complex Reaction Mechanisms: A New Method for the Identification of Rate-Limiting Steps. Molecules, 24(9), 1683. https://doi.org/10.3390/molecules24091683