
Nickel-Catalyzed Decarbonylative Stannylation of Acyl Fluorides under Ligand-Free Conditions



Abstract
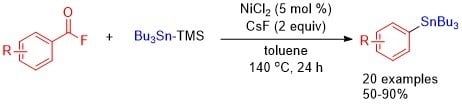
1. Introduction
2. Results and Discussion
3. Experimental Sections
3.1. General
3.2. Experimental Method
3.2.1. Representative Procedure for the Synthesis of Acyl Fluorides from Acyl Chlorides
3.2.2. Representative Procedure for the Synthesis of Acyl Fluorides from Carboxylic Acids
3.2.3. Synthesis of Trimethyl(tributylstannyl)silane (2)
3.2.4. Representative Procedure for Ni-catalyzed Decarbonylative Stannylation of Acyl Fluorides
3.2.5. One-Pot Decarbonylative Stannylation/Migita-Kosugi-Stille Cross-Coupling Reaction of 1a
3.3. Characterization Data of Starting Materials and Products
4. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bunton, C.A.; Fendler, J.H. The Hydrolysis of Acetyl Fluoride. J. Org. Chem. 1966, 31, 2307–2312. [Google Scholar] [CrossRef]
- Carpino, L.A.; Beyermann, M.; Wenschuh, H.; Bienert, M. Peptide Synthesis via Amino Acid Halides. Acc. Chem. Res. 1996, 29, 268–274. [Google Scholar] [CrossRef]
- Blanchard, N.; Bizet, V. Acid Fluorides in Transition-Metal Catalysis: A Good Balance between Stability and Reactivity. Angew. Chem. Int. Ed. 2019, 58, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rovis, T. A Unique Catalyst Effects the Rapid Room-Temperature Cross-Coupling of Organozinc Reagents with Carboxylic Acid Fluorides, Chlorides, Anhydrides, and Thioesters. J. Am. Chem. Soc. 2004, 126, 15964–15965. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Maegawa, Y.; Sakino, D.; Sakai, N. Palladium-Catalyzed Coupling of Benzoyl Halides with Aryltrifluorosilanes Leading to Diaryl Ketones. Chem. Lett. 2016, 45, 790–792. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Sakino, D.; Sakurai, Y.; Sakai, N. Acid Fluorides as Acyl Electrophiles in Suzuki-Miyaura Coupling. Eur. J. Org. Chem. 2017, 4324–4327. [Google Scholar] [CrossRef]
- Liu, C.; Szostak, M. Decarbonylative cross-coupling of amides. Org. Biomol. Chem. 2018, 16, 7998–8010. [Google Scholar] [CrossRef]
- Zhao, Q.; Szostak, M. Redox-Neutral Decarbonylative Cross-Couplings Coming of Age. ChemSusChem 2019, 12. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Sakai, N. Acyl Fluorides in Late Transition-Metal Catalysis. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Sakurai, Y.; Hattori, H.; Sakai, N. Palladium-Catalyzed Reductive Conversion of Acyl Fluorides via Ligand-Controlled Decarbonylation. Org. Lett. 2018, 20, 4204–4208. [Google Scholar] [CrossRef] [PubMed]
- Keaveney, S.T.; Schoenebeck, F. Palladium-Catalyzed Decarbonylative Trifluoromethylation of Acid Fluorides. Angew. Chem. Int. Ed. 2018, 57, 4073–4077. [Google Scholar] [CrossRef]
- Malapit, C.A.; Bour, J.R.; Brigham, C.E.; Sanford, M.S. Base-free nickel-catalysed decarbonylative Suzuki-Miyaura coupling of acid fluorides. Nature 2018, 563, 100–104. [Google Scholar] [CrossRef]
- Sakurai, S.; Yoshida, T.; Tobisu, M. Iridium-catalyzed decarbonylative coupling of acyl fluorides with arenes and heteroarenes via C-H activation. Chem. Lett. 2019, 48, 94–97. [Google Scholar] [CrossRef]
- Okuda, Y.; Xu, J.; Ishida, T.; Wang, C.; Nishihara, Y. Nickel-Catalyzed Decarbonylative Alkylation of Aroyl Fluorides Assisted by Lewis-Acidic Organoboranes. ACS Omega 2018, 3, 13129–13140. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Asanuma, Y.; Nishihara, Y. Synthesis of 2-Substituted Propenes by Bidentate Phosphine-Assisted Methylenation of Acyl Fluorides and Acyl Chlorides with AlMe3. Org. Lett. 2019. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Nishihara, Y. Nickel-catalysed decarbonylative borylation of aroyl fluorides. Chem. Commun. 2018, 54, 13969–13972. [Google Scholar] [CrossRef]
- Liu, C.; Ji, C.L.; Hong, X.; Szostak, M. Palladium-Catalyzed Decarbonylative Borylation of Carboxylic Acids: Tuning Reaction Selectivity by Computation. Angew. Chem. Int. Ed. 2018, 57, 16721–16726. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Decarbonylative Borylation of Amides by Palladium Catalysis. ACS Omega 2019, 4, 4901–4907. [Google Scholar] [CrossRef]
- Hassan, J.; Sevignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl-Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef]
- Cordovilla, C.; Bartolom, C.; Martinez-Ilarduya, J.M.; Espinet, P. The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053. [Google Scholar] [CrossRef]
- Mailhol, D.; Willwacher, J.; Kausch-Busies, N.; Rubitski, E.E.; Sobol, Z.; Schuler, M.; Lam, M.H.; Musto, S.; Loganzo, F.; Maderna, A.; et al. Synthesis, Molecular Editing, and Biological Assessment of the Potent Cytotoxin Leiodermatolide. J. Am. Chem. Soc. 2014, 136, 15719–15729. [Google Scholar] [CrossRef]
- Li, J.; Yang, P.; Yao, M.; Deng, J.; Li, A. Total Synthesis of Rubriflordilactone A. J. Am. Chem. Soc. 2014, 136, 16477–16480. [Google Scholar] [CrossRef]
- Logan, M.M.; Toma, T.; Thomas-Tran, R.; Du Bois, J. Asymmetric synthesis of batrachotoxin: Enantiomeric toxins show functional divergence against NaV. Science 2016, 354, 865–869. [Google Scholar] [CrossRef]
- Wursthorn, K.R.; Kuivila, H.G. Synthesis of substituted aryltrimethylstannanes by the reaction of trimethylstannylsodium with aryl bromides. J. Organomet. Chem. 1977, 140, 29–33. [Google Scholar] [CrossRef]
- Knochel, P.; Singer, R.D. Preparation and Reactions of Polyfunctional Organozinc Reagents in Organic Synthesis. Chem. Rev. 1993, 93, 2117–2188. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Sun, B.; Peng, L.Z.; Li, Y.; Li, Y.L. Novel Access to Organostannane Compounds under Ultrasound Irradiation. Chin. J. Chem. 2004, 22, 1382–1383. [Google Scholar] [CrossRef]
- Murata, M.; Watanabe, S.; Masuda, Y. Palladium-Catalyzed Cross-Coupling Reaction of Tributyltin Hydride with Aryl Iodides: Formation of A Tin-Carbon Bond. Synlett 2000, 7, 1043–1045. [Google Scholar] [CrossRef]
- Komeyama, K.; Asakura, R.; Takaki, K. A Sn atom-economical approach toward arylstannanes: Ni-catalysed stannylation of aryl halides using Bu3SnOMe. Org. Biomol. Chem. 2015, 13, 8713–8716. [Google Scholar] [CrossRef]
- Azizian, H.; Eaborn, C.; Pidcock, A. Synthesis of organotrialkylstannanes. The reaction between organic halides and hexaalkyldistannanes in the presence of palladium complexes. J. Organomet. Chem. 1981, 215, 49–58. [Google Scholar] [CrossRef]
- Gribanov, P.S.; Golenko, Y.D.; Topchiy, M.A.; Minaeva, L.I.; Asachenko, A.F.; Nechaev, M.S. Stannylation of Aryl Halides, Stille Cross-Coupling, and One-Pot, Two-Step Stannylation/Stille Cross-Coupling Reactions under Solvent-Free Conditions. Eur. J. Org. Chem. 2018, 120–125. [Google Scholar] [CrossRef]
- Gu, Y.; Martin, R. Ni-Catalyzed Stannylation of Aryl Esters via C–O Bond Cleavage. Angew. Chem. Int. Ed. 2017, 56, 3187–3190. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhu, C.; Rueping, M. Catalytic Ester to Stannane Functional Group Interconversion via Decarbonylative Cross-Coupling of Methyl Esters. Org. Lett. 2018, 20, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Fahey, D.R.; Mahan, J.E. Oxidative Additions of Aryl, Vinyl, and Acyl Halides to Triethylphosphinenickel(0) Complexes. J. Am. Chem. Soc. 1977, 99, 2501–2508. [Google Scholar] [CrossRef]
- Lee, L.; Shim, C.S.; Chung, S.Y.; Kim, H.Y.; Lee, H.W. Cross-interaction constants as a measure of the transition-state structure. Part 1. The degree of bond formation in nucleophilic substitution reactions. J. Chem. Soc. Perkin. Trans. 2 1988, 0, 1919–1923. [Google Scholar] [CrossRef]
- Lal, G.S.; Pez, G.P.; Pesaresi, R.J.; Prozonic, F.M.; Cheng, H.S. Bis(2-methoxyethyl)aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability. J. Org. Chem. 1999, 64, 7048–7054. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Wang, C.; Uchiyama, M. Stannyl-Lithium: A Facile and Efficient Synthesis Facilitating Further Applications. J. Am. Chem. Soc. 2015, 137, 10488–10491. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- Akram, M.O.; Shinde, P.S.; Chintawarc, C.C.; Patil, N.T. Gold(I)-catalyzed cross-coupling reactions of aryldiazonium salts with organostannanes. Org. Biomol. Chem. 2018, 16, 2865–2869. [Google Scholar] [CrossRef]
- Abe, M.; Mori, T.; Osaka, I.; Sugimoto, K.; Takimiya, K. Thermally, Operationally, and Environmentally Stable Organic Thin-Film Transistors Based on Bis[1]benzothieno[2,3-d:2′,3′-d′]naphtho[2,3-b:6,7-b′]dithiophene Derivatives: Effective Synthesis, Electronic Structures, and Structure–Property Relationship. Chem. Mater. 2015, 27, 5049–5057. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3a–3t are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | [M] | Base | 2 (equiv) | Time (h) | Yield of 3a (%) 1 |
|---|---|---|---|---|---|
| 1 | FeCl2 | CsF | 1.5 | 24 | 29 |
| 2 | CoCl2 | CsF | 1.5 | 24 | 21 |
| 3 | NiCl2 | CsF | 1.5 | 24 | 90 |
| 4 | NiBr2 | CsF | 1.5 | 24 | 68 |
| 5 | Ni(cod)2 | CsF | 1.5 | 24 | 16 |
| 6 | PdCl2 | CsF | 1.5 | 24 | 4 |
| 7 | NiCl2 | Cs2CO3 | 1.5 | 24 | 48 |
| 8 | NiCl2 | KF | 1.5 | 24 | 0 |
| 9 | NiCl2 | CsF | 1.2 | 24 | 94 (90) |
| 10 | NiCl2 | CsF | 1.0 | 24 | 58 |
| 11 | NiCl2 | CsF | 1.2 | 18 | 91 |
| 12 | NiCl2 | CsF | 1.2 | 12 | 87 |
| 13 | NiCl2 | CsF | 1.2 | 6 | 76 |

 3a, 90% |  3b, 81% |  3c, 63% |  3d, 67% |
 3e, 82% |  3f, 56% |  3g, 85% |  3h, 64% |
 3i, 61% |  3j, 50% |  3k, 87% |  3l, 61% |
 3m, 86% |  3n, 72% |  3o, 90% |  3p, 51% |
 3q, 79% |  3r, 82% |  3s, 62% |  3t, 84% |

| Entry | Deviation from Standard Conditions | GC Yield (%) 1 | ||
|---|---|---|---|---|
| 2 | 3a | 5 | ||
| 1 | none | 0 | 94 | 13 |
| 2 | without CsF | 120 | 0 | 0 |
| 3 | without NiCl2 | 0 | 0 | 63 |
| 4 2 | 5 instead of 2 | - | 0 | >99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, Z.; Liu, L.; Asanuma, Y.; Nishihara, Y. Nickel-Catalyzed Decarbonylative Stannylation of Acyl Fluorides under Ligand-Free Conditions. Molecules 2019, 24, 1671. https://doi.org/10.3390/molecules24091671
Wang X, Wang Z, Liu L, Asanuma Y, Nishihara Y. Nickel-Catalyzed Decarbonylative Stannylation of Acyl Fluorides under Ligand-Free Conditions. Molecules. 2019; 24(9):1671. https://doi.org/10.3390/molecules24091671
Chicago/Turabian StyleWang, Xiu, Zhenhua Wang, Li Liu, Yuya Asanuma, and Yasushi Nishihara. 2019. "Nickel-Catalyzed Decarbonylative Stannylation of Acyl Fluorides under Ligand-Free Conditions" Molecules 24, no. 9: 1671. https://doi.org/10.3390/molecules24091671
APA StyleWang, X., Wang, Z., Liu, L., Asanuma, Y., & Nishihara, Y. (2019). Nickel-Catalyzed Decarbonylative Stannylation of Acyl Fluorides under Ligand-Free Conditions. Molecules, 24(9), 1671. https://doi.org/10.3390/molecules24091671