
Ni/Co-Catalyzed Homo-Coupling of Alkyl Tosylates



Abstract
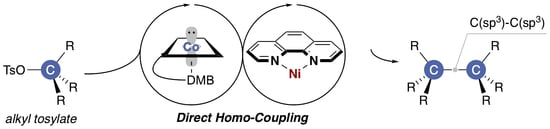
1. Introduction
2. Results and Discussion
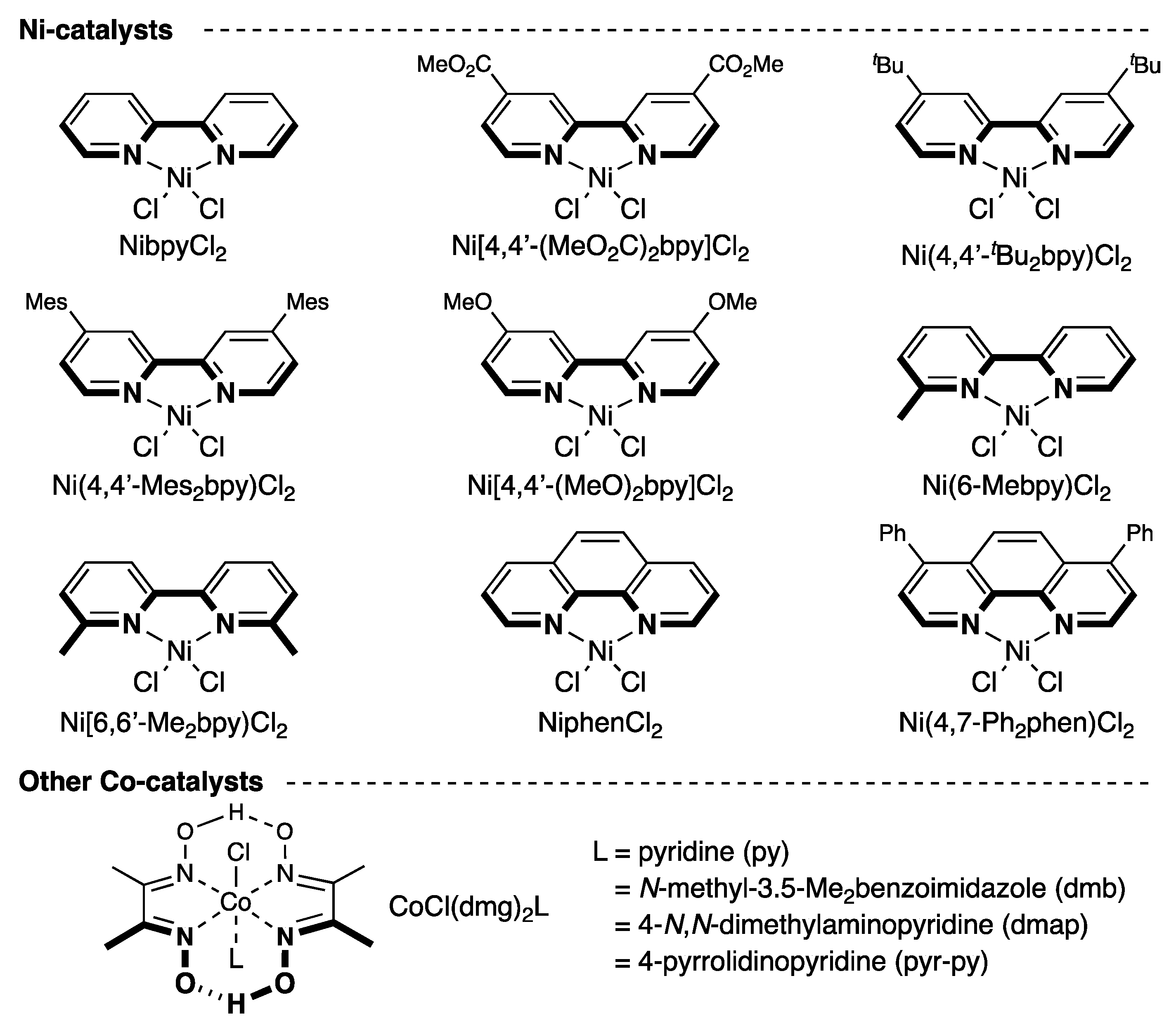
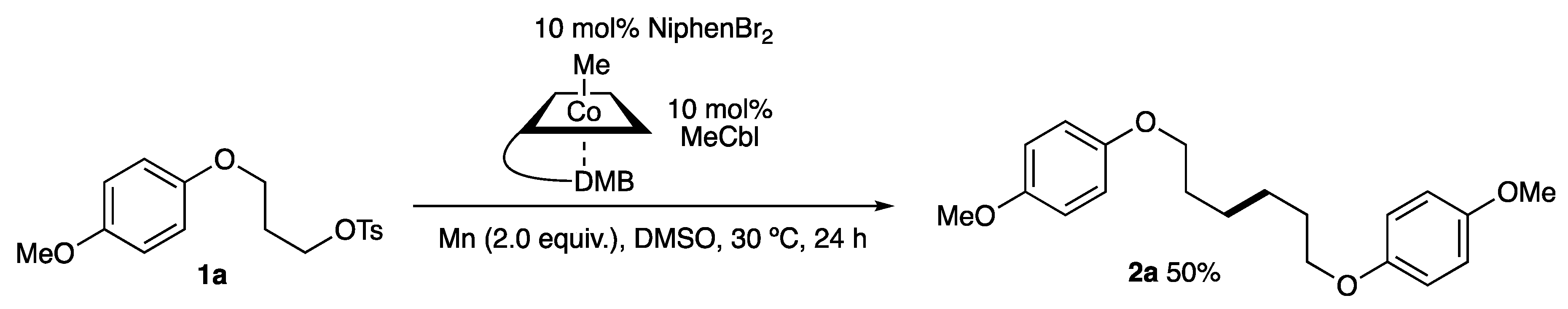
2.1. Screening of Reaction Conditions
2.2. Substrate Scope
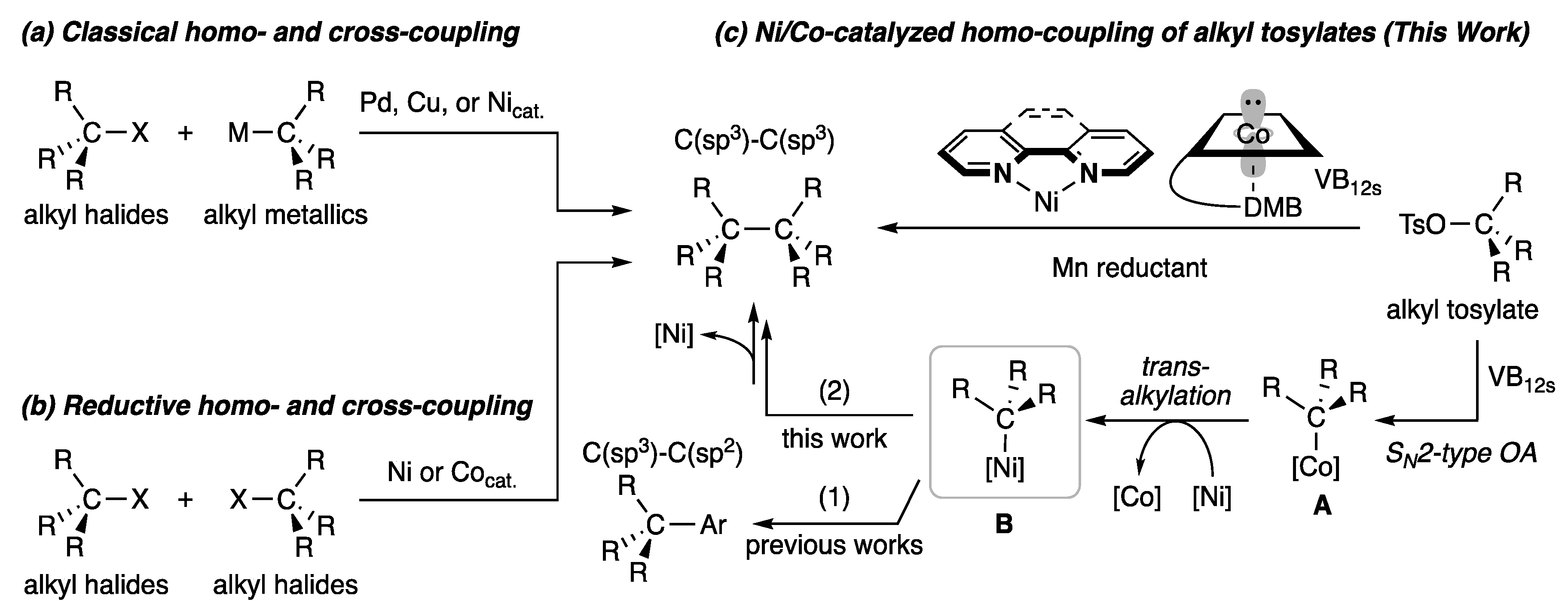
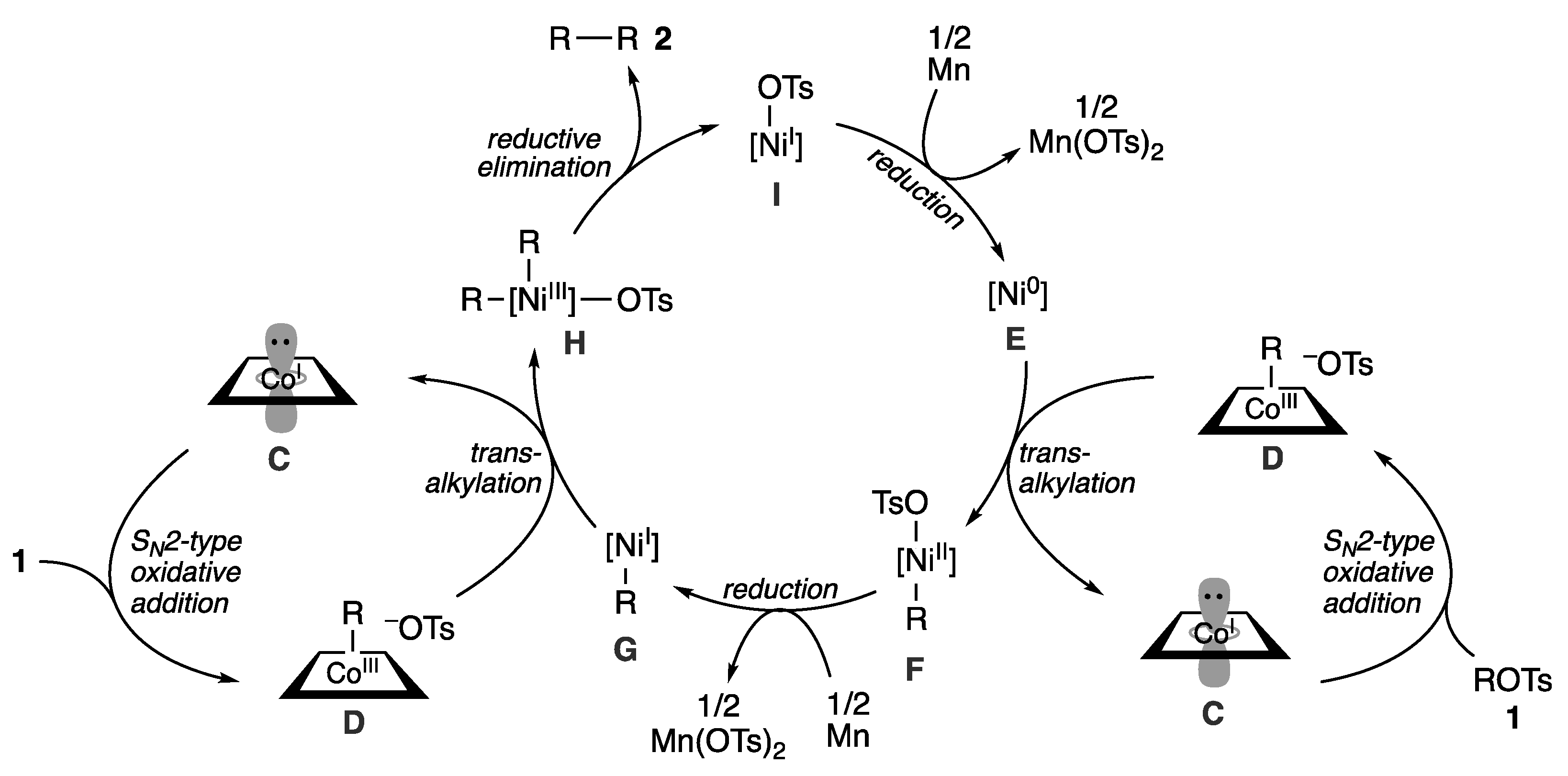
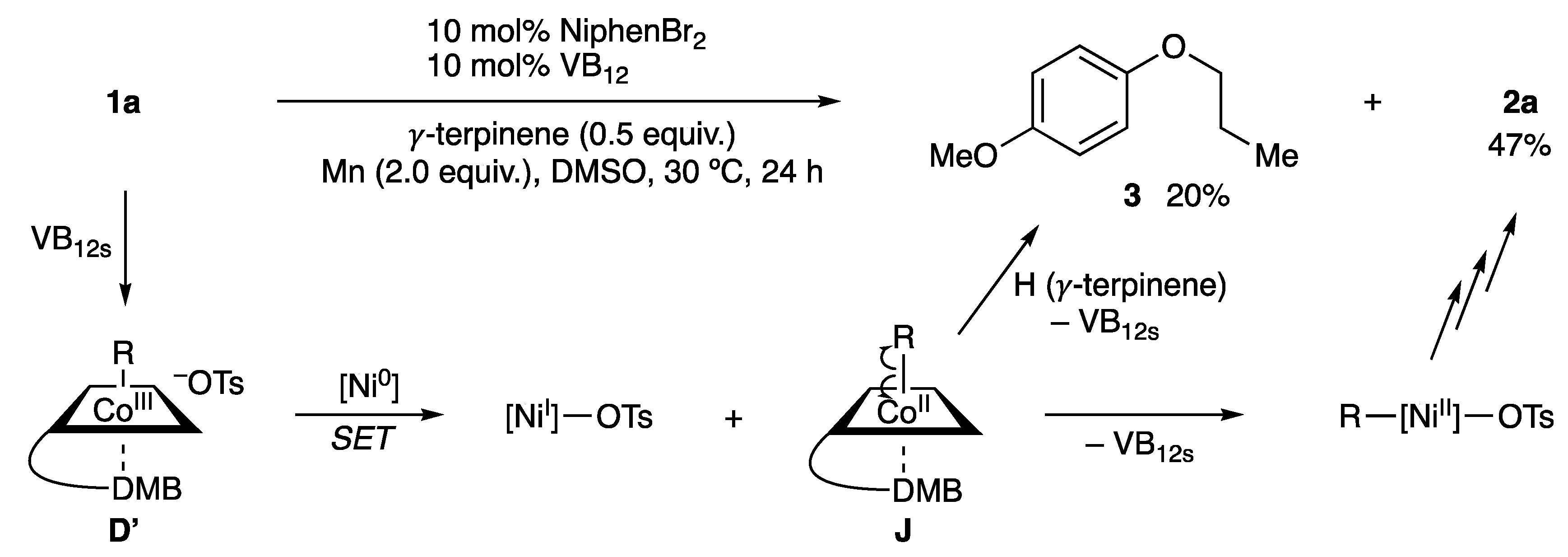
2.3. Plausible Reaction Mechanism
3. Materials and Methods
3.1. General Information
3.2. General Procedure of the NiBr2phen/VB12-Catalyzed Homo-Coupling of Alkyl Tosylates
3.3. Product Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef]
- Netherton, M.R.; Dai, C.Y.; Neuschütz, K.; Fu, G.C. Room-temperature alkyl-alkyl Suzuki cross-coupling of alkyl bromides that possess beta hydrogens. J. Am. Chem. Soc. 2001, 123, 10099–10100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fu, G.C. Palladium-Catalyzed Negishi Cross-Coupling Reactions of Unactivated Alkyl Iodides, Bromides, Chlorides, and Tosylates. J. Am. Chem. Soc. 2003, 125, 12527–12530. [Google Scholar] [CrossRef]
- Hadei, N.; Kantchev, E.A.B.; O’Brien, C.J.; Organ, M.G. Room-temperature Negishi cross-coupling of unactivated alkyl bromides with alkyl organozinc reagents utilizing a Pd/N-heterocyclic carbene catalyst. J. Org. Chem. 2005, 70, 8503–8507. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.H.; Miller, J.D.; Chan, H.-K.; Delaney, M.O. Scope and Utility of a New Soluble Copper Catalyst [CuBr−LiSPh−LiBr−THF]: A Comparison with Other Copper Catalysts in Their Ability to Couple One Equivalent of a Grignard Reagent with an Alkyl Sulfonate. J. Am. Chem. Soc. 1997, 119, 2125–2133. [Google Scholar] [CrossRef]
- Terao, J.; Todo, H.; Begum, S.A.; Kuniyasu, H.; Kambe, N. Copper-catalyzed cross-coupling reaction of grignard reagents with primary-alkyl halides: Remarkable effect of 1-phenylpropyne. Angew. Chem. Int. Ed. 2007, 46, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- Devasagayaraj, A.; Stüdemann, T.; Knochel, P. A New Nickel-Catalyzed Cross-Coupling Reaction between sp3 Carbon Centers. Angew. Chem. Int. Ed. 1995, 34, 2723–2725. [Google Scholar] [CrossRef]
- Terao, J.; Ikumi, A.; Kuniyasu, H.; Kambe, N. Ni- or Cu-catalyzed cross-coupling reaction of alkyl fluorides with Grignard reagents. J. Am. Chem. Soc. 2003, 125, 5646–5647. [Google Scholar] [CrossRef]
- Saito, B.; Fu, G.C. Alkyl-alkyl suzuki cross-couplings of unactivated secondary alkyl halides at room temperature. J. Am. Chem. Soc. 2007, 129, 9602–9603. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Gagné, M.R. Diastereoselective Ni-Catalyzed Negishi Cross-Coupling Approach to Saturated, Fully Oxygenated C-Alkyl and C-Aryl Glycosides. J. Am. Chem. Soc. 2008, 130, 12177–12183. [Google Scholar] [CrossRef]
- Vechorkin, O.; Hu, X. Nickel-catalyzed cross-coupling of non-activated and functionalized alkyl halides with alkyl Grignard reagents. Angew. Chem. Int. Ed. 2009, 48, 2937–2940. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Cornella, J.; Li, C.; Malins, L.R.; Edwards, J.T.; Kawamura, S.; Maxwell, B.D.; Eastgate, M.D.; Baran, P.S. A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Choi, J.; Liu, A.T.; Slusarczyk, M.; Fu, G.C. A general, modular method for the catalytic asymmetric synthesis of alkylboronate esters. Science 2016, 354, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Auffrant, A.; Felouat, A.; Gosmini, C. Cobalt-Catalyzed Reductive Allylation of Alkyl Halides with Allylic Acetates or Carbonates. Angew. Chem. Int. Ed. 2011, 50, 10402–10405. [Google Scholar] [CrossRef]
- Yu, X.; Yang, T.; Wang, S.; Xu, H.; Gong, H. Nickel-catalyzed reductive cross-coupling of unactivated alkyl halides. Org. Lett. 2011, 13, 2138–2141. [Google Scholar] [CrossRef]
- Dai, Y.; Wu, F.; Zang, Z.; You, H.; Gong, H. Ni-Catalyzed Reductive Allylation of Unactivated Alkyl Halides with Allylic Carbonates. Chem. Eur. J. 2012, 18, 808–812. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, C.; Qian, Q.; Deng, W.; Gong, H. Nickel-catalyzed cross-coupling of unactivated alkyl halides using bis(pinacolato)diboron as reductant. Chem. Sci. 2013, 4, 4022. [Google Scholar] [CrossRef]
- Chen, H.; Jia, X.; Yu, Y.; Qian, Q.; Gong, H. Nickel-Catalyzed Reductive Allylation of Tertiary Alkyl Halides with Allylic Carbonates. Angew. Chem. Int. Ed. 2017, 129, 13283–13286. [Google Scholar] [CrossRef]
- Smith, R.T.; Zhang, X.; Rincon, J.A.; Agejas, J.; Mateos, C.; Barberis, M.; García-Cerrada, S.; de Frutos, O.; MacMillan, D.W.C. Metallaphotoredox-Catalyzed Cross-Electrophile Csp3-Csp3 Coupling of Aliphatic Bromides. J. Am. Chem. Soc. 2018, 140, 17433–17438. [Google Scholar] [CrossRef]
- Goldup, S.M.; Leigh, D.A.; McBurney, R.T.; McGonigal, P.R.; Plant, A. Ligand-assisted nickel-catalysed sp3–sp3 homocoupling of unactivated alkyl bromides and its application to the active template synthesis of rotaxanes. Chem. Sci. 2010, 1, 383. [Google Scholar] [CrossRef]
- Prinsell, M.R.; Everson, D.A.; Weix, D.J. Nickel-catalyzed, sodium iodide-promoted reductive dimerization of alkyl halides, alkyl pseudohalides, and allylic acetates. Chem. Commun. 2010, 46, 5743–5745. [Google Scholar] [CrossRef]
- Peng, Y.; Luo, L.; Yan, C.S.; Zhang, J.-J.; Wang, Y.W. Ni-Catalyzed Reductive Homocoupling of Unactivated Alkyl Bromides at Room Temperature and Its Synthetic Application. J. Org. Chem. 2013, 78, 10960–10967. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Qian, X.; Gosmini, C. Cobalt-Catalyzed Csp3−Csp3 Homocoupling. Adv. Synth. Catal. 2016, 358, 2427–2430. [Google Scholar] [CrossRef]
- Yang, C.-T.; Zhang, Z.-Q.; Liang, J.; Liu, J.-H.; Lu, X.-Y.; Chen, H.-H.; Liu, L. Copper-catalyzed cross-coupling of nonactivated secondary alkyl halides and tosylates with secondary alkyl Grignard reagents. J. Am. Chem. Soc. 2012, 134, 11124–11127. [Google Scholar] [CrossRef]
- Powell, D.A.; Fu, G.C. Nickel-Catalyzed Cross-Couplings of Organosilicon Reagents with Unactivated Secondary Alkyl Bromides. J. Am. Chem. Soc. 2004, 126, 7788–7789. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.A.; Maki, T.; Fu, G.C. Stille cross-couplings of unactivated secondary alkyl halides using monoorganotin reagents. J. Am. Chem. Soc. 2005, 127, 510–511. [Google Scholar] [CrossRef] [PubMed]
- González-Bobes, F.; Fu, G.C. Amino Alcohols as Ligands for Nickel-Catalyzed Suzuki Reactions of Unactivated Alkyl Halides, Including Secondary Alkyl Chlorides, with Arylboronic Acids. J. Am. Chem. Soc. 2006, 128, 5360–5361. [Google Scholar] [CrossRef]
- Dudnik, A.S.; Fu, G.C. Nickel-catalyzed coupling reactions of alkyl electrophiles, including unactivated tertiary halides, to generate carbon-boron bonds. J. Am. Chem. Soc. 2012, 134, 10693–10697. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Fu, G.C. Nickel-Catalyzed Alkyl-Alkyl Cross-Couplings of Fluorinated Secondary Electrophiles: A General Approach to the Synthesis of Compounds having a Perfluoroalkyl Substituent. Angew. Chem. Int. Ed. 2015, 54, 9047–9051. [Google Scholar] [CrossRef]
- Lévêque, C.; Corcé, V.; Chenneberg, L.; Ollivier, C.; Fensterbank, L. Photoredox/Nickel Dual Catalysis for the C(sp3)–C(sp3) Cross-Coupling of Alkylsilicates with Alkyl Halides. Eur. J. Org. Chem. 2017, 2017, 2118–2121. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Y.; Zhang, B.; Pi, J.-J.; Wang, X.-X.; Gong, T.-J.; Xiao, B.; Fu, Y. Nickel-Catalyzed Defluorinative Reductive Cross-Coupling of gem-Difluoroalkenes with Unactivated Secondary and Tertiary Alkyl Halides. J. Am. Chem. Soc. 2017, 139, 12632–12637. [Google Scholar] [CrossRef] [PubMed]
- Komeyama, K.; Ohata, R.; Kiguchi, S.; Osaka, I. Highly nucleophilic vitamin B12-assisted nickel-catalysed reductive coupling of aryl halides and non-activated alkyl tosylates. Chem. Commun. 2017, 53, 6401–6404. [Google Scholar] [CrossRef] [PubMed]
- Komeyama, K.; Yamahata, Y.; Osaka, I. Nickel and Nucleophilic Cobalt-Catalyzed Trideuteriomethylation of Aryl Halides Using Trideuteriomethyl p-Toluenesulfonate. Org. Lett. 2018, 20, 4375–4378. [Google Scholar] [CrossRef]
- Ito, S.; Fujiwara, Y.-I.; Nakamura, E.; Nakamura, M. Iron-Catalyzed Cross-Coupling of Alkyl Sulfonates with Arylzinc Reagents. Org. Lett. 2009, 11, 4306–4309. [Google Scholar] [CrossRef] [PubMed]
- Closson, W.D.; Wriede, P.; Bank, S. Reductive Cleavage of Toluenesulfonates with Sodium Naphthalene 1. J. Am. Chem. Soc. 1966, 88, 1581–1583. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Wilhelm, R.S.; Nugent, S.T.; Little, R.D.; Baizer, M.M. Electrochemical peak potentials of typical substrates used for coupling reactions with organocuprates: Effects of solvent. J. Org. Chem. 1983, 48, 3306–3308. [Google Scholar] [CrossRef]
- Sridhar, M.; Kumar, B.A.; Narender, R. Expedient and simple method for regeneration of alcohols from toluenesulfonates using Mg-MeOH. Tetrahedron Lett. 1998, 39, 2847–2850. [Google Scholar] [CrossRef]
- Kotsuki, H.; Kadota, I.; Ochi, M. A new expeditious synthesis of (+)-exo-brevicomin via efficient C-C bond formation of triflates. Tetrahedron Lett. 1989, 30, 3999–4000. [Google Scholar] [CrossRef]
- O’Donnel, C.J.; Burke, S.D. Selective Mesylation of Vicinal Diols: A Systematic Case Study. J. Org. Chem. 1998, 63, 8614–8616. [Google Scholar] [CrossRef]
- Bouzide, A.; Sauvé, G. Silver(I) Oxide Mediated Highly Selective Monotosylation of Symmetrical Diols. Application to the Synthesis of Polysubstituted Cyclic Ethers. Org. Lett. 2002, 4, 2329–2332. [Google Scholar] [CrossRef] [PubMed]
- Ram, M.S.; Riordan, C.G. Methyl transfer from a cobalt complex to Ni(tmc)+ yielding Ni(tmc)Me+: A model for methylcobalamin alkylation of CO dehydrogenase. J. Am. Chem. Soc. 1995, 117, 2365–2366. [Google Scholar] [CrossRef]
- Eckert, N.A.; Dougherty, W.G.; Yap, G.P.A.; Riordan, C.G. Methyl Transfer from Methylcobaloxime to (Triphos)Ni(PPh3): Relevance to the Mechanism of Acetyl Coenzyme A Synthase. J. Am. Chem. Soc. 2007, 129, 9286–9287. [Google Scholar] [CrossRef]
- Gansäuer, A.; Fleckhaus, A.; Lafont, M.A.; Okkel, A.; Kotsis, K.; Anoop, A.; Neese, F. Catalysis via Homolytic Substitutions with C−O and Ti−O Bonds: Oxidative Additions and Reductive Eliminations in Single Electron Steps. J. Am. Chem. Soc. 2009, 131, 16989–16999. [Google Scholar] [CrossRef]
- Martin, B.D.; Finke, R.G. Cobalt-carbon homolysis and bond dissociation energy studies of biological alkylcobalamins: Methylcobalamin, including a > 1015 Co-CH3 homolysis rate enhancement at 25 °C following one-electron reduction. J. Am. Chem. Soc. 1990, 112, 2419–2420. [Google Scholar] [CrossRef]
- Birke, R.L.; Huang, Q.; Spataru, T.; Gosser, D.K. Electroreduction of a Series of Alkylcobalamins: Mechanism of Stepwise Reductive Cleavage of the Co−C Bond. J. Am. Chem. Soc. 2006, 128, 1922–1936. [Google Scholar] [CrossRef]
- Bialek, M.; Cramail, H.; Deffieux, A.; Guillaume, S.M. Styrene polymerization using nickel(II) complexes as catalysts. Eur. Polym. J. 2005, 41, 2678–2684. [Google Scholar] [CrossRef]
- Panagiotopoulos, A.; Ladomenou, K.; Sun, D.; Artero, V.; Coutsolelos, A.G. Photochemical hydrogen production and cobaloximes: The influence of the cobalt axial N-ligand on the system stability. Dalton Trans. 2016, 45, 6732–6738. [Google Scholar] [CrossRef]
- Bissember, A.C.; Levina, A.; Fu, G.C. A Mild, Palladium-Catalyzed Method for the Dehydrohalogenation of Alkyl Bromides: Synthetic and Mechanistic Studies. J. Am. Chem. Soc. 2012, 134, 14232–14237. [Google Scholar] [CrossRef]
- Komber, H.; Müllers, S.; Lombeck, F.; Held, A.; Walter, M.; Sommer, M. Soluble and stable alternating main-chain merocyanine copolymers through quantitative spiropyran–merocyanine conversion. Polym. Chem. 2013, 5, 443–453. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ni catalysts | Yield of 2a (%) a | Conversion of 1a (%) a |
|---|---|---|---|
| 1 | NibpyCl2 | 72 | 100 |
| 2 b | NibpyCl2 | 0 | 20 |
| 3 | None | 0 | 32 |
| 4 c | NibpyCl2 | 0 | 0 |
| 5 d | NibpyCl2 | 38 | 65 |
| 6 | Ni(4,4’-(MeO2C)2bpy)Cl2 | 38 | 76 |
| 7 | Ni(4,4’-tBu2bpy)Cl2 | 18 | 67 |
| 8 | Ni(4,4’-Mes2bpy)Cl2 e | 38 | 78 |
| 9 | Ni[4,4’-(MeO)2bpy]Cl2 | 10 | 66 |
| 10 | Ni(6-Mebpy)Cl2 | 42 | 92 |
| 11 | Ni(6,6’-Me2bpy)Cl2 | 17 | 95 |
| 12 | Ni(1,10-phen)Cl2 | 75 | 100 |
| 13 | Ni(4,7-Ph2phen)Cl2 | 60 | 100 |
| 14 f | NiphenCl2 | 82 | 100 |
| 15 g | NiphenCl2 | 0 | 0 |
| 16 f | NiphenBr2 | 93 | 100 |
| 17 h | NiphenBr2 | 21–28 | 51–59 |

| Entry | Alkyl Tosylates 1 | Product 2 and Yield (%) a | |||
|---|---|---|---|---|---|
| 1 |  | 1b |  | 2b | 86 |
| 2 |  | 1c |  | 2c | 70 |
| 3 |  | 1d |  | 2d | 67 |
| 4 |  | 1e |  | 2e | 63 |
| 5 |  | 1f |  | 2f | 65 |
| 6 |  | 1g |  | 2g | 75 |
| 7 |  | 1h |  | 2h | 70 |
| 8 b |  | 1i |  | 2i | 60 |
| 9 |  | 1j |  | 2j | 73 |
| 10 |  | 1k |  | 2k | 90 [1:1] c |
| 11 d,e |  | 1l |  | 2l | 80 [1:1] c |
| 12 e, f |  | 1m |  | 2m | 65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komeyama, K.; Tsunemitsu, R.; Michiyuki, T.; Yoshida, H.; Osaka, I. Ni/Co-Catalyzed Homo-Coupling of Alkyl Tosylates. Molecules 2019, 24, 1458. https://doi.org/10.3390/molecules24081458
Komeyama K, Tsunemitsu R, Michiyuki T, Yoshida H, Osaka I. Ni/Co-Catalyzed Homo-Coupling of Alkyl Tosylates. Molecules. 2019; 24(8):1458. https://doi.org/10.3390/molecules24081458
Chicago/Turabian StyleKomeyama, Kimihiro, Ryusuke Tsunemitsu, Takuya Michiyuki, Hiroto Yoshida, and Itaru Osaka. 2019. "Ni/Co-Catalyzed Homo-Coupling of Alkyl Tosylates" Molecules 24, no. 8: 1458. https://doi.org/10.3390/molecules24081458
APA StyleKomeyama, K., Tsunemitsu, R., Michiyuki, T., Yoshida, H., & Osaka, I. (2019). Ni/Co-Catalyzed Homo-Coupling of Alkyl Tosylates. Molecules, 24(8), 1458. https://doi.org/10.3390/molecules24081458