
Modern Methods of Sample Preparation for the Analysis of Oxylipins in Biological Samples



Abstract
:
1. Introduction
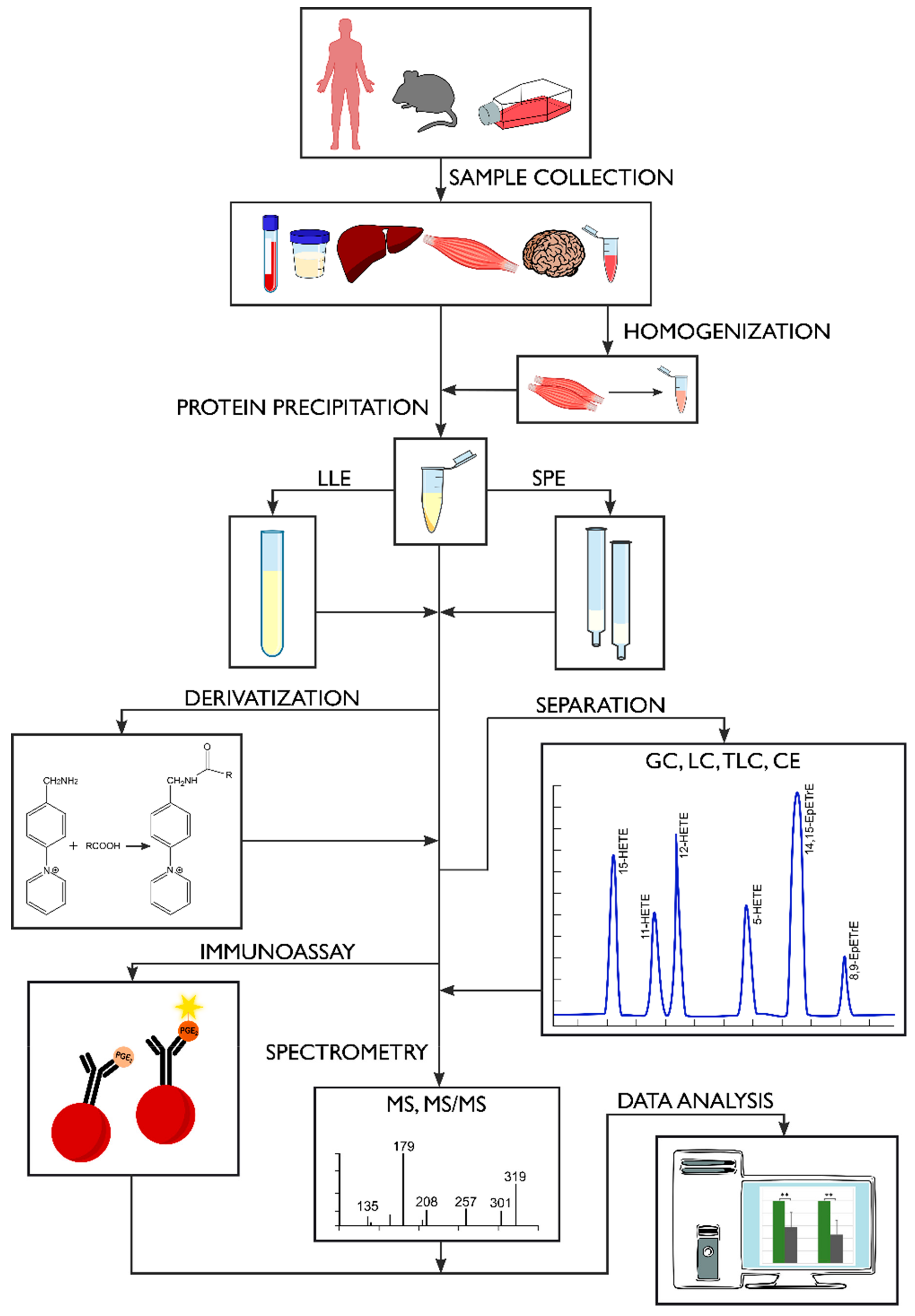
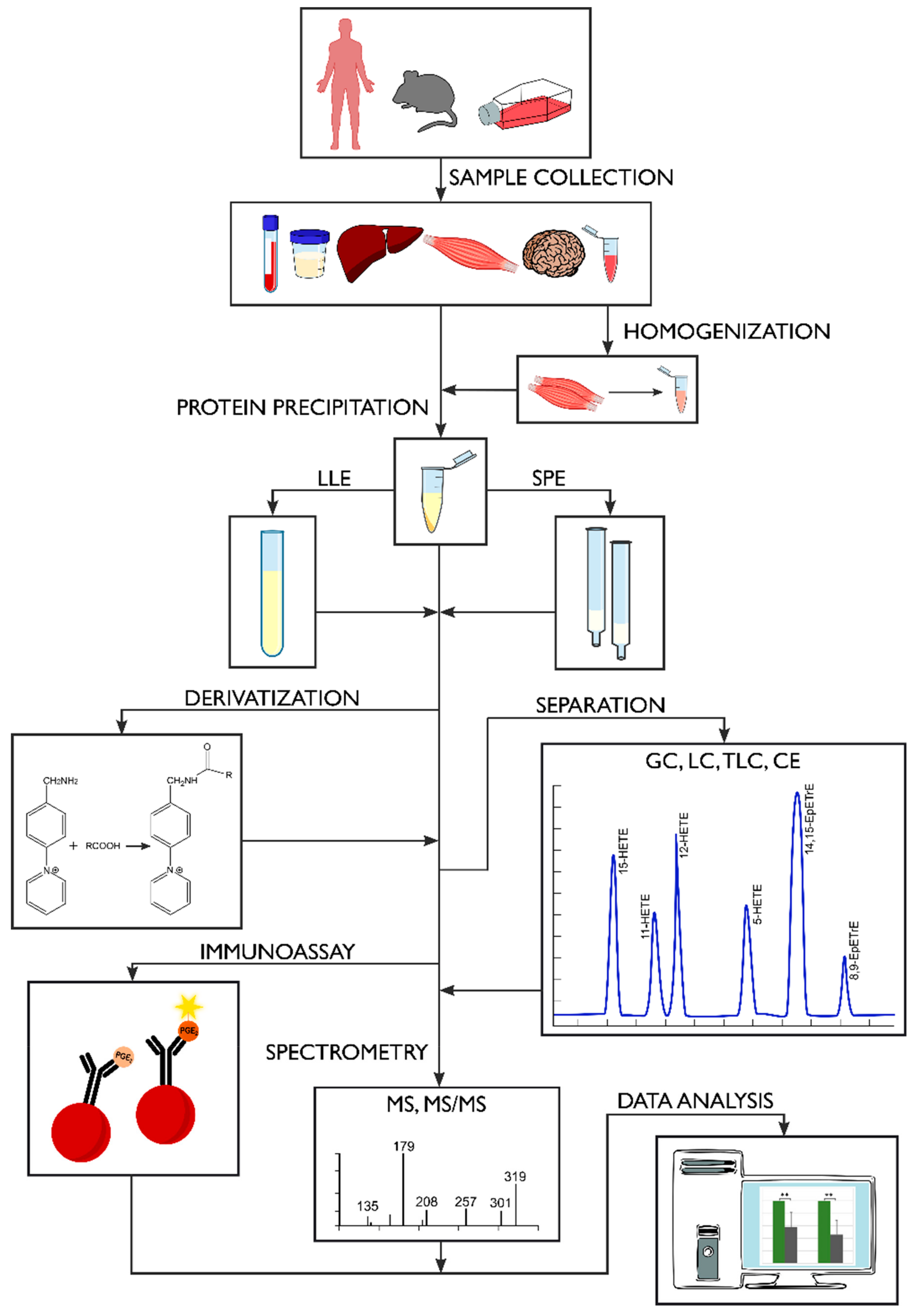
2. Sample Preparation
2.1. Sample Collection and Storage
2.2. Pre-Extraction Additives
2.2.1. Antioxidants
2.2.2. Standards
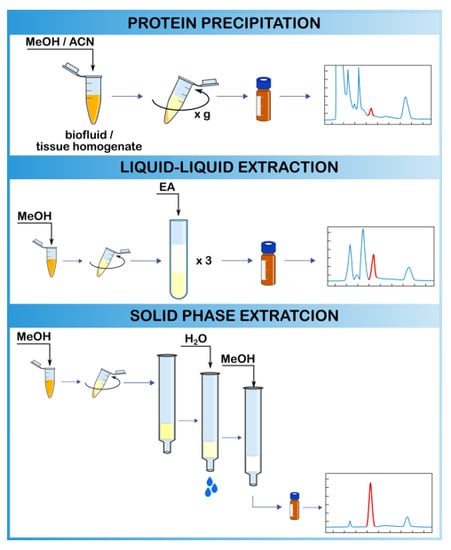
2.3. Extraction Methods
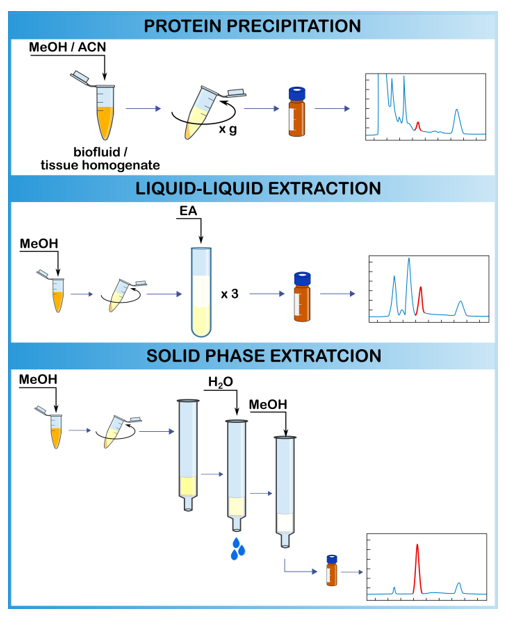
2.3.1. Protein Precipitation
2.3.2. Liquid–Liquid Extraction
Biofluids
Solid Tissues
Cell Cultures
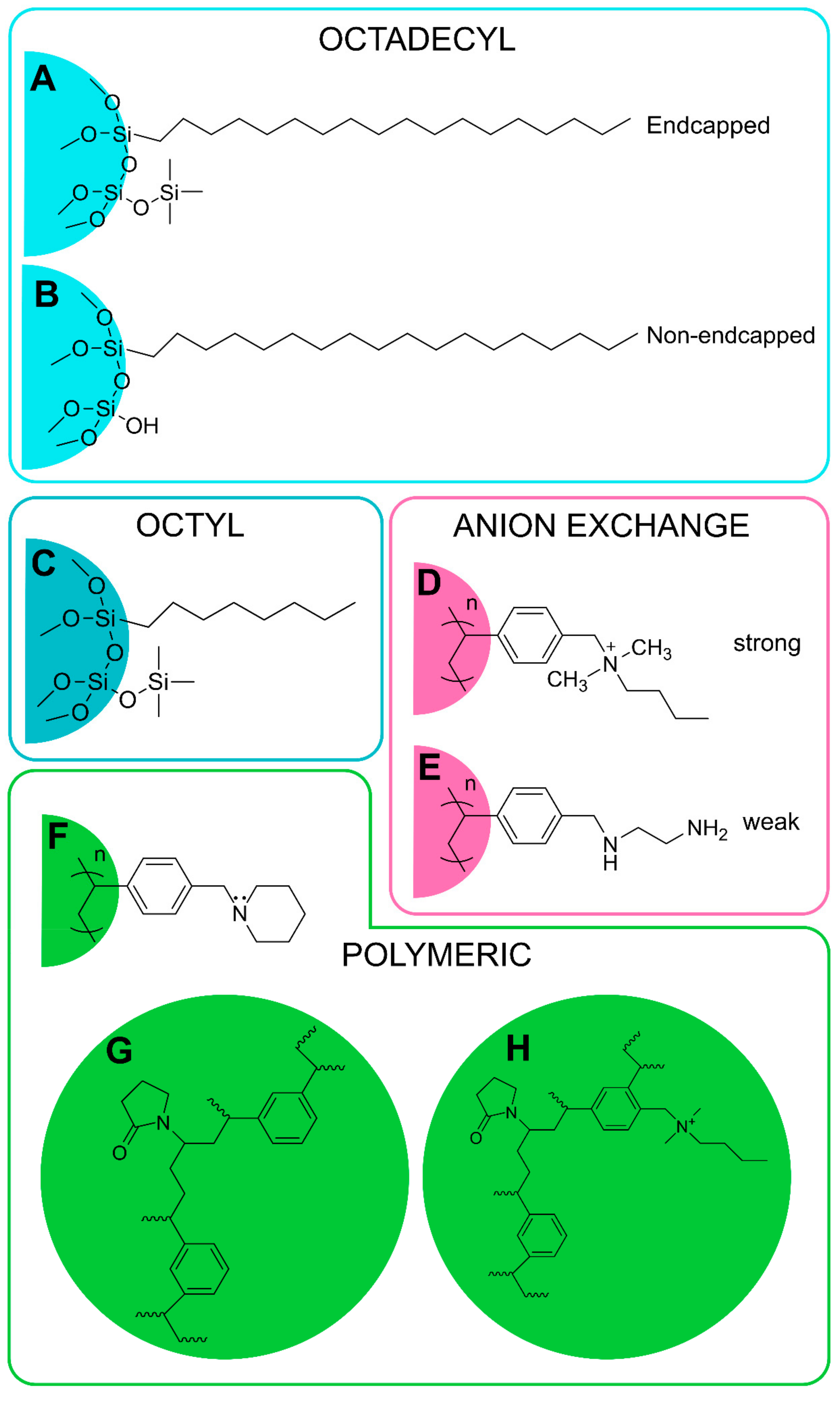
2.3.3. Solid-Phase Extraction
Biofluids
Blood/Serum/Plasma
Urine
Other Biofluids
Solid Tissues
Cell Cultures
2.3.4. LLE or SPE?
2.3.5. New Approaches in Oxylipin Extraction
2.4. Derivatization Process
3. Methods of Oxylipin Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. An Int. Rev. J. 2015, 6, 513–540. [Google Scholar] [CrossRef] [PubMed]
- Vigor, C.; Bertrand-Michel, J.; Pinot, E.; Oger, C.; Vercauteren, J.; Le Faouder, P.; Galano, J.M.; Lee, J.C.Y.; Durand, T. Non-enzymatic lipid oxidation products in biological systems: ASSESSMENT of the metabolites from polyunsaturated fatty acids. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 964, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Shearer, G.C.; Walker, R.E. An overview of the biologic effects of omega-6 oxylipins in humans. Prostaglandins Leukot. Essent. Fat. Acids 2018, 137, 26–38. [Google Scholar] [CrossRef]
- Khan, S.A.; Ali, A.; Khan, S.A.; Zahran, S.A.; Damanhouri, G.; Azhar, E.; Qadri, I. Unraveling the Complex Relationship Triad between Lipids, Obesity, and Inflammation. Mediators Inflamm. 2014, 2014, 1–16. [Google Scholar] [CrossRef]
- Yeung, J.; Hawley, M.; Holinstat, M. The expansive role of oxylipins on platelet biology. J. Mol. Med. 2017, 95, 575–588. [Google Scholar] [CrossRef]
- Christophersen, O.A.; Haug, A. Animal products, diseases and drugs: a plea for better integration between agricultural sciences, human nutrition and human pharmacology. Lipids Health Dis. 2011, 10, 16. [Google Scholar] [CrossRef]
- Spector, A.A.; Kim, H.-Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dong, H.; Hammock, B.D. Profiling the regulatory lipids: Another systemic way to unveil the biological mystery. Curr. Opin. Lipidol. 2011, 22, 197–203. [Google Scholar] [CrossRef]
- Arnold, C.; Markovic, M.; Blossey, K.; Wallukat, G.; Fischer, R.; Dechend, R.; Konkel, A.; Von Schacky, C.; Luft, F.C.; Muller, D.N.; et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of ω-3 fatty acids. J. Biol. Chem. 2010, 285, 32720–32733. [Google Scholar] [CrossRef]
- Austin, C.; Sordillo, L.M.; Zhang, C.; Fenton, J.I. Obesity is positively associated with arachidonic acid-derived 5- and 11-hydroxyeicosatetraenoic acid (HETE). Metabolism 2017, 70, 177–191. [Google Scholar]
- Zhou, J.; Chen, L.; Liu, Z.; Sang, L.; Li, Y.; Yuan, D. Changes in erythrocyte polyunsaturated fatty acids and plasma eicosanoids level in patients with asthma. Lipids Health Dis. 2018, 17, 206. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Yang, J.; Eiserich, J.P.; Cross, C.E.; Morrissey, B.M.; Hammock, B.D. Metabolomic profiling of regulatory lipid mediators in sputum from adult cystic fibrosis patients. Free Radic. Biol. Med. 2012, 53, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Gupta, G.; Anilkumar, K.; Fatima, N.; Karnati, R.; Reddy, G.V.; Giri, P.V.; Reddanna, P. 15-Lipoxygenase metabolites of α-linolenic acid, [13-(S)-HPOTrE and 13-(S)-HOTrE], mediate anti-inflammatory effects by inactivating NLRP3 inflammasome. Sci. Rep. 2016, 6, 31649. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.-J.; Croft, K.D.; Mori, T.A.; Falck, J.R.; Beilin, L.J.; Puddey, I.B.; Barden, A.E. 20-HETE and F2-isoprostanes in the metabolic syndrome: The effect of weight reduction. Free Radic. Biol. Med. 2009, 46, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Shishehbor, M.H.; Zhang, R.; Medina, H.; Brennan, M.-L.; Brennan, D.M.; Ellis, S.G.; Topol, E.J.; Hazen, S.L. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic. Biol. Med. 2006, 41, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Foegh, M.L.; Zhao, Y.; Madren, L.; Rolnick, M.; Stair, T.O.; Huang, K.S.; Ramwell, P.W. Urinary thromboxane A 2 metabolites in patients presenting in the emergency room with acute chest pain. J. Intern. Med. 1994, 235, 153–161. [Google Scholar] [CrossRef]
- Tsikas, D. Application of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry to assess in vivo synthesis of prostaglandins, thromboxane, leukotrienes, isoprostanes and related compounds in humans. J. Chromatogr. B Biomed. Appl. 1998, 717, 201–245. [Google Scholar] [CrossRef]
- Berkecz, R.; Lísa, M.; Holčapek, M. Analysis of oxylipins in human plasma: Comparison of ultrahigh-performance liquid chromatography and ultrahigh-performance supercritical fluid chromatography coupled to mass spectrometry. J. Chromatogr. A 2017, 1511, 107–121. [Google Scholar] [CrossRef]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef]
- Xi, S.; Pham, H.; Ziboh, V.A. 15-Hydroxyeicosatrienoic acid (15-HETrE) suppresses epidermal hyperproliferation via the modulation of nuclear transcription factor (AP-1) and apoptosis. Arch. Dermatol. Res. 2000, 292, 397–403. [Google Scholar] [CrossRef]
- Möller, K.; Ostermann, A.I.; Rund, K.; Thoms, S.; Blume, C.; Stahl, F.; Hahn, A.; Schebb, N.H.; Schuchardt, J.P. Influence of weight reduction on blood levels of C-reactive protein, tumor necrosis factor-α, interleukin-6, and oxylipins in obese subjects. Prostaglandins Leukot. Essent. Fat. Acids 2016, 106, 39–49. [Google Scholar] [CrossRef]
- Virtue, S.; Masoodi, M.; de Weijer, B.A.M.; van Eijk, M.; Mok, C.Y.L.; Eiden, M.; Dale, M.; Pirraco, A.; Serlie, M.J.; Griffin, J.L.; et al. Prostaglandin profiling reveals a role for haematopoietic prostaglandin D synthase in adipose tissue macrophage polarisation in mice and humans. Int. J. Obes. 2015, 39, 1151–1160. [Google Scholar] [CrossRef]
- Hoopes, S.L.; Garcia, V.; Edin, M.L.; Schwartzman, M.L.; Zeldin, D.C. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat. 2015, 120, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Xu, Z.; Yin, X.; Zheng, F.; Lin, X.; Pan, Q.; Li, H. Inverse Relationship between Serum Lipoxin A4 Level and the Risk of Metabolic Syndrome in a Middle-Aged Chinese Population. PLoS ONE 2015, 10, e0142848. [Google Scholar] [CrossRef]
- Liu, J.-B.; Li, W.-J.; Fu, F.-M.; Zhang, X.-L.; Jiao, L.; Cao, L.-J.; Chen, L. Inverse correlation between serum adiponectin and 8-iso-prostaglandin F2α in newly diagnosed type 2 diabetes patients. Int. J. Clin. Exp. Med. 2015, 8, 6085–6090. [Google Scholar]
- Mukhtar, M.H.; El-Emshaty, H.M.; Alamodi, H.S.; Nasif, W.A. The Activity of Serum 8-Iso-Prostaglandin F2α as Oxidative Stress Marker in Patients with Diabetes Mellitus Type 2 and Associated Dyslipidemic Hyperglycemia. J. Diabetes Mellit. 2016, 6, 318–332. [Google Scholar] [CrossRef]
- Grapov, D.; Adams, S.H.; Pedersen, T.L.; Garvey, W.T.; Newman, J.W. Type 2 Diabetes Associated Changes in the Plasma Non-Esterified Fatty Acids, Oxylipins and Endocannabinoids. PLoS ONE 2012, 7, e48852. [Google Scholar] [CrossRef]
- Altmann, R.; Hausmann, M.; Spöttl, T.; Gruber, M.; Bull, A.W.; Menzel, K.; Vogl, D.; Herfarth, H.; Schölmerich, J.; Falk, W.; et al. 13-Oxo-ODE is an endogenous ligand for PPARγ in human colonic epithelial cells. Biochem. Pharmacol. 2007, 74, 612–622. [Google Scholar] [CrossRef]
- Caligiuri, S.P.B.; Parikh, M.; Stamenkovic, A.; Pierce, G.N.; Aukema, H.M. Dietary modulation of oxylipins in cardiovascular disease and aging. Am. J. Physiol. Circ. Physiol. 2017, 313, H903–H918. [Google Scholar] [CrossRef]
- Yao, X.; Sa, R.; Ye, C.; Zhang, D.; Zhang, S.; Xia, H.; Wang, Y.; Jiang, J.; Yin, H.; Ying, H. Effects of thyroid hormone status on metabolic pathways of arachidonic acid in mice and humans: A targeted metabolomic approach. Prostaglandins Other Lipid Mediat. 2015, 118–119, 11–18. [Google Scholar] [CrossRef]
- Bruegel, M.; Ludwig, U.; Kleinhempel, A.; Petros, S.; Kortz, L.; Ceglarek, U.; Holdt, L.M.; Thiery, J.; Fiedler, G.M. Sepsis-associated changes of the arachidonic acid metabolism and their diagnostic potential in septic patients*. Crit. Care Med. 2012, 40, 1478–1486. [Google Scholar] [CrossRef]
- Gouveia-Figueira, S.; Nording, M.L.; Gaida, J.E.; Forsgren, S.; Alfredson, H.; Fowler, C.J. Serum levels of oxylipins in achilles tendinopathy: An exploratory study. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Jira, W.; Spiteller, G.; Carson, W.; Schramm, A. Strong increase in hydroxy fatty acids derived from linoleic acid in human low density lipoproteins of atherosclerotic patients. Chem. Phys. Lipids 1998, 91, 1–11. [Google Scholar] [CrossRef]
- Ozawa, T.; Sugiyama, S.; Hayakawa, M.; Satake, T.; Taki, F.; Iwata, M.; Taki, K. Existence of Leukotoxin 9,10-Epoxy-12-Octadecenoate in Lung Lavages from Rats Breathing Pure Oxygen and from Patients with the Adult Respiratory Distress Syndrome. Am. Rev. Respir. Dis. 1988, 137, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Yoshida, Y.; Yoshikawa, A.; Kinumi, T.; Ogawa, Y.; Saito, Y.; Ohara, K.; Yamamoto, H.; Imai, Y.; Niki, E. Hydroxyoctadecadienoic acid and oxidatively modified peroxiredoxins in the blood of Alzheimer’s disease patients and their potential as biomarkers. Neurobiol. Aging 2009, 30, 174–185. [Google Scholar] [CrossRef]
- Dietrich-Muszalska, A.; Olas, B. Isoprostenes as indicators of oxidative stress in schizophrenia. World J. Biol. Psychiatry 2009, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Chocholoušková, M.; Jirásko, R.; Vrána, D.; Gatěk, J.; Melichar, B.; Holčapek, M. Reversed phase UHPLC/ESI-MS determination of oxylipins in human plasma: a case study of female breast cancer. Anal. Bioanal. Chem. 2019, 411, 1239–1251. [Google Scholar] [CrossRef]
- Kapadia, R.; Yi, J.-H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Gorman, R.R. Chemotactic and chemokinetic stimulation of human eosinophil and neutrophil polymorphonuclear leukocytes by 12-L-hydroxy-5,8,10-heptadecatrienoic acid (HHT). J. Immunol. 1978, 120, 526–531. [Google Scholar] [PubMed]
- Zhang, L.; Chen, B.; Zhang, J.; Li, J.; Yang, Q.; Zhong, Q.; Zhan, S.; Liu, H.; Cai, C. Serum polyunsaturated fatty acid metabolites as useful tool for screening potential biomarker of colorectal cancer. Prostaglandins Leukot. Essent. Fat. Acids 2017, 120, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, M.-Y.; Ma, L.T.; Hsin, M.K.Y.; Mok, T.S.K.; Underwood, M.J.; Chen, G.G. 15-Lipoxygenases and its metabolites 15(S)-HETE and 13(S)-HODE in the development of non-small cell lung cancer. Thorax 2010, 65, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Larré, S.; Tran, N.; Fan, C.; Hamadeh, H.; Champigneulles, J.; Azzouzi, R.; Cussenot, O.; Mangin, P.; Olivier, J.L. PGE2 and LTB4 tissue levels in benign and cancerous prostates. Prostaglandins Other Lipid Mediat. 2008, 87, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Spickett, C.M.; Pitt, A.R. Oxidative Lipidomics Coming of Age: Advances in Analysis of Oxidized Phospholipids in Physiology and Pathology. Antioxid. Redox Signal. 2015, 22, 1646–1666. [Google Scholar] [CrossRef] [PubMed]
- Lundström, S.L.; Levänen, B.; Nording, M.; Klepczynska-Nyström, A.; Sköld, M.; Haeggström, J.Z.; Grunewald, J.; Svartengren, M.; Hammock, B.D.; Larsson, B.-M.; et al. Asthmatics Exhibit Altered Oxylipin Profiles Compared to Healthy Individuals after Subway Air Exposure. PLoS ONE 2011, 6, e23864. [Google Scholar] [CrossRef]
- Yang, J.; Schmelzer, K.; Georgi, K.; Hammock, B.D. Quantitative Profiling Method for Oxylipin Metabolome by Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem. 2009, 81, 8085–8093. [Google Scholar] [CrossRef]
- Colas, R.A.; Shinohara, M.; Dalli, J.; Chiang, N.; Serhan, C.N. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. AJP Cell Physiol. 2014, 307, C39–C54. [Google Scholar] [CrossRef]
- Golovko, M.Y.; Murphy, E.J. An improved LC-MS/MS procedure for brain prostanoid analysis using brain fixation with head-focused microwave irradiation and liquid-liquid extraction. J. Lipid Res. 2008, 49, 893–902. [Google Scholar] [CrossRef]
- Willenberg, I.; Ostermann, A.I.; Schebb, N.H. Targeted metabolomics of the arachidonic acid cascade: current state and challenges of LC-MS analysis of oxylipins. Anal. Bioanal. Chem. 2015, 407, 2675–2683. [Google Scholar] [CrossRef]
- Strassburg, K.; Huijbrechts, A.M.L.; Kortekaas, K.A.; Lindeman, J.H.; Pedersen, T.L.; Dane, A.; Berger, R.; Brenkman, A.; Hankemeier, T.; Van Duynhoven, J.; et al. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis: Application in cardiac surgery. Anal. Bioanal. Chem. 2012, 404, 1413–1426. [Google Scholar] [CrossRef]
- Brose, S.A.; Baker, A.G.; Golovko, M.Y. A Fast One-Step Extraction and UPLC–MS/MS Analysis for E2/D2 Series Prostaglandins and Isoprostanes. Lipids 2013, 48, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-X.; Majchrzak-Hong, S.; Keyes, G.S.; Iadarola, M.J.; Mannes, A.J.; Ramsden, C.E. Lipidomic profiling of targeted oxylipins with ultra-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2018, 410, 6009–6029. [Google Scholar] [CrossRef] [PubMed]
- Drake, S.K.; Bowen, R.A.R.; Remaley, A.T.; Hortin, G.L. Potential Interferences from Blood Collection Tubes in Mass Spectrometric Analyses of Serum Polypeptides. Clin. Chem. 2004, 50, 2398–2401. [Google Scholar] [CrossRef]
- Ito, R.; Miura, N.; Iguchi, H.; Nakamura, H.; Ushiro, M.; Wakui, N.; Nakahashi, K.; Iwasaki, Y.; Saito, K.; Suzuki, T.; et al. Determination of tris(2-ethylhexyl)trimellitate released from PVC tube by LC–MS/MS. Int. J. Pharm. 2008, 360, 91–95. [Google Scholar] [CrossRef]
- Schauer, K.L.; Broccardo, C.J.; Webb, K.M.; Covey, P.A.; Prenni, J.E. Mass Spectrometry Contamination from Tinuvin 770, a Common Additive in Laboratory Plastics. J. Biomol. Tech. 2013, 24, jbt.13-2402-004. [Google Scholar] [CrossRef]
- Haned, Z.; Moulay, S.; Lacorte, S. Migration of plasticizers from poly(vinyl chloride) and multilayer infusion bags using selective extraction and GC–MS. J. Pharm. Biomed. Anal. 2018, 156, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. Measurement of Eicosanoids in Cancer Tissues. Methods Enzymol. 2007, 433, 27–50. [Google Scholar] [PubMed]
- Rund, K.M.; Ostermann, A.I.; Kutzner, L.; Galano, J.M.; Oger, C.; Vigor, C.; Wecklein, S.; Seiwert, N.; Durand, T.; Schebb, N.H. Development of an LC-ESI(-)-MS/MS method for the simultaneous quantification of 35 isoprostanes and isofurans derived from the major n3- and n6-PUFAs. Anal. Chim. Acta 2018, 1037, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Falck, J.R.; McGiff, J.C.; Carroll, M.A.; Quilley, J. A method for the determination of 5,6-EET using the lactone as an intermediate in the formation of the diol. J. Lipid Res. 1998, 39, 1713–1721. [Google Scholar] [PubMed]
- Araujo, P.; Mengesha, Z.; Lucena, E.; Grung, B. Development and validation of an extraction method for the determination of pro-inflammatory eicosanoids in human plasma using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2014, 1353, 57–64. [Google Scholar] [CrossRef]
- Mueller, M.J.; Mène-Saffrané, L.; Grun, C.; Karg, K.; Farmer, E.E. Oxylipin analysis methods. Plant J. 2006, 45, 472–489. [Google Scholar] [CrossRef]
- Balvers, M.G.J.; Verhoeckx, K.C.M.; Bijlsma, S.; Rubingh, C.M.; Meijerink, J.; Wortelboer, H.M.; Witkamp, R.F. Fish oil and inflammatory status alter the n-3 to n-6 balance of the endocannabinoid and oxylipin metabolomes in mouse plasma and tissues. Metabolomics 2012, 8, 1130–1147. [Google Scholar] [CrossRef]
- Hennebelle, M.; Otoki, Y.; Yang, J.; Hammock, B.D.; Levitt, A.J.; Taha, A.Y.; Swardfager, W. Altered soluble epoxide hydrolase-derived oxylipins in patients with seasonal major depression: An exploratory study. Psychiatry Res. 2017, 252, 94–101. [Google Scholar] [CrossRef]
- Hewawasam, E.; Liu, G.; Jeffery, D.W.; Muhlhausler, B.S.; Gibson, R.A. A stable method for routine analysis of oxylipins from dried blood spots using ultra-high performance liquid chromatography–tandem mass spectrometry. Prostaglandins Leukot. Essent. Fat. Acids 2018, 137, 12–18. [Google Scholar] [CrossRef]
- Yang, P.; Felix, E.; Madden, T.; Fischer, S.M.; Newman, R.A. Quantitative high-performance liquid chromatography/electrospray ionization tandem mass spectrometric analysis of 2- and 3-series prostaglandins in cultured tumor cells. Anal. Biochem. 2002, 308, 168–177. [Google Scholar] [CrossRef]
- Newman, J.W.; Watanabe, T.; Hammock, B.D. The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. J. Lipid Res. 2002, 43, 1563–1578. [Google Scholar] [CrossRef]
- Morgan, A.H.; Hammond, V.J.; Morgan, L.; Thomas, C.P.; Tallman, K.A.; Garcia-Diaz, Y.R.; McGuigan, C.; Serpi, M.; Porter, N.A.; Murphy, R.C.; et al. Quantitative assays for esterified oxylipins generated by immune cells. Nat. Protoc. 2010, 5, 1919–1931. [Google Scholar] [CrossRef]
- Dumlao, D.S.; Buczynski, M.W.; Norris, P.C.; Harkewicz, R.; Dennis, E.A. High-throughput lipidomic analysis of fatty acid derived eicosanoids and N-acylethanolamines. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2011, 1811, 724–736. [Google Scholar] [CrossRef]
- Mesaros, C.; Lee, S.H.; Blair, I.A. Analysis of epoxyeicosatrienoic acids by chiral liquid chromatography/electron capture atmospheric pressure chemical ionization mass spectrometry using [13C]-analog internal standards. Rapid Commun. Mass Spectrom. 2010, 24, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Cui, L.; Fang, J.; Chern, B.S.M.; Tan, H.H.; Chan, J.K.Y. Limited value of pro-inflammatory oxylipins and cytokines as circulating biomarkers in endometriosis - A targeted ’omics study. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Armando, A.M.; Quehenberger, O.; Yan, C.; Dennis, E.A. Comprehensive ultra-performance liquid chromatographic separation and mass spectrometric analysis of eicosanoid metabolites in human samples. J. Chromatogr. A 2014, 1359, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Shearer, G.C.; Harris, W.S.; Pedersen, T.L.; Newman, J.W. Detection of omega-3 oxylipins in human plasma and response to treatment with omega-3 acid ethyl esters. J. Lipid Res. 2010, 51, 2074–2081. [Google Scholar] [CrossRef]
- Gouveia-Figueira, S.; Späth, J.; Zivkovic, A.M.; Nording, M.L. Profiling the oxylipin and endocannabinoid metabolome by UPLC-ESI-MS/MS in human plasma to monitor postprandial inflammation. PLoS ONE 2015, 10, 1–29. [Google Scholar] [CrossRef]
- Gouveia-Figueira, S.; Nording, M.L. Validation of a tandem mass spectrometry method using combined extraction of 37 oxylipins and 14 endocannabinoid-related compounds including prostamides from biological matrices. Prostaglandins Other Lipid Mediat. 2015, 121, 110–121. [Google Scholar] [CrossRef]
- Schuchardt, J.P.; Schmidt, S.; Kressel, G.; Dong, H.; Willenberg, I.; Hammock, B.D.; Hahn, A.; Schebb, N.H. Comparison of free serum oxylipin concentrations in hyper- vs. normolipidemic men. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hellström, F.; Gouveia-Figueira, S.; Nording, M.L.; Björklund, M.; Fowler, C.J. Association between plasma concentrations of linoleic acid-derived oxylipins and the perceived pain scores in an exploratory study in women with chronic neck pain. BMC Musculoskelet. Disord. 2016, 17, 103. [Google Scholar] [CrossRef] [PubMed]
- Astarita, G.; Kendall, A.C.; Dennis, E.A.; Nicolaou, A. Targeted lipidomic strategies for oxygenated metabolites of polyunsaturated fatty acids. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2015, 1851, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Maskrey, B.H.; O’Donnell, V.B. Analysis of eicosanoids and related lipid mediators using mass spectrometry. Biochem. Soc. Trans. 2008, 36, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D.; Zoerner, A.A. Analysis of eicosanoids by LC-MS/MS and GC-MS/MS: A historical retrospect and a discussion. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 964, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Schweer, H.; Kammer, J.; Kühl, P.G.; Seyberth, H.W. Determination of peripheral plasma prostanoid concentration: an unreliable index of “in vivo” prostanoid activity. Eur. J. Clin. Pharmacol. 1986, 31, 303–305. [Google Scholar] [CrossRef]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef]
- Tan, Z.-R.; Ouyang, D.-S.; Zhou, G.; Wang, L.-S.; Li, Z.; Wang, D.; Zhou, H.-H. Sensitive bioassay for the simultaneous determination of pseudoephedrine and cetirizine in human plasma by liquid-chromatography–ion trap spectrometry. J. Pharm. Biomed. Anal. 2006, 42, 207–212. [Google Scholar] [CrossRef]
- Satomi, Y.; Hirayama, M.; Kobayashi, H. One-step lipid extraction for plasma lipidomics analysis by liquid chromatography mass spectrometry. J. Chromatogr. B 2017, 1063, 93–100. [Google Scholar] [CrossRef]
- Martin-Venegas, R.; Jáuregui, O.; Moreno, J.J. Liquid chromatography-tandem mass spectrometry analysis of eicosanoids and related compounds in cell models. J. Chromatogr. B 2014, 964, 41–49. [Google Scholar] [CrossRef]
- Heemskerk, M.M.; Dharuri, H.K.; van den Berg, S.A.A.; Jónasdóttir, H.S.; Kloos, D.-P.; Giera, M.; van Dijk, K.W.; van Harmelen, V. Prolonged niacin treatment leads to increased adipose tissue PUFA synthesis and anti-inflammatory lipid and oxylipin plasma profile. J. Lipid Res. 2014, 55, 2532–2540. [Google Scholar] [CrossRef]
- Polson, C.; Sarkar, P.; Incledon, B.; Raguvaran, V.; Grant, R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography-tandem mass spectrometry. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2003, 785, 263–275. [Google Scholar] [CrossRef]
- Zein Elabdeen, H.R.; Mustafa, M.; Szklenar, M.; Rühl, R.; Ali, R.; Bolstad, A.I. Ratio of Pro-Resolving and Pro-Inflammatory Lipid Mediator Precursors as Potential Markers for Aggressive Periodontitis. PLoS ONE 2013, 8, e70838. [Google Scholar] [CrossRef]
- Wang, W.; Qin, S.; Li, L.; Chen, X.; Wang, Q.; Wei, J. An Optimized High Throughput Clean-Up Method Using Mixed-Mode SPE Plate for the Analysis of Free Arachidonic Acid in Plasma by LC-MS/MS. Int. J. Anal. Chem. 2015, 2015, 1–6. [Google Scholar] [CrossRef]
- Kortz, L.; Helmschrodt, C.; Ceglarek, U. Fast liquid chromatography combined with mass spectrometry for the analysis of metabolites and proteins in human body fluids. Anal. Bioanal. Chem. 2011, 399, 2635–2644. [Google Scholar] [CrossRef]
- Kortz, L.; Dorow, J.; Becker, S.; Thiery, J.; Ceglarek, U. Fast liquid chromatography-quadrupole linear ion trap-mass spectrometry analysis of polyunsaturated fatty acids and eicosanoids in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 927, 209–213. [Google Scholar] [CrossRef]
- Klawitter, J.; Haschke, M.; Shokati, T.; Klawitter, J.; Christians, U. Quantification of 15-F2t-isoprostane in human plasma and urine: results from enzyme-linked immunoassay and liquid chromatography/tandem mass spectrometry cannot be compared. Rapid Commun. Mass Spectrom. 2011, 25, 463–468. [Google Scholar] [CrossRef]
- Bessonneau, V.; Zhan, Y.; De Lannoy, I.A.M.; Saldivia, V.; Pawliszyn, J. In vivo solid-phase microextraction liquid chromatography-tandem mass spectrometry for monitoring blood eicosanoids time profile after lipopolysaccharide-induced inflammation in Sprague-Dawley rats. J. Chromatogr. A 2015, 1424, 134–138. [Google Scholar] [CrossRef]
- Tumanov, S.; Kamphorst, J.J. Recent advances in expanding the coverage of the lipidome. Curr. Opin. Biotechnol. 2017, 43, 127–133. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Physiol. Pharmacol. 1959, 37, 911–917. [Google Scholar]
- Puppolo, M.; Varma, D.; Jansen, S.A. A review of analytical methods for eicosanoids in brain tissue. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 964, 50–64. [Google Scholar] [CrossRef]
- Yang, Y.; Cruickshank, C.; Armstrong, M.; Mahaffey, S.; Reisdorph, R.; Reisdorph, N. New sample preparation approach for mass spectrometry-based profiling of plasma results in improved coverage of metabolome. J. Chromatogr. A 2013, 1300, 217–226. [Google Scholar] [CrossRef]
- Fromel, T.; Jungblut, B.; Hu, J.; Trouvain, C.; Barbosa-Sicard, E.; Popp, R.; Liebner, S.; Dimmeler, S.; Hammock, B.D.; Fleming, I. Soluble epoxide hydrolase regulates hematopoietic progenitor cell function via generation of fatty acid diols. Proc. Natl. Acad. Sci. 2012, 109, 9995–10000. [Google Scholar] [CrossRef]
- Pier, B.; Edmonds, J.W.; Wilson, L.; Arabshahi, A.; Moore, R.; Bates, G.W.; Prasain, J.K.; Miller, M.A. Comprehensive profiling of prostaglandins in human ovarian follicular fluid using mass spectrometry. Prostaglandins Other Lipid Mediat. 2018, 134, 7–15. [Google Scholar] [CrossRef]
- Larose, J.; Julien, P.; Bilodeau, J.-F. Analysis of F 2 -isoprostanes in plasma of pregnant women by HPLC-MS/MS using a column packed with core-shell particles. J. Lipid Res. 2013, 54, 1505–1511. [Google Scholar] [CrossRef]
- Hall, L.M.; Murphy, R.C. Electrospray mass spectrometric analysis of 5-hydroperoxy and 5-hydroxyeicosatetraenoic acids generated by lipid peroxidation of red blood cell ghost phospholipids. J. Am. Soc. Mass Spectrom. 1998, 9, 527–532. [Google Scholar] [CrossRef]
- Brose, S.A.; Thuen, B.T.; Golovko, M.Y. LC/MS/MS method for analysis of E 2 series prostaglandins and isoprostanes. J. Lipid Res. 2011, 52, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Enot, D.P.; Dallmann, G.; Körner, L.; Forcher, V.; Enoh, P.; Koal, T.; Keller, M.; Deigner, H.P. Complexity and pitfalls of mass spectrometry-based targeted metabolomics in brain research. Anal. Biochem. 2010, 406, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kempen, E.C.; Yang, P.; Felix, E.; Madden, T.; Newman, R.A. Simultaneous Quantification of Arachidonic Acid Metabolites in Cultured Tumor Cells Using High-Performance Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry. Anal. Biochem. 2001, 297, 183–190. [Google Scholar] [CrossRef]
- Schroeder, C.P.; Yang, P.; Newman, R.A.; Lotan, R. Eicosanoid metabolism in squamous cell carcinoma cell lines derived from primary and metastatic head and neck cancer and its modulation by celecoxib. Cancer Biol. Ther. 2004, 3, 847–852. [Google Scholar] [CrossRef]
- Michaelis, U.R.; Xia, N.; Barbosa-Sicard, E.; Falck, J.R.; Fleming, I. Role of cytochrome P450 2C epoxygenases in hypoxia-induced cell migration and angiogenesis in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xiao, L.; Park, G.; Wang, X.; Azim, A.C.; Christman, J.W.; van Breemen, R.B. An improved LC–MS/MS method for the quantification of prostaglandins E2 and D2 production in biological fluids. Anal. Biochem. 2008, 372, 41–51. [Google Scholar] [CrossRef]
- Guide to Solid Phase Extraction. SUPELCO Bull. 910 1998, 1–12.
- Späth, J. Oxylipins in human plasma – method development and dietary effects on levels. Master’s Thesis, Umea Universitet, Umea, Sweden, 2014. [Google Scholar]
- VanRollins, M.; VanderNoot, V.A. Simultaneous resolution of underivatized regioisomers and stereoisomers of arachidonate epoxides by capillary electrophoresis. Anal. Biochem. 2003, 313, 106–116. [Google Scholar] [CrossRef]
- Ostermann, A.I.; Willenberg, I.; Schebb, N.H. Comparison of sample preparation methods for the quantitative analysis of eicosanoids and other oxylipins in plasma by means of LC-MS/MS. Anal. Bioanal. Chem. 2015, 407, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, M.; Mir, A.A.; Petasis, N.A.; Serhan, C.N.; Nicolaou, A. Simultaneous lipidomic analysis of three families of bioactive lipid mediators leukotrienes, resolvins, protectins and related hydroxy-fatty acids by liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 75–83. [Google Scholar] [CrossRef]
- Galvão, A.F.; Petta, T.; Flamand, N.; Bollela, V.R.; Silva, C.L.; Jarduli, L.R.; Malmegrim, K.C.R.; Simões, B.P.; de Moraes, L.A.B.; Faccioli, L.H. Plasma eicosanoid profiles determined by high-performance liquid chromatography coupled with tandem mass spectrometry in stimulated peripheral blood from healthy individuals and sickle cell anemia patients in treatment. Anal. Bioanal. Chem. 2016, 408, 3613–3623. [Google Scholar] [CrossRef]
- Okemoto, K.; Maekawa, K.; Tajima, Y.; Tohkin, M.; Saito, Y. Cross-Classification of Human Urinary Lipidome by Sex, Age, and Body Mass Index. PLoS ONE 2016, 11, e0168188. [Google Scholar] [CrossRef] [PubMed]
- Balgoma, D.; Larsson, J.; Rokach, J.; Lawson, J.A.; Daham, K.; Dahlén, B.; Dahlén, S.-E.; Wheelock, C.E. Quantification of Lipid Mediator Metabolites in Human Urine from Asthma Patients by Electrospray Ionization Mass Spectrometry: Controlling Matrix Effects. Anal. Chem. 2013, 85, 7866–7874. [Google Scholar] [CrossRef] [PubMed]
- Sterz, K.; Scherer, G.; Ecker, J. A simple and robust UPLC-SRM/MS method to quantify urinary eicosanoids. J. Lipid Res. 2012, 53, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.; Domínguez-Perles, R.; Gil, J.I.; Ferreres, F.; García-Viguera, C.; Martínez-Sanz, J.M.; Gil-Izquierdo, A. A ultra-pressure liquid chromatography/triple quadrupole tandem mass spectrometry method for the analysis of 13 eicosanoids in human urine and quantitative 24 hour values in healthy volunteers in a controlled constant diet. Rapid Commun. Mass Spectrom. 2012, 26, 1249–1257. [Google Scholar] [CrossRef]
- Gouveia-Figueira, S.; Karimpour, M.; Bosson, J.A.; Blomberg, A.; Unosson, J.; Pourazar, J.; Sandström, T.; Behndig, A.F.; Nording, M.L. Mass spectrometry profiling of oxylipins, endocannabinoids, and N-acylethanolamines in human lung lavage fluids reveals responsiveness of prostaglandin E2 and associated lipid metabolites to biodiesel exhaust exposure. Anal. Bioanal. Chem. 2017, 409, 2967–2980. [Google Scholar] [CrossRef] [PubMed]
- Panthi, S.; Chen, J.; Wilson, L.; Nichols, J.J. Detection of Lipid Mediators of Inflammation in the Human Tear Film. Eye Contact Lens Sci. Clin. Pract. 2018, 0, 1. [Google Scholar] [CrossRef] [PubMed]
- Giera, M.; Ioan-Facsinay, A.; Toes, R.; Gao, F.; Dalli, J.; Deelder, A.M.; Serhan, C.N.; Mayboroda, O.A. Lipid and lipid mediator profiling of human synovial fluid in rheumatoid arthritis patients by means of LC–MS/MS. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2012, 1821, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Rauzi, F.; Kirkby, N.S.; Edin, M.L.; Whiteford, J.; Zeldin, D.C.; Mitchell, J.A.; Warner, T.D. Aspirin inhibits the production of proangiogenic 15( S )-HETE by platelet cyclooxygenase-1. FASEB J. 2016, 30, 4256–4266. [Google Scholar] [CrossRef]
- Wang, C.; Colas, R.A.; Dalli, J.; Arnardottir, H.H.; Nguyen, D.; Hasturk, H.; Chiang, N.; Van Dyke, T.E.; Serhan, C.N. Maresin 1 Biosynthesis and Proresolving Anti-infective Functions with Human-Localized Aggressive Periodontitis Leukocytes. Infect. Immun. 2016, 84, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gouveia-Figueira, S.; Domellöf, M.; Zivkovic, A.M.; Nording, M.L. Oxylipins, endocannabinoids, and related compounds in human milk: Levels and effects of storage conditions. Prostaglandins Other Lipid Mediat. 2016, 122, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.T.; Palac, H.L.; Baillif, V.; Van Goethem, E.; Dubourdeau, M.; Van Horn, L.; Martin, C.R. Long chain fatty acids and related pro-inflammatory, specialized pro-resolving lipid mediators and their intermediates in preterm human milk during the first month of lactation. Prostaglandins Leukot. Essent. Fat. Acids 2017, 121, 1–6. [Google Scholar] [CrossRef]
- Yue, H.; Strauss, K.I.; Borenstein, M.R.; Barbe, M.F.; Rossi, L.J.; Jansen, S.A. Determination of bioactive eicosanoids in brain tissue by a sensitive reversed-phase liquid chromatographic method with fluorescence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 803, 267–277. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.B.; Maskrey, B.; Taylor, G.W. Eicosanoids: Generation and detection in mammalian cells. Methods Mol Biol 2009, 462, 5–23. [Google Scholar]
- Blewett, A.J.; Varma, D.; Gilles, T.; Libonati, J.R.; Jansen, S.A. Development and validation of a high-performance liquid chromatography-electrospray mass spectrometry method for the simultaneous determination of 23 eicosanoids. J. Pharm. Biomed. Anal. 2008, 46, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Le Faouder, P.; Baillif, V.; Spreadbury, I.; Motta, J.-P.; Rousset, P.; Chêne, G.; Guigné, C.; Tercé, F.; Vanner, S.; Vergnolle, N.; et al. LC–MS/MS method for rapid and concomitant quantification of pro-inflammatory and pro-resolving polyunsaturated fatty acid metabolites. J. Chromatogr. B 2013, 932, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Weylandt, K.H.; Krause, L.F.; Gomolka, B.; Chiu, C.Y.; Bilal, S.; Nadolny, A.; Waechter, S.F.; Fischer, A.; Rothe, M.; Kang, J.X. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-α. Carcinogenesis 2011, 32, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Jelińska, M.; Białek, A.; Mojska, H.; Gielecińska, I.; Tokarz, A. Effect of conjugated linoleic acid mixture supplemented daily after carcinogen application on linoleic and arachidonic acid metabolites in rat serum and induced tumours. Biochim. Biophys. Acta - Mol. Basis Dis. 2014, 1842, 2230–2236. [Google Scholar] [CrossRef]
- Deems, R.; Buczynski, M.W.; Bowers-Gentry, R.; Harkewicz, R.; Dennis, E.A. Detection and Quantitation of Eicosanoids via High Performance Liquid Chromatography-Electrospray Ionization-Mass Spectrometry. In Methods in Enzymology; Academic Press: New York, NY, USA, 2007; ISBN 9780123738950. [Google Scholar]
- Takabatake, M.; Hishinuma, T.; Suzuki, N.; Chiba, S.; Tsukamoto, H.; Nakamura, H.; Saga, T.; Tomioka, Y.; Kurose, A.; Sawai, T.; et al. Simultaneous quantification of prostaglandins in human synovial cell-cultured medium using liquid chromatography/tandem mass spectrometry. Prostaglandins Leukot. Essent. Fat. Acids 2002, 67, 51–56. [Google Scholar] [CrossRef]
- Masoodi, M.; Nicolaou, A. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 3023–3029. [Google Scholar] [CrossRef] [PubMed]
- Tajima, Y.; Ishikawa, M.; Maekawa, K.; Murayama, M.; Senoo, Y.; Nishimaki-Mogami, T.; Nakanishi, H.; Ikeda, K.; Arita, M.; Taguchi, R.; et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Rago, B.; Fu, C. Development of a high-throughput ultra performance liquid chromatography–mass spectrometry assay to profile 18 eicosanoids as exploratory biomarkers for atherosclerotic diseases. J. Chromatogr. B 2013, 936, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Margalit, A.; Duffin, K.L.; Isakson, P.C. Rapid Quantitation of a Large Scope of Eicosanoids in Two Models of Inflammation: Development of an Electrospray and Tandem Mass Spectrometry Method and Application to Biological Studies. Anal. Biochem. 1996, 235, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [PubMed]
- Sanaki, T.; Fujihara, T.; Iwamoto, R.; Yoshioka, T.; Higashino, K.; Nakano, T.; Numata, Y. Improvements in the High-Performance Liquid Chromatography and Extraction Conditions for the Analysis of Oxidized Fatty Acids Using a Mixed-Mode Spin Column. Mod. Chem. Appl. 2015, 3, 1000161. [Google Scholar]
- Petta, T.; Moraes, L.A.B.; Faccioli, L.H. Versatility of tandem mass spectrometry for focused analysis of oxylipids. J. Mass Spectrom. 2015, 50, 879–890. [Google Scholar] [CrossRef]
- Shinde, D.D.; Kim, K.-B.; Oh, K.-S.; Abdalla, N.; Liu, K.-H.; Bae, S.K.; Shon, J.-H.; Kim, H.-S.; Kim, D.-H.; Shin, J.G. LC–MS/MS for the simultaneous analysis of arachidonic acid and 32 related metabolites in human plasma: Basal plasma concentrations and aspirin-induced changes of eicosanoids. J. Chromatogr. B 2012, 911, 113–121. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Q. Comprehensive analysis of oxylipins in human plasma using reversed-phase liquid chromatography-triple quadrupole mass spectrometry with heatmap-assisted selection of transitions. Anal. Bioanal. Chem. 2019, 411, 367–385. [Google Scholar] [CrossRef]
- Yasumoto, A.; Tokuoka, S.M.; Kita, Y.; Shimizu, T.; Yatomi, Y. Multiplex quantitative analysis of eicosanoid mediators in human plasma and serum: Possible introduction into clinical testing. J. Chromatogr. B 2017, 1068–1069, 98–104. [Google Scholar] [CrossRef]
- Thakare, R.; Chhonker, Y.S.; Gautam, N.; Nelson, A.; Casaburi, R.; Criner, G.; Dransfield, M.T.; Make, B.; Schmid, K.K.; Rennard, S.I.; et al. Simultaneous LC-MS/MS analysis of eicosanoids and related metabolites in human serum, sputum and BALF. Biomed. Chromatogr. 2018, 32, e4102. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Donnelly, M.K.; Crago, E.A.; Roman, D.M.; Sherwood, P.R.; Horowitz, M.B.; Poloyac, S.M. Rapid, simultaneous quantitation of mono and dioxygenated metabolites of arachidonic acid in human CSF and rat brain. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3991–4000. [Google Scholar] [CrossRef]
- Bollinger, J.G.; Thompson, W.; Lai, Y.; Oslund, R.C.; Hallstrand, T.S.; Sadilek, M.; Turecek, F.; Gelb, M.H. Improved Sensitivity Mass Spectrometric Detection of Eicosanoids by Charge Reversal Derivatization. Anal. Chem. 2010, 82, 6790–6796. [Google Scholar] [CrossRef]
- Lee, C.-Y.J.; Jenner, A.; Halliwell, B. Rapid preparation of human urine and plasma samples for analysis of F2-isoprostanes by gas chromatography-mass spectrometry. Biochem. Biophys. Res. Commun. 2004, 320, 696–702. [Google Scholar] [CrossRef]
- Caligiuri, S.P.B.; Aukema, H.M.; Ravandi, A.; Guzman, R.; Dibrov, E.; Pierce, G.N. Flaxseed consumption reduces blood pressure in patients with hypertension by altering circulating oxylipins via an α-linolenic acid-induced inhibition of soluble epoxide hydrolase. Hypertension 2014, 64, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, X.; Wu, J.; Meehan, M.J.; Blevitt, J.M.; Dorrestein, P.C.; Milla, M.E. A highly efficient, high-throughput lipidomics platform for the quantitative detection of eicosanoids in human whole blood. Anal. Biochem. 2013, 433, 181–188. [Google Scholar] [CrossRef]
- Lebold, K.M.; Kirkwood, J.S.; Taylor, A.W.; Choi, J.; Barton, C.L.; Miller, G.W.; La Du, J.; Jump, D.B.; Stevens, J.F.; Tanguay, R.L.; et al. Novel liquid chromatography–mass spectrometry method shows that vitamin E deficiency depletes arachidonic and docosahexaenoic acids in zebrafish (Danio rerio) embryos. Redox Biol. 2014, 2, 105–113. [Google Scholar] [CrossRef] [PubMed]
- García-Flores, L.A.; Medina, S.; Gómez, C.; Wheelock, C.E.; Cejuela, R.; Martínez-Sanz, J.M.; Oger, C.; Galano, J.M.; Durand, T.; Hernández-Sáez, Á.; et al. Aronia - Citrus juice (polyphenol-rich juice) intake and elite triathlon training: A lipidomic approach using representative oxylipins in urine. Food Funct. 2018, 9, 463–475. [Google Scholar] [CrossRef]
- Mizuno, K.; Kataoka, H. Analysis of urinary 8-isoprostane as an oxidative stress biomarker by stable isotope dilution using automated online in-tube solid-phase microextraction coupled with liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2015, 112, 36–42. [Google Scholar] [CrossRef]
- Rodríguez Patiño, G.; Castillo Rodríguez, M.A.; Ramírez Bribiesca, J.E.; Ramírez Noguera, P.; Gonsebatt Bonaparte, M.E.; López-Arellano, R. Development of a method for the determination of 8-iso-PGF2α in sheep and goat plasma using solid-phase microextraction and ultra-performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 1675–1682. [Google Scholar] [CrossRef]
- Hyötyläinen, T. Critical evaluation of sample pretreatment techniques. Anal. Bioanal. Chem. 2009, 394, 743–758. [Google Scholar] [CrossRef]
- Prosen, H. Applications of liquid-phase microextraction in the sample preparation of environmental solid samples. Molecules 2014, 19, 6776–6808. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, R.; Silva, C.L.; Câmara, J.S. Determination of urinary levels of leukotriene B4 using ad highly specific and sensitive methodology based on automatic MEPS combined with UHPLC-PDA analysis. Talanta 2015, 144, 382–389. [Google Scholar] [CrossRef]
- Suhr, A.C.; Bruegel, M.; Maier, B.; Holdt, L.M.; Kleinhempel, A.; Teupser, D.; Grimm, S.H.; Vogeser, M. Ferromagnetic particles as a rapid and robust sample preparation for the absolute quantification of seven eicosanoids in human plasma by UHPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1022, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Balashova, E.E.; Trifonova, O.P.; Maslov, D.L.; Lokhov, P.G. Application of dried blood spot for analysis of low molecular weight fraction ( metabolome ) of blood. Heal. Prim. Care 2018, 2, 1–11. [Google Scholar]
- Ferreiro-Vera, C.; Mata-Granados, J.M.; Priego-Capote, F.; Quesada-Gómez, J.M.; Luque de Castro, M.D. Automated targeting analysis of eicosanoid inflammation biomarkers in human serum and in the exometabolome of stem cells by SPE–LC–MS/MS. Anal. Bioanal. Chem. 2011, 399, 1093–1103. [Google Scholar] [CrossRef]
- Wagner, B.M. Entwicklung Eines Multidimensionalen bioanalytischen Modellsystems für die Gesicherte Identifikation der Auswirkungen von Oxidativen Stressfaktoren auf den Organismus. Ph.D. Thesis, Medical University of Graz, Graz, Austria, 2014. [Google Scholar]
- Kita, Y.; Takahashi, T.; Uozumi, N.; Shimizu, T. A multiplex quantitation method for eicosanoids and platelet-activating factor using column-switching reversed-phase liquid chromatography–tandem mass spectrometry. Anal. Biochem. 2005, 342, 134–143. [Google Scholar] [CrossRef]
- Parkinson, D.R. Analytical Derivatization Techniques. In Comprehensive Sampling and Sample Preparation; Elsevier Science: Saint Louis, MO, USA, 2012; ISBN 9780123813749. [Google Scholar]
- Moraes, L.A.; Giner, R.M.; Paul-Clark, M.J.; Perretti, M.; Perrett, D. An isocratic HPLC method for the quantitation of eicosanoids in human platelets. Biomed. Chromatogr. 2004, 18, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Nithipatikom, K.; Pratt, P.F.; Campbell, W.B. Determination of EETs using microbore liquid chromatography with fluorescence detection. Am. J. Physiol. Circ. Physiol. 2000, 279, H857–H862. [Google Scholar] [CrossRef]
- Chavis, C.; Fraissinet, L.; Chanez, P.; Thomas, E.; Bousquet, J. A Method for the Measurement of Plasma Hydroxyeicosatetraenoic Acid Levels. Anal. Biochem. 1999, 271, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh-Habashi, A.; Asghar, W.; Jamali, F. Simultaneous determination of selected eicosanoids by reversed-phase HPLC method using fluorescence detection and application to rat and human plasma, and rat heart and kidney samples. J. Pharm. Biomed. Anal. 2015, 110, 12–19. [Google Scholar] [CrossRef]
- Meckelmann, S.W.; Hellhake, S.; Steuck, M.; Krohn, M.; Schebb, N.H. Comparison of derivatization/ionization techniques for liquid chromatography tandem mass spectrometry analysis of oxylipins. Prostaglandins Other Lipid Mediat. 2017, 130, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B. Oxylipins and their involvement in plant response to biotic and abiotic stress. Ph.D. Thesis, Friedrich-Schiller-Universität Jena, Jena, Germany, 18 October 2005. [Google Scholar]
- Mesaros, C.; Blair, I.A. Targeted Chiral Analysis of Bioactive Arachidonic Acid Metabolites Using Liquid-Chromatography-Mass Spectrometry. Metabolites 2012, 2, 337–365. [Google Scholar] [CrossRef]
- Knott, I.; Dieu, M.; Burton, M.; Lecomte, V.; Remacle, J.; Raes, M. Differential effects of interleukin-1α and β on the arachidonic acid cascade in human synovial cells and chondrocytes in culture. Agents Actions 1993, 39, 126–131. [Google Scholar] [CrossRef]
- Jira, W.; Spitellera, G.; Richter, A. Increased Levels of Lipid Oxidation Products in Rheumatically Destructed Bones of Patients Suffering from Rheumatoid Arthritis. Zeitschrift Naturforsch. Sect. C J. Biosci. 1998, 53, 1061–1071. [Google Scholar] [CrossRef]
- Molnár-Perl, I. AMINO ACIDS | Liquid Chromatography. In Encyclopedia of Separation Science; Wilson, I., Poole, C., Cooke, M., Eds.; Elsevier: Oxford, 2000; ISBN 978-0-12-226770-3. [Google Scholar]
- Prinsen, H.C.M.T.; Schiebergen-Bronkhorst, B.G.M.; Roeleveld, M.W.; Jans, J.J.M.; de Sain-van der Velden, M.G.M.; Visser, G.; van Hasselt, P.M.; Verhoeven-Duif, N.M. Rapid quantification of underivatized amino acids in plasma by hydrophilic interaction liquid chromatography (HILIC) coupled with tandem mass-spectrometry. J. Inherit. Metab. Dis. 2016, 39, 651–660. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef]
- VanderNoot, V.A.; VanRollins, M. Capillary Electrophoresis of Cytochrome P-450 Epoxygenase Metabolites of Arachidonic Acid. 2. Resolution of Stereoisomers. Anal. Chem. 2002, 74, 5866–5870. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
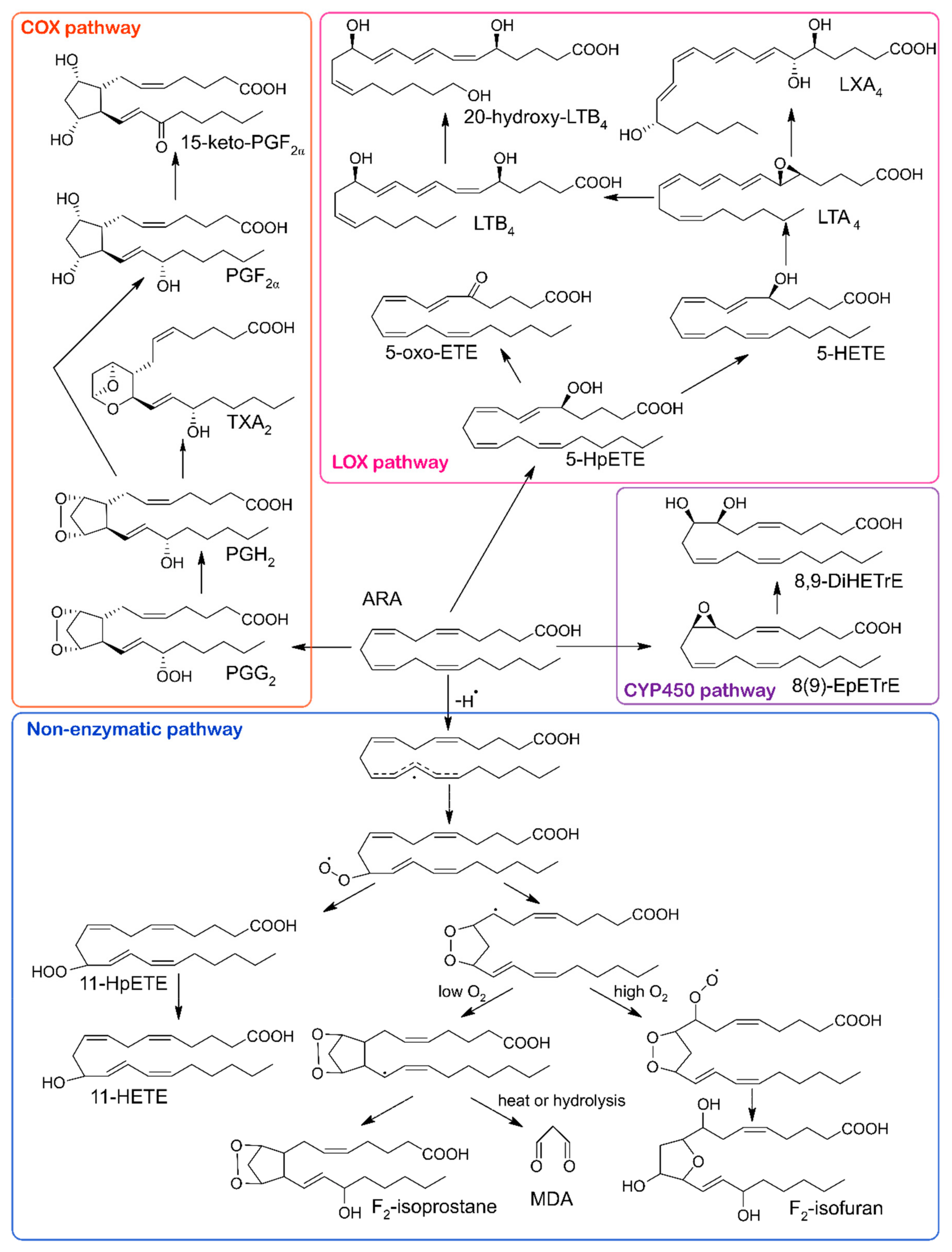
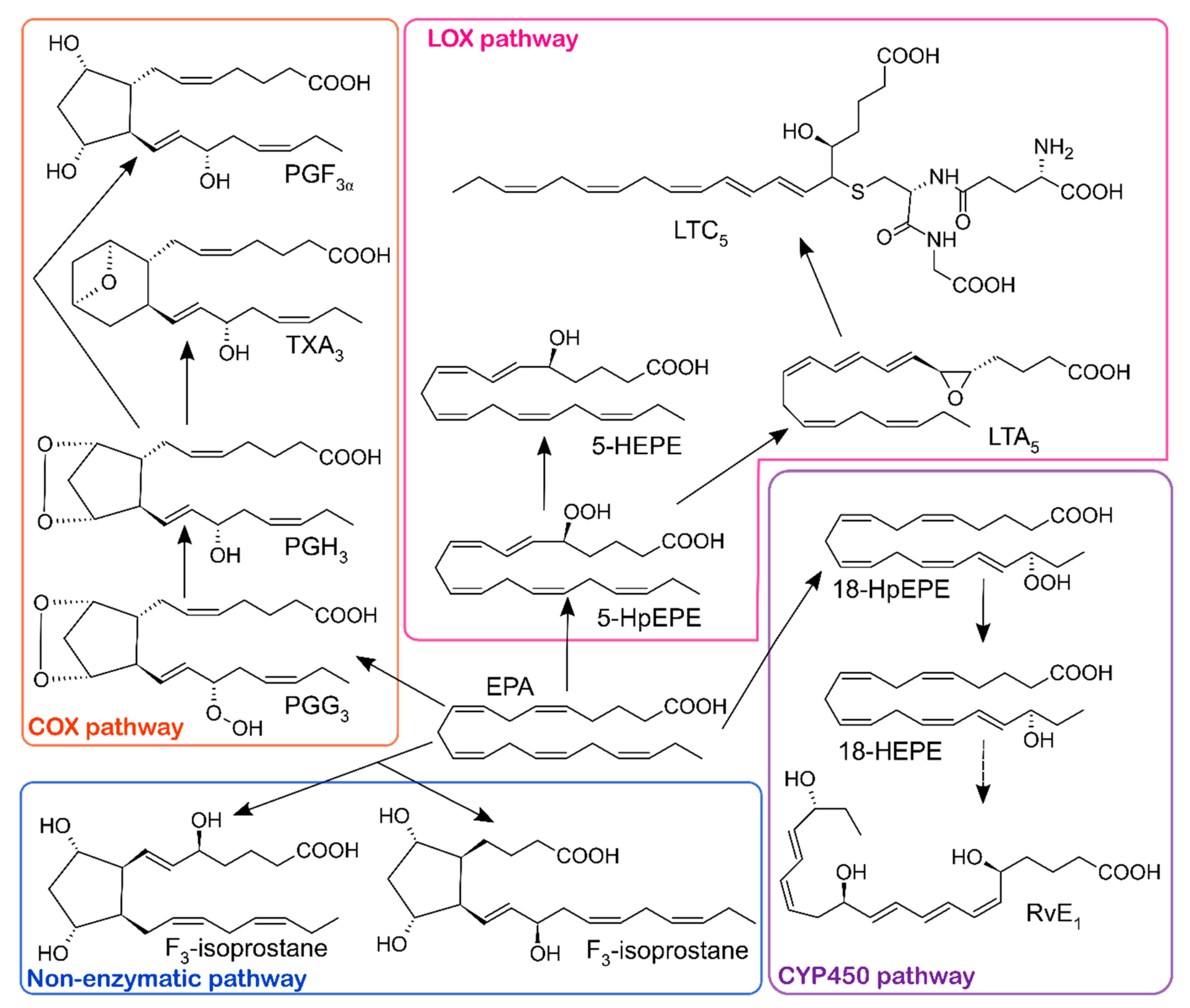
| Disease | Oxylipin | Precursor | Direction of Change | Function | Ref. |
|---|---|---|---|---|---|
| Obesity | 5-, 11-, 20-HETE | ARA | ↑ | promotion of inflammation, blood pressure regulation | [10,20,21] |
| 15-HETE | ARA | ↑ | substrates for lipoxins synthesis | ||
| 12,13-DiHOME | LA | ↓ | brown adipose tissue lipid uptake activation | ||
| 12,13-Di/EpOME | LA | ↓ | putative markers of adipose lipolysis | ||
| 15-HETrE | DGLA | ↑ | antiproliferative function | ||
| 5-, 8-, 12-HETE | ARA | ↑ | associated with low-grade inflammation | [22] | |
| PGD2 | ARA | ↑ | polarization of adipose tissue macrophage against inflammation | [23] | |
| Metabolic syndrome | 20-HETE | ARA | ↑ | vascular inflammation, angiogenesis | [15,24] |
| F2-isoprostanes | ARA | ↑ | oxidative stress marker | ||
| LXA4 | ARA | ↓ | promotion of inflammation resolution | [25] | |
| Type II diabetes | 8-iso-PGF2α | ARA | ↑ | oxidative stress marker | [26,27] |
| 11,12-, 14,15-DiHETrE | ARA | ↑ | EpETrE’s less active metabolites | [28,29,30] | |
| 13-oxo-ODE | LA | ↑ | inhibition of inflammation | ||
| 11(12)-, 14(15)-EpETrE | ARA | ↑ | vasodilation | ||
| 9(10)-EpOME | LA | ↑ | leukotoxin | ||
| 12(13)-EpOME | LA | ↑ | putative marker of adipose lipolysis | ||
| 9(10)-EpODE | ALA | ↑ | putative markers of adipose lipolysis | ||
| Hypothyroidism | PGI2 | ARA | ↑ | platelet activation inhibitor | [31] |
| PGE2 | ARA | ↓ | promotion of arterial thrombosis | ||
| 12-HETE | ARA | ↓ | blood pressure regulation | ||
| Hyperthyroidism | 12-HETE | ARA | ↑ | blood pressure regulation | [31] |
| 20-HETE | ARA | ↑ | vasoconstriction | ||
| Sepsis | 11-HETE | ARA | ↓ | promotion of inflammation | [32] |
| PGE2 | ARA | ↓ | vasodilation | ||
| TXB2 | ARA | ↓ | downstream metabolite of TXA2 which is involved in platelet aggregation and vasoconstriction | ||
| Achilles tendopathy | 13-HODE | LA | ↑ | association with pain | [33] |
| 12,13-DiHOME | LA | ↑ | association with pain | ||
| Coronary artery disease | 9-HETE | ARA | ↑ | oxidative stress marker | [16] |
| F2-isoprostanes | ARA | ↑ | oxidative stress marker | ||
| Myocardial infraction | 11-dehydro-TXB2 | ARA | ↑ | oxidative stress marker | [17] |
| 2,3-dinor-TXB2 | ARA | ↑ | oxidative stress marker | ||
| Atherosclerosis | 9-HODE in LDL | LA | ↑ | lipid peroxidation marker | [34] |
| Acute respiratory distress syndrome | 9(10)-EpOME | LA | ↑ | leukotoxin | [35] |
| Asthma | PGE2 | ARA | ↑ | promotion of inflammation | [11,36] |
| PGI2 | ARA | ↑ | inhibition of thrombosis and inflammation | ||
| TXB2 | ARA | ↑ | promotion of inflammation | ||
| PGF2α | ARA | ↓ | promotion of inflammation | ||
| 6-keto-PGF1α | ARA | ↓ | oxidative stress marker | ||
| Cystic fibrosis | LXA4 | ARA | ↓ | inhibition of inflammation | [13] |
| RvE1 | EPA | ↓ | promotion of inflammation resolution, positively associated with better lung function | ||
| Alzheimer’s disease | total HODE | LA | ↑ | in vivo lipid peroxidation marker | [37] |
| Schizophrenia | 8-iso-PGF2α | ARA | ↑ | oxidative stress marker | [38] |
| Breast cancer | 9-, 13-HODE | LA | ↑ | PPAR-γ ligand | [14,39,40,41] |
| 9-, 13-HOTrE | LA | ↑ | inhibition of inflammation | ||
| 12-HHTrE | ARA | ↑ | polymorphonuclear leucocytes (PMN) chemotaxis enhancer | ||
| Colorectal cancer | 2,3-dinor-PGF2α | ARA | ↑ | oxidative stress marker | [42] |
| 19-HETE | ARA | ↑ | possible competitive antagonist of 20-HETE | ||
| 12-keto-LTB4 | ARA | ↑ | inactive metabolite of pro-inflammatory LTB4 | ||
| 9-HODE | LA | ↓ | PPAR-γ ligand | ||
| 13-HODE | LA | ↓ | promotion of apoptosis | ||
| PGE2 | ARA | ↑ | promotion of inflammation | [12,36] | |
| PGI2 | ARA | ↓ | inhibition of thrombosis and inflammation | ||
| Non-small cell lung cancer | 15S-HETE | ARA | ↓ | induction of apoptosis | [43] |
| 13S-HODE | LA | ↓ | induction of apoptosis | ||
| Prostate cancer | LTB4 | ARA | ↑ | promotion of survival and proliferation of cancerous cells | [44] |
| Sample | Stationary Phase | Sample Preparation | Column Precondition | Sample Wash | Elution | Reference |
|---|---|---|---|---|---|---|
| OCTADECYL PHASES | ||||||
| Human plasma (1 mL) Rat Carrageenan-induced air pouch fluid (1 mL) | Bond Elut C18 | Pouch fluid: + 1 mL heparinized saline centrifugation at 1000× g, 4 °C, 10 min | 2 mL MeOH 2 mL H2O | 2 mL MeOH 2 mL petroleum ether | 1 mL methyl formate | [137] |
| Rat brain (~500 mg) rat liver (~500 mg) plasma (500 µL) | C18-E (6 mL, 500 mg) | Tissues: homogenization in H2O, adjusted to 15% MeOH to 3 mL incubation on ice, 30 min centrifugation at 3000 rpm, 5 min + 0.025 mM HCL (pH 3) Plasma: dilution with H2O, adjusted to 15% MeOH to 3 mL incubation on ice, 30 min centrifugation at 3000 rpm, 5 min + 0.025 mM HCl (pH 3) | 20 mL MeOH 20 mL H2O | 20 mL 15% MeOH 20 mL H2O 10 mL Hex | 15 mL methyl formate | [113] |
| Rat serum (400 µL) rat mammary tumor (~200 mg) | Bakerbond C18 (3 mL, 500 mg) | Serum: + 0.5 mL MeOH + 4 mL MeOH Tumor: homogenization with 2 mL H2O on ice 4 °C, 30 min centrifugation at 3000 rpm, 5 min | 10 mL MeOH 10 mL H2O | 2 mL H2O 2 mL 10% MeOH | 3 × 0.5 mL MeOH | [131] |
| Human phagocytes culture supernatant | Waters C18 | 1:10 dilution with H2O (pH ~3.5) | 1× MeOH 2× H2O | 1× H2O 1× Hex | 6 mL methyl formate | [138] |
| Human plasma (300 µL) | Sep-Pak C18 (2.8 mL, 500 mg) | + 1.5 mL MeOH/ACN (1/1, v/v) 4 °C, overnight centrifugation at 400× g, 20 mindilution with H2O to 10% MeOH/ACN | 2 mL MeOH 2 mL 0.1% HAc | sample + 2 mL 0.1% HAc | 2 mL 0.1% HAc in MeOH | [114] |
| 10 mL ethanol 20 mL H2O | sample acidified with 0.1 M HCl 10 mL H2O 1 mL 35% ethanol | 2 mL ethanol | ||||
| 10 mL methanol 10 mL H2O | sample acidified with 0.1 M HCl 5 mL 15% MeOH 5 mL H2O 2.5 mL Hex | 2 mL Hex | ||||
| Mouse brain tissue (20 mg) | Sep-Pak C18 | microwave processing homogenization in 3 mL 15% MeOH, 0.005% BHT (pH 3) centrifugation at 2000× g, 4 °C, 10 min | 20 mL 15% MeOH 20 mL H2O dried with syringe air | 10 mL methyl formate | [49] | |
| Mouse lung homogenate (50 µL) | Sep-Pak tC18 (6 mL, 500 mg) | + 0.45 mL MeOH, 10 min vortex 0.5 mL supernatant collected + 4.5 mL HCL in H2O (pH 3.5) | 12 mL MeOH 12 mL H2O | 12 mL H2O 6 mL Hex | 9 mL methyl formate | [139] |
| MonoSpin™ C18 | + 0.45 mL MeOH, 10 min vortex centrifugation at 9000× g, 4 °C, 5min evaporation with N2, 40 °C reconstituted in 0.1 mL MeOH + 0.9 mL HCl in H2O (pH 3.5) | 0.3 mL MeoH 0.3 mL H2O | centrifugation at 9000× g, 4 °C, 1 min 0.3 mL H2O 0.3 mL Hex centrifugation at 9000× g, 4 °C, 1 min | 2 × 0.3 mL methyl formate | ||
| Rat hypothalamus (95 mg) | Hypersep C18 (3 mL, 500 mg) | homogenization in 2 mL water acidified with 1 M HCL to pH 3.0 centrifugation at 7000 rpm | 6 mL MeOH 6 mL H2O | 2 mL 2% HAc | 2 mL MeOH | [140] |
| OCTYL PHASES | ||||||
| Human plasma (1 mL) | Discovery DSC-C8 | 1 mL MeOH 1 mL 0.1% FAc | 1 mL 0.1% FAc | 1 mL MeOH | [141] | |
| POLYMERIC PHASES | ||||||
| Human plasma (500 µL) | Bond Elut Certify II (3 mL, 200 mg) | + 10 µL 0.2 mg/mL BHT, EDTA, 100 µM indomethacin, 100 µM soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid in 50% MeOH + 1.4 mL ice cold MeOH, −80 °C, 30 min 10 min, 4 °C, 20,000× g N2 to final volume <1 mL + 2 mL 0.1 M disodium hydrogen phosphate buffer (pH 5.5) | 1 × 1% HAc in EA/n-Hex (75/25, v/v) 1× MeOH 1 × 0.1 M disodium hydrogen phosphate buffer (pH 6) | 3 mL H2O 3 mL 50% MeOH dried under N2, 1 min | 2 mL 1% HAc in EA/n-Hex (75/25, v/v) | [59] |
| Rat brain/heart/kidney/liver/lung/pancreas/red blood cells (50 mg) plasma (200 μL) | Bond Elut Certify II | + 0.5 mL distilled H2O + 0.5 mL MeOH + 300 μL (100 μL for plasma) 10 M NaOH, 60 °C, 20 min + 300 μL 60% HAc + 2 mL 1 M sodium acetate buffer (pH 6) adjusted to pH 6 centrifugation | 2 mL MeOH 2 mL 5% MeOH, 0.1 M sodium acetate (pH 7) | 2 mL MeOH/H2O (1/1, v/v) | 2 mL Hex/EA (75/25, v/v) | [9] |
| Human whole-blood and platelet-rich plasma (250 μL) | HyperSep Retain PEP | dilution with 5% MeOH, 0.1% HAc with 0.009 mM BHT | 5% MeOH, 0.1% HAc | 2 × 5% MeOH, 0.1% HAc | 1 mL EA 1 mL MeOH | [122] |
| Human milk (1.5–2 mL) | Oasis HLB (60 mg, 30 µm) | centrifugation + 10 µL 0.2 mg/mL BHT/EDTA | 4 mL EA 8 mL MeOH 8 mL 5% MeOH, 0.1% HAc | 8 mL 5% MeOH, 0.1% HAc | 2 mL MeOH 3 mL ACN 2 mL EA | [124] |
| Human plasma (250 µL) | Oasis HLB (60 mg, 30 µm) | + 0.2 mg BHT/EDTA | 0.5 mL MeOH | 2 mL EA | [51] | |
| Human plasma (50–200 μL) | Oasis HLB (3 mL, 60 mg) | Free oxylipins: 200 µL plasma + 10 µL 0.2 mg/mL of BHT and TPP and 1 mg/mL EDTA in 50% MeOH + 1 mL 5% MeOH, 0.1% FAc Total oxylipins: 50–100 µL plasma + 10 µL 0.2 mg/mL of BHT and TPP and 1 mg/mL EDTA in 50% MeOH + 0.1 mL 0.1% acetic acid and 0.1% BHT in MeOH 80 °C, overnight + 0.2 mL of 0.25 M sodium carbonate solution, 60 °C, 30 min, constant shaking + 25 µL HAc, 1.575 mL H2O to pH 4–6 | 1× EA 1× MeOH 2 × 5% MeOH, 0.1% FAc | 2 × 5% MeOH, 0.1% FAc dried under ~20 psi, 20 min | 0.5 mL MeOH 1.5 mL EA into 6 µL of 30% glycerol in MeOH | [64] |
| Human plasma (200 µL) | Oasis HLB 96-well plates (30 mg) | 1 mL MeOH 1 mL H2O | 1.5 mL 5% MeOH | 1.2 mL MeOH | [142] | |
| Human serum/plasma/washed platelets (200 μL) | Oasis HLB (1 mL, 10 mg) | + 1 mL MeOH centrifugation at 20,000× g, 4 °C, 10 min | 0.15 mL MeOH 1 mL 0.03% FAc in H2O | 1 mL 0.03% FAc in H2O 1 mL 15% ethanol, 0.03% FAc in H2O 3 mL petroleum ether | 0.2 mL MeOH | [143] |
| Human serum (500 µL) Human sputum (500 µL) Human bronchoalveolar lavage fluid (500 µL) | Oasis HLB 3 mL, 60 mg | + 1.5 mL 5% HAc | 2 mL MeOH 2 mL 0.1% HAc | 2 mL 0.1% HAc | 2 mL MeOH | [144] |
| Human serum (950 µL) | Oasis HLB (60 mg) | + 10 mL 0.2 mg/mL BHT and EDTA | 1× EA 1× MeOH | 2 × 5% MeOH, 0.1% HAc dried under 0.2 bar | 500 μL MeOH 1500 μL EA | [76] |
| Human urine | Oasis HLB (3 mL, 60 mg) | dilution with H2O to 2 mL + 0.5 mL 0.5% HAc | 3 mL H2O 3 mL 0.1% HAc | 3 mL 0.1% HAc dried at −30 kPa, 30 min | 3 mL ACN | [116] |
| dilution with H2O to 2.7 mL + 0.3 mL 1% HAc | 3 mL H2O 3 mL 0.1% HAc | 1 mL 50% MeOH 3 mL 0.1% HAc dried at −30 kPa, 30 min | 1 mL MeOH | |||
| Human cerebrospinal fluid (0.11–1 mL) Rat cortical brain tissue | Oasis HLB (30 mg) | cerebrospinal fluid: dilution with 0.12 M potassium phosphate buffer with 5 mM magnesium chloride, 0.113 mM BHT Tissue: + 0.12 M potassium phosphate buffer with 5 mM magnesium chloride, 0.113 mM BHT centrifugation at 10,000 rpm, 30 min | 1 mL MeOH 1 mL H2O | 3 × 1 mL 5% MeOH | MeOH | [145] |
| Mouse serum (1–10 µL) Human lung epithelial cells (5 × 104 cells/well) Rat fibroblast cell line culture medium (50 µL) | Oasis HLB (10 mg) | + 2× MeOH vortex, 10 s dilution with H2O to 10% MeOH | 1 mL MeoH 2 × 0.75 mL 5% MeOH | 2 × 1 mL 5% MeOH | 1 mL MeOH | [146] |
| Cow heart (~130 mg) cow liver (~320 mg) Pig/elk/cow brain (~80–180 mg) Human plasma (250 µL) Human milk (500 µL) Cell medium 2 mL | Oasis HLB (60 mg, 30 µm) | Tissues: homogenization in 1 mL MeOH with 10 µL 0.2 mg/mL BHT/EDTA in 50% MeOH centrifugation at 2125× g, 10 min diluted to 5% MeOH Plasma, milk, cell medium: + 10 µL 0.2 mg/mL BHT/EDTA in 50% MeOH | 2 mL EA 2 × 2 mL MeOH 2 mL 5% MeOH, 0.1% HAc | 2 × 2 mL 5% MeOH, 0.1% HAc | 3 mL ACN 2 mL MeOH 1 mL EA | [75] |
| Mouse serum/bronchoalveolar lavage fluid (250 µL) | Oasis HLB (60 mg) | + 0.2% w/w TPP/BHT | 2 mL EA 2 × 2 mL MeOH 2 mL 5% MeOH, 0.1% HAc | 1.5 mL 5% MeOH, 0.1% HAc dried under vacuum, 20 min | 0.5 mL MeOH 2 mL EA | [47] |
| Rat cortical brain tissue (20 mg) | Oasis HLB (30 mg) | homogenization with 0.2 mL MeOH, 0.4 µL FAc on ice centrifugation at 14,000 rpm, 0 °C, 10 min + 1.8 mL H2O | 1 mL MeOH 1 mL acetone 2 mL Hex 1 mL acetone 1 mL MeOH 2 mL H2O | 3 mL H2O 1 mL 10% MeOH dried under Ar pressure, 10 min | 2 mL ACN | [126] |
| Rat kidney (100 mg) | Oasis | homogenization in 0.2 mL MeOH with 0.01 M BHT and 5 µL FAc centrifugation at 14,000 rpm, 0 °C, 15 min dilution with H2O to 2 mL | 2 mL 0.1% FAc 2 mL MeOH 2 mL EA | 2 mL 0.1% FAc 2 mL 10% MeOH, 0.1% FAc | 1.5 mL 0.1% FAc with 0.01 BHT 0.5 mL MeOH, 0.2% FAc with 0.01 BHT | [128] |
| Human plasma/urine (1 mL) | Oasis MAX | Urine: + 1 mL 40 mM FAc (pH 2.6) Plasma: + 1 mL 1M KOH in MeOH, 37 °C, 30 min + 1 mL MeOH + 0.2 mL 5 M HCL + 1.7 mL 40 mM FAc to pH 2.6 centrifugation at 10,000× g, 4 °C, 10 min | 2 mL MeOH 2 mL 20 mM FAc | 2 mL 2% ammonium hydroxide 2 mL MeOH/20 mM FAc (40/60, v/v) 2 mL Hex 2 mL Hex/EA (70/30, v/v) | EA | [147] |
| Human plasma (200 µL) | Strata-X (3 mL, 60 mg, 33 µm) | + 0.8 mL H2O, pH 3 acidified with HCL to pH 3 | 2 mL MeOH 2 mL H2O, pH 3 | 10% MeOH in H2O pH 3 | 2 mL MeOH | [148] |
| Human plasma (500 µL) | Strata-X (3 mL, 200 mg, 33 µm) | + 1.5 mL 90% MeOH centrifugation at 6000 rpm, 10 min | 3 mL MeOH 3 mL H2O | 3 mL H2O | 3 mL MeOH | [19] |
| Human plasma (200 µL) | Strata-X (6 mL, 200 mg, 33 µm) | + 0.5 mL cold MeOH with 20 mg/mL BHT/EDTA −80 °C, 30 min centrifugation at 14,000 rpm, 4 °C, 10 min | 6 mL MeOH 6 mL H2O | 6 mL 10% MeOH air-dried, 2 min | 6 mL MeOH with 0.0004% w/v BHT | [53] |
| Human whole blood | Strata-X 96-well plates (60 mg/well, 33 μm) | 1:1 dilution with RPMI-1640 medium (+ 25 mM Hepes and l-glutamine) + calcium ionophore A23187 (final concentration 30 μM), 37 °C, 30 min centrifugation at 1300 rpm, 10 min + 10% MeOH to 1 mL | MeOH H2O | 10% MeOH | 1.0 mL MeOH | [149] |
| Control human plasma (20 µL) Mouse and human tissue: adipose, liver, muscle (2 mg) | Strata-X (3 mL, 60 mg) | Plasma: + 1 mL phosphate salt buffer Tissues: homogenization in 10% MeOH | 3 mL MeOH 3 mL H2O | 10% MeOH | 1 mL MeOH | [72] |
| Cell culture, cell medium (2 mL) | Strata-X | Medium: + 0.1 mL ethanol centrifugation at 3000 rpm, 5 min Cells: + 0.5 mL MeOH + 1 mL phosphate-buffered saline centrifugation at 3000 rpm, 5 min | 2 mL MeOH 2 mL H2O | 1 mL 10% MeOH | 1 mL MeOH | [132] |
| ANION-EXCHANGE PHASES | ||||||
| Zebrafish embryo | Strata-X-A (3 mL, 200 mg, 22 µm) | saponification | 3 mL MeOH 3 mL H2O | 4 mL MeOH—cholesterol 3 × 6 mL air 4 mL ACN—α-tocopherol | 4 mL FAc/MeOH/ACN (5/47.5/47.5) | [150] |
| Human urine | Strata-X-AW (3 mL, 100 mg) | + 200 mM MeOH/HCl centrifugation at 10,000 rpm, 5 min | 2 mL MeOH 2 mL H2O | 4 mL H2O | 1 mL MeOH | [151] |
| VARIOUS PHASES | ||||||
| Human urine (2 mL) | Bond Elut C18 (3 mL, 500 mg) | + 11.25 mL MeOH/CH3Cl (2:1, v/v), room temperature, 1 h + 3.75 mL CH3Cl + 3.75 mL water centrifugation at 2500 rpm, 10 min CH3Cl phase evaporated and reconstituted in 100 μL MeOH | 5 mL MeOH 5 mL H2O | 5 mL H2O 3 mL 5% MeOH dried under vacuum | 4 mL MeOH | [117] |
| Oasis HLB (6 mL, 500 mg) | 5 mL MeOH 5 mL ACN 5 mL H2O | 3 mL 5% ACN dried under vacuum | 4 mL ACN | |||
| Strata-X (6 mL, 200 mg) | 5 mL MeOH 5 mL H2O | 3 mL 10% MeOH | 4 mL MeOH | |||
| Chromabond Easy (6 mL, 200 mg) Oasis MAX (6 mL, 500 mg) | 5 mL 2% FAc in MeOH 5 mL H2O | 5 mL H2O 3 mL 25% MeOH 3 mL ACN dried under vacuum | 2 × 2 mL MeOH | |||
| Mouse colon tissue Human epithelial colorectal adenocarcinoma cells Cell supernatant | Marchery Nagel C18 (15 mL, 200 mg) | Colon tissue: homogenization in 0.5 mL of HBSS + 1 mL MeOH centrifugation at 900× g, 4 °C, 15 min Cells/supernatant: + 1 mL MeOH centrifugation at 900× g, 4 °C, 15 min | 10 mL MeOH 10 mL 0.02 M HCL/MeOH (90/10, v/v) | 5 mL 0.02 M HCl/MeOH (90/10, v/v) dried under aspiration | 5 mL methyl formate | [129] |
| Foam macrophages supernatant Mouse peritoneal exudate | Oasis HLB 96-well plates | centrifugation at 20,000× g, 4 °C, 20 min | 4 mL MeOH 4 mL H2O | 2 mL 5% MeOH 2 mL 10% MeOH 2 mL H2O dried under aspiration, 15 min centrifugation at 200× g, 2 min | 4 mL methyl formate | |
| Human plasma (500 μL) | Oasis HLB (3 mL, 60 mg, 30 μm) | 1:1 dilution with 5% MeOH acidified with 0.1% HAc centrifugation at 20,000× g, 4 °C, 10 min | 1× EA 1× MeOH 2 × 5% MeOH, 0.1% HAc | 2 × 5% MeOH, 0.1% HAc | 0.5 mL MeOH 1.5 mL EA | [112] |
| 1:1 dilution with 40% MeOH centrifugation at 20,000× g, 4 °C, 10 min | 1× EA 1× MeOH 1 × 20% MeOH, 0.1% FAc | 1 × 20% MeOH, 0.1% FAc | 2.0 mL MeOH | |||
| SepPak tC18 (6 mL, 500 mg, 37–55 µm) | + 1.5 mL 20% MeOH centrifugation at 20,000× g, 4 °C, 10 min + 80 µL conc. HAc to pH 3 | 3× MeOH 3× H2O | 10 mL H2O 6 mL Hex | 8 mL methyl formate | ||
| Bond Elut Certify II (3 mL, 200 mg, 47–60 µm) | + 500 μL 1 M sodium acetate buffer (pH 6) centrifugation at 20,000× g, 4 °C, 10 min | 1× MeOH 1 × 0.1 M sodium acetate buffer, 5% MeOH | 1× MeOH/H2O (50/50, v/v) | 2.0 mL n-Hex/EA (25/75, v/v) | ||
| 2.0 mL n-Hex/EA (75/25, v/v) | ||||||
| Strata-X (3 mL, 100 mg, 33 µm) | 1:1 dilution with 20% MeOH centrifugation at 20,000× g, 4 °C, 10 min | 3.5 mL MeOH 3.5 mL H2O | 3.5 mL 10% MeOH | 1.0 mL MeOH | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liakh, I.; Pakiet, A.; Sledzinski, T.; Mika, A. Modern Methods of Sample Preparation for the Analysis of Oxylipins in Biological Samples. Molecules 2019, 24, 1639. https://doi.org/10.3390/molecules24081639
Liakh I, Pakiet A, Sledzinski T, Mika A. Modern Methods of Sample Preparation for the Analysis of Oxylipins in Biological Samples. Molecules. 2019; 24(8):1639. https://doi.org/10.3390/molecules24081639
Chicago/Turabian StyleLiakh, Ivan, Alicja Pakiet, Tomasz Sledzinski, and Adriana Mika. 2019. "Modern Methods of Sample Preparation for the Analysis of Oxylipins in Biological Samples" Molecules 24, no. 8: 1639. https://doi.org/10.3390/molecules24081639
APA StyleLiakh, I., Pakiet, A., Sledzinski, T., & Mika, A. (2019). Modern Methods of Sample Preparation for the Analysis of Oxylipins in Biological Samples. Molecules, 24(8), 1639. https://doi.org/10.3390/molecules24081639