Future Prospects in the Treatment of Parasitic Diseases: 2-Amino-1,3,4-Thiadiazoles in Leishmaniasis

Abstract

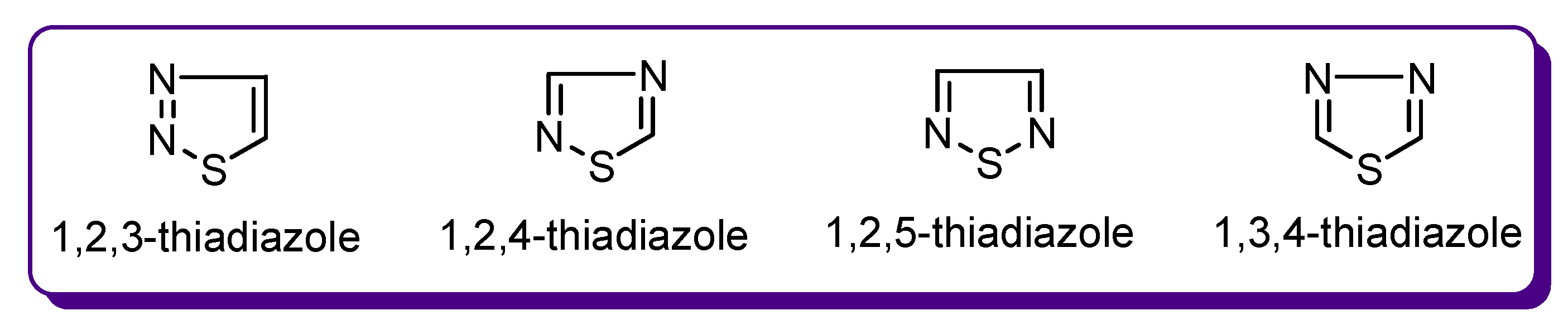
1. The Significance of Thiadiazole Derivatives for Medicinal Chemistry
1.1. Biological Significance of Heterocyclic Compounds
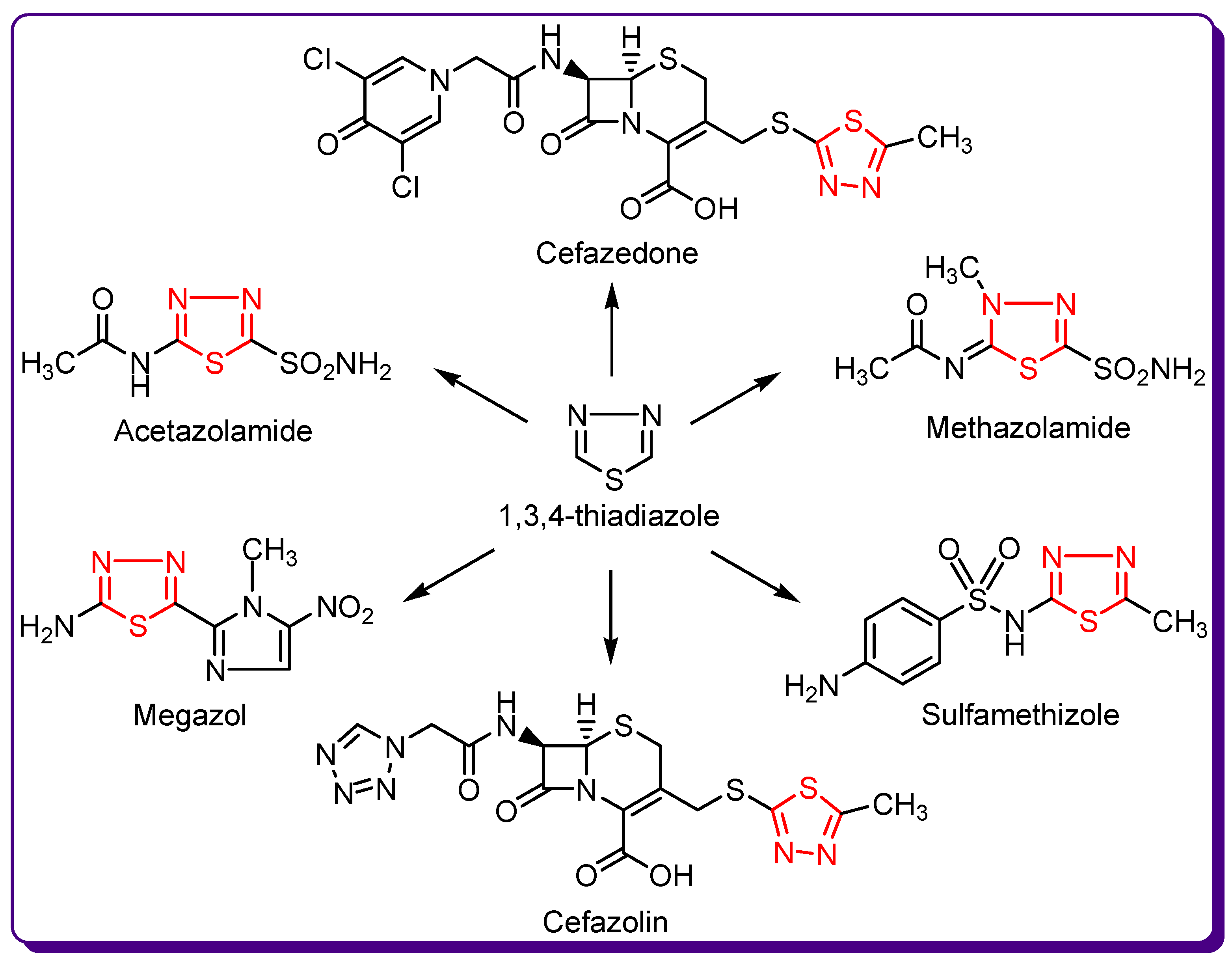
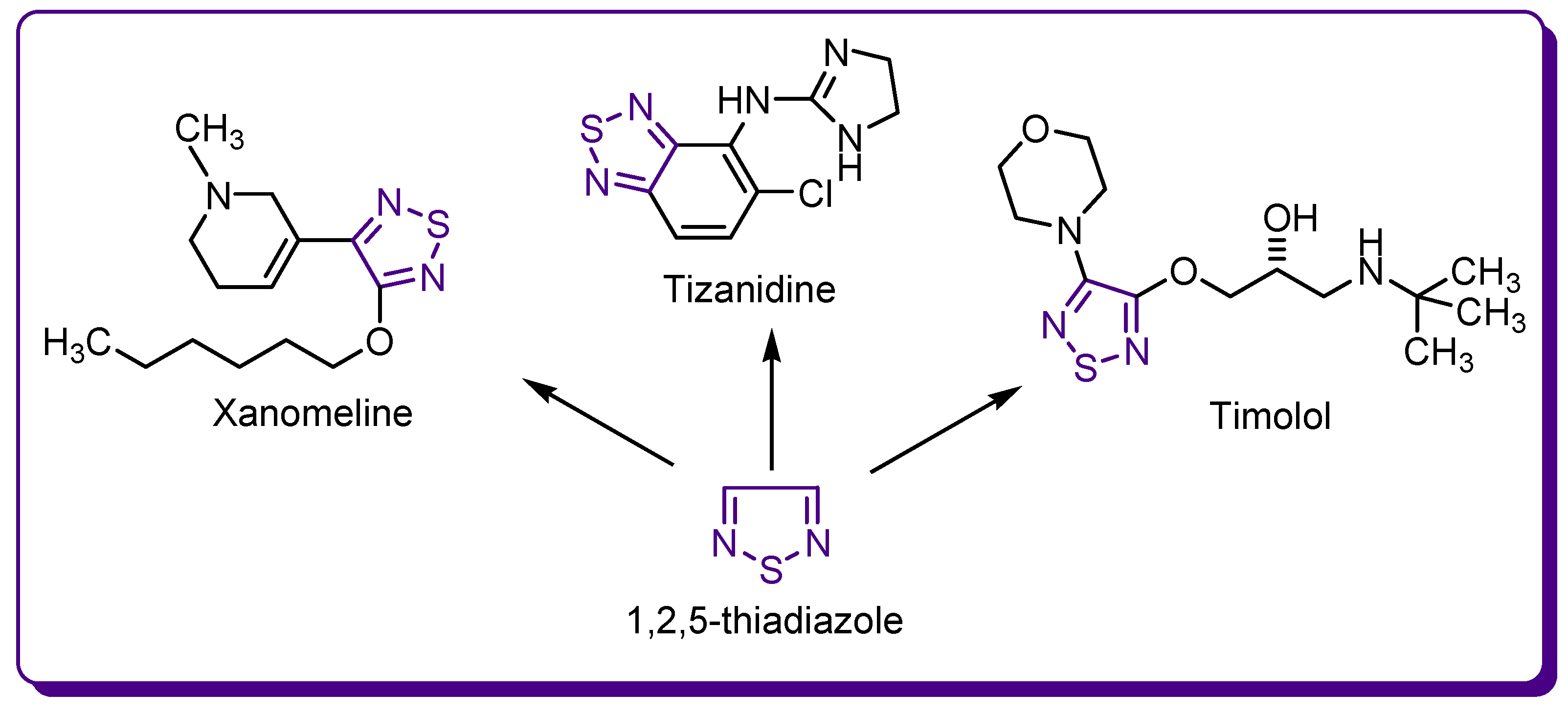
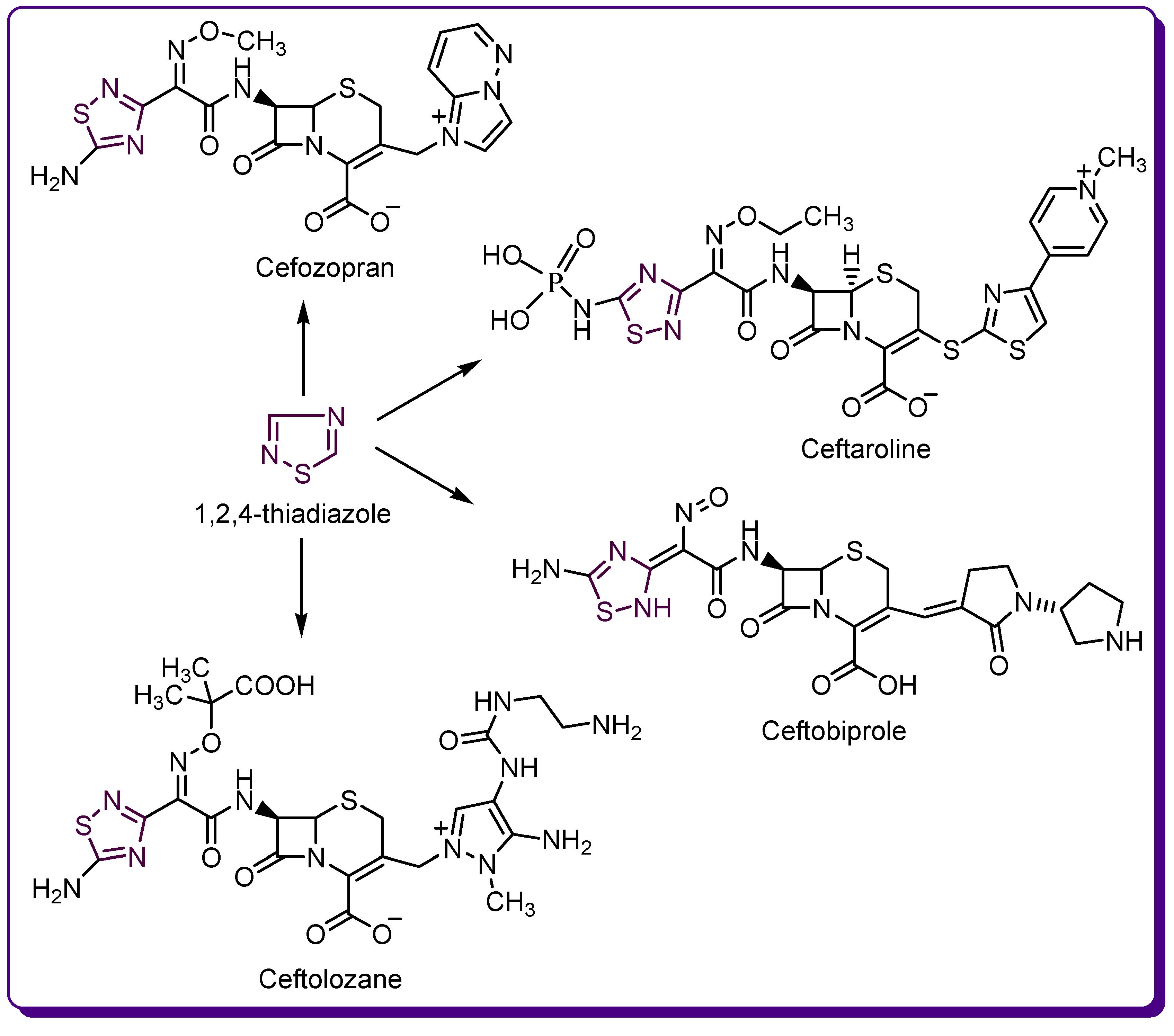
1.2. Thiadiazole Derivatives with Medical Significance
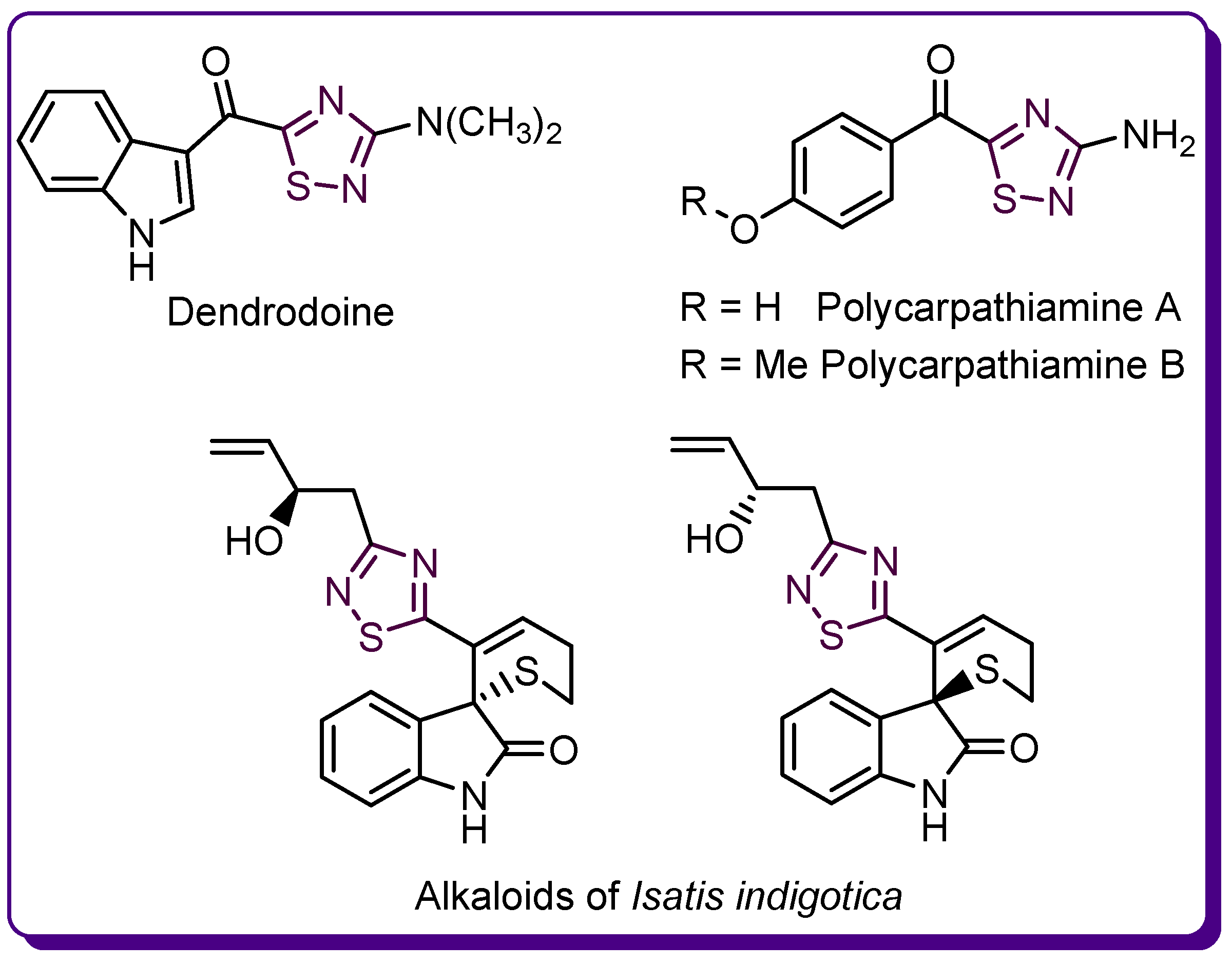
1.3. Studies on the 2-Amino-1,3,4-thiadiazole Series
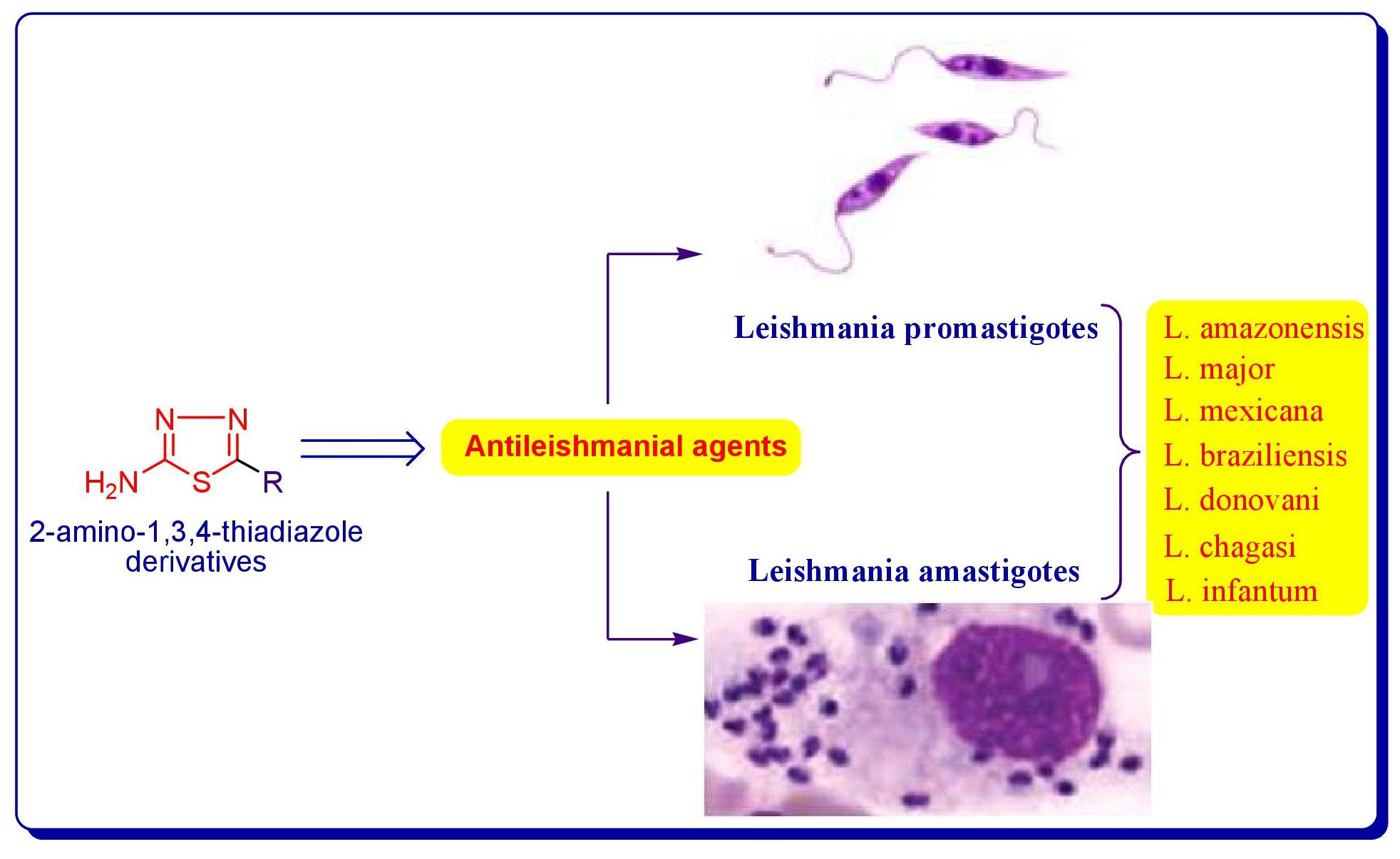
2. Kinetoplastea Parasites
2.1. Kinetoplastid Infections
2.2. Leishmania Parasites and Leishmaniasis
2.3. Current Pharmacotherapy for Leishmaniasis
3. Antileishmanial Activity Associated with 2-Amino-1,3,4-thiadiazole System
3.1. The Significance of Hydroxyl, Oxyalkyl and Aminoalkyl Side Chains for Antileishmanial Activity


3.2. The Significance of a Supplementary Heterocycle for the Antileishmanial Activity


3.3. Mesoionic Derivatives of 2-amino-1,3,4-thiadiazole Moiety

3.4. 2-Amino-1,3,4-thiadiazole Derivatives as Parasite Enzyme Inhibitors
3.4.1. Nitric Oxide Synthase Inhibitors

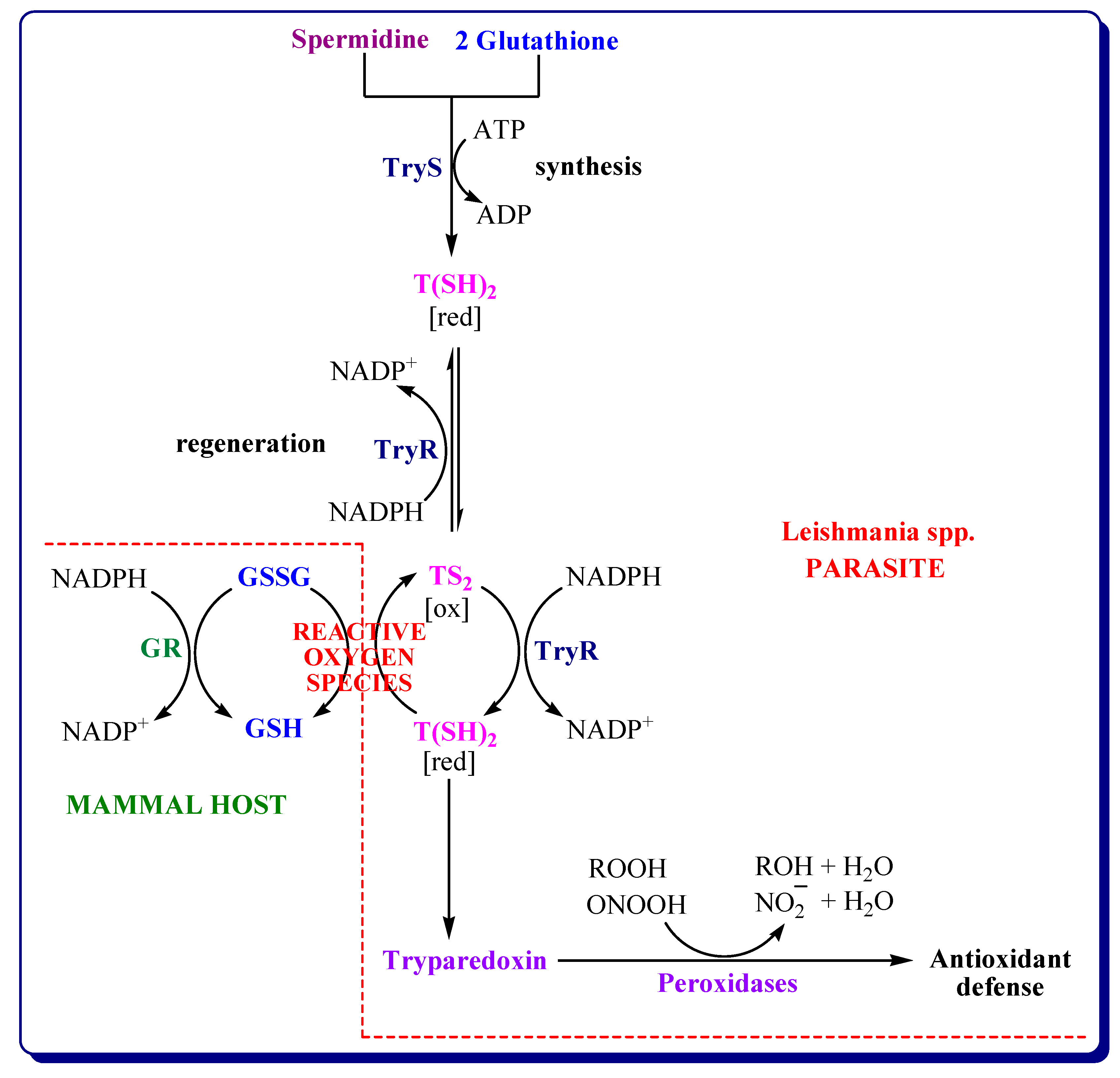
3.4.2. Inhibitors of Trypanothione Reductase/Tryparedoxin/Tryparedoxin Peroxidase System

3.4.3. Pteridine Reductase-1 Inhibitors

4. Conclusions
Funding
Conflicts of Interest
References
- Shukla, P.K.; Verma, A.; Mishra, P. Significance of nitrogen heterocyclic nuclei in the search of pharmacological active compounds. In New Perspective in Agricultural and Human Health; Shukla, R.P., Mishra, R.S., Tripathi, A.D., Yadav, A.K., Tiwari, M., Mishra, R.R., Eds.; Bharti Publication: New Delhi, India, 2017; pp. 100–126. [Google Scholar]
- Pearce, S. The importance of heterocyclic compounds in anti-cancer drug design. DDW 2017, Summer 2017, 66–70. Available online: https://www.ddw-online.com/therapeutics/p320375-the-importance-of -heterocyclic-compounds-in-anti-cancer-drug-design.html (accessed on 17 January 2019).
- Diaba, F.; Montiel, J.A.; Serban, G.; Bonjoch, J. Synthesis of normorphans through an efficient intramolecular carbamoylation of ketones. Org. Lett. 2015, 17, 3860–3863. [Google Scholar] [CrossRef] [PubMed]
- Serban, G.; Abe, H.; Takeuchi, Y. Synthetic studies of substituted pyridine aldehydes as intermediates for the synthesis of toddaquinoline, its derivatives and other natural products. Heterocycles 2011, 83, 1989–2000. [Google Scholar] [CrossRef]
- Serban, G.; Abe, H.; Takeuchi, Y.; Harayama, T. A new approach to the benzopyridoxepine core by metal mediated intramolecular biaryl ether formation. Heterocycles 2008, 75, 2949–2958. [Google Scholar] [CrossRef]
- Serban, G.; Shigeta, Y.; Nishioka, H.; Abe, H.; Takeuchi, Y.; Harayama, T. Studies toward the synthesis of toddaquinoline by intramolecular cyclization. Heterocycles 2007, 71, 1623–1630. [Google Scholar] [CrossRef]
- Dawood, K.M.; Farghaly, T.A. Thiadiazole inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 477–505. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Alam, M.S.; Hamid, H. 1,3,4-Thiadiazoles: A potent multi targeted pharmacological scaffold. Eur. J. Med. Chem. 2015, 92, 156–177. [Google Scholar] [CrossRef]
- Serban, G.; Stanasel, O.; Serban, E.; Bota, S. 2-Amino-1,3,4-thiadiazole as a potential scaffold for promising antimicrobial agents. Drug Des. Dev. Ther. 2018, 12, 1545–1566. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. Adrenergic agents. In Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry, 12th ed.; Beale, J.M., Jr., Block, J.H., Eds.; Lippincott Williams and Wilkins, Wolters Kluwer: Philadelphia, PA, USA, 2011; Chapter 16; p. 538. [Google Scholar]
- Shekhar, A.; Potter, W.Z.; Lightfoot, J.; Lienemann, J.; Dube, S.; Mallinckrodt, C.; Bymaster, F.P.; McKinzie, D.L.; Felder, C.C. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry 2008, 165, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.R.; Peters, D.; Sparks, R.G. Xanomeline and the antipsychotic potential of muscarinic receptor subtype selective agonists. CNS Drug Rev. 2003, 9, 158–186. [Google Scholar] [CrossRef]
- Karuna Announces First Patient Dosed in Phase 2 Study of Lead Product Candidate KarXT for the Treatment of Schizophrenia, 15 October 2018. Available online: https://www.businesswire.com/news/ home/20181014005040/en/Karuna-Announces-Patient-Dosed-Phase-2-Study (accessed on 18 January 2019).
- Proshin, A.N.; Serkov, I.V.; Bachurin, S.O. 5-Amino-1,2,4-thiadiazole derivatives. European Patent Application, EP 2 628 734 A2, 21 August 2013. [Google Scholar]
- Pandeya, S.N.; Yadav, M.K. Synthesis and in-vivo analgesic activity of 1,2,4-thiadiazole derivatives. Int. J. ChemTech Res. 2012, 4, 618–624. [Google Scholar]
- Zalewski, P.; Skibinski, R.; Szymanowska-Powalowska, D.; Piotrowska, H.; Bednarski, W.; Cielecka-Piontek, J. Radiolytic studies of cefozopran hydrochloride in the solid state. Electron. J. Biotechnol. 2017, 25, 28–32. [Google Scholar] [CrossRef]
- Long, T.E.; Williams, J.T. Cephalosporins currently in early clinical trials for the treatment of bacterial infections. Expert Opin. Investig. Drugs 2014, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kanafani, Z.A.; Corey, G.R. Ceftaroline: A cephalosporin with expanded Gram-positive activity. Future Microbiol. 2009, 4, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, T.W.L. Ceftobiprole medocaril in the treatment of hospital-acquired pneumonia. Future Microbiol. 2015, 10, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Davison, E.K.; Sperry, J. Synthesis of the 1,2,4-thiadiazole alkaloids polycarpathiamines A and B. Org. Chem. Front. 2016, 3, 38–42. [Google Scholar] [CrossRef]
- Heitz, S.; Durgeat, M.; Guyot, M.; Brassy, C.; Bachet, B. Nouveau derive indolique du thiadiazole-1,2,4, isole d’un tunicier (Dendrodoagrossularia). Tetrahedron Lett. 1980, 21, 1457–1458. [Google Scholar] [CrossRef]
- Pham, C.D.; Weber, H.; Hartmann, R.; Wray, V.; Lin, W.; Lai, D.; Proksch, P. New cytotoxic 1,2,4-thiadiazole alkaloids from the ascidian Polycarpaaurata. Org. Lett. 2013, 15, 2230–2233. [Google Scholar] [CrossRef]
- Chen, M.; Lin, S.; Li, L.; Zhu, C.; Wang, X.; Wang, Y.; Jiang, B.; Wang, S.; Li, Y.; Jiang, J.; et al. Enantiomers of an indole alkaloid containing unusual dihydrothiopyran and 1,2,4-thiadiazole rings from the root of Isatisindigotica. Org. Lett. 2012, 14, 5668–5671. [Google Scholar] [CrossRef]
- Hipler, F.; Winter, M.; Fischer, R.A. N-H···S hydrogen bonding in 2-mercapto-5-methyl-1,3,4-thiadiazole. Synthesis and crystal structures of mercapto functionalized 1,3,4-thiadiazoles. J. Mol. Struct. 2003, 658, 179–191. [Google Scholar] [CrossRef]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole—A promising structure in Medicinal Chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, C.Y.; Wang, X.M.; Yang, Y.H.; Zhu, H.L. 1,3,4-Thiadiazole: Synthesis, reactions and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef]
- Bhuva, H.; Sahu, D.; Shah, B.N.; Modi, D.C.; Patel, M.B. Biological profile of thiadiazole. Pharmacologyonline 2011, 1, 528–543. [Google Scholar]
- Wermuth, C.G.; Aldous, D.; Raboisson, P.; Rognan, D. The Practice of Medicinal Chemistry, 4th ed.; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2015; p. 196. [Google Scholar]
- Holla, B.S.; Poorjary, K.N.; Rao, B.S.; Shivananda, M.K. New bis-aminomercaptotriazoles and bis-triazolothiadiazoles as possible anticancer agents. Eur. J. Med. Chem. 2002, 37, 511–517. [Google Scholar] [CrossRef]
- Senff-Ribeiro, A.; Echevarria, A.; Silva, E.F.; Franco, C.R.C.; Veiga, S.S.; Oliveira, M.B.M. Cytotoxic effect of a new 1,3,4-thiadiazolium mesoionic compound (MI-D) on cell lines of human mellanoma. Br. J. Cancer 2004, 91, 297–304. [Google Scholar] [CrossRef]
- Ciotti, M.M.; Humphreys, S.R.; Venditti, J.M.; Kaplan, N.O.; Goldin, A. The antileukemic action of two thiadiazole derivatives. Cancer Res. 1960, 20, 1195–1201. [Google Scholar]
- Juszczak, M.; Matysiak, J.; Brzana, W.; Niewiadomy, A.; Rzeski, W. Evaluation of antiproliferative activity of 2-(monohalogenophenylamino)-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazoles. Arzneim. Forsch. Drug Res. 2008, 58, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J. Evaluation of antiproliferative effect in vitro of some 2-amino-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazole derivatives. Chem. Pharm. Bull. 2006, 54, 988–991. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matysiak, J.; Opolski, A. Synthesis and antiproliferative activity of N-substituted 2-amino-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazoles. Bioorg. Med. Chem. 2006, 14, 4483–4489. [Google Scholar] [CrossRef]
- Rzeski, W.; Matysiak, J.; Kandefer-Szerszen, M. Anticancer, neuroprotective activities and computational studies of 2-amino-1,3,4-thiadiazole based compound. Bioorg. Med.Chem. 2007, 15, 3201–3207. [Google Scholar] [CrossRef]
- Asbury, R.; Blessing, J.A.; Moore, D. A phase II trial of aminothiadiazole in patients with mixed mesodermal tumors of the uterine corpus: A gynecologic oncology group study. Am. J. Clin. Oncol. 1996, 19, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Elson, P.L.; Kvols, L.K.; Vogl, S.E.; Glover, D.J.; Hahn, R.G.; Trump, D.L. Phase II trials of 5-day vinblastine infusion (NSC 49842), L-alanosine (NSC153353), acivicin (NSC 163501), and aminothiadiazole (NSC 4728) in patientswith recurrent or metastatic renal cell carcinoma. Investig. New Drugs 1988, 6, 97–103. [Google Scholar] [CrossRef]
- Engstrom, P.F.; Ryan, L.M.; Falkson, G.; Haller, D.G. Phase II study of aminothiadiazole in advanced squamous cell carcinoma of the esophagus. Am. J. Clin. Oncol. 1991, 14, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Locker, G.Y.; Kilton, L.; Khandekar, J.D.; Lad, T.E.; Knop, R.H.; Albain, K.; Blough, R.; French, S.; Benson, A.B. High-dose aminothiadiazole in advanced colorectal cancer. An Illinois Cancer Center phase II trial. Investig. New Drugs 1994, 12, 299–301. [Google Scholar] [CrossRef]
- Serban, G. 5-Arylamino-1,3,4-thiadiazol-2-yl acetic acid esters as intermediates for the synthesis of new bisheterocyclic compounds. Farmacia 2015, 63, 146–149. [Google Scholar]
- Horvath, T.; Serban, G.; Cuc, S. Synthesis of new 2-phenylamino-5-[(α-acylamino)-p-X-stiryl]-1,3,4-thiadiazole compounds. Farmacia 2014, 62, 422–427. [Google Scholar]
- Serban, G.; Suciu, A.; Coman, M.; Curea, E. Synthesis and physical-chemical study of some 3-(5-arylamino-1,3,4-thiadiazol-2-yl)coumarins. Farmacia 2002, 50, 50–54. [Google Scholar]
- Serban, G.; Coman, M.; Curea, E. Synthesis of some heterocyclic nitrocoumarins by Knoevenagel condensation. Farmacia 2005, 53, 78–84. [Google Scholar]
- Serban, G.; Matinca, D.; Bradea, O.; Gherman, L.; Coman, M.; Curea, E. The study of the biological activity of some heterocyclic coumarins. Farmacia 2005, 53, 91–99. [Google Scholar]
- Serban, G.; Coman, M.; Curea, E.; Proinov, L. Synthesis and description of some heterocyclic coumarins. Farmacia 2001, 49, 45–52. [Google Scholar]
- Lemke, T.L. Antiparasitic agents. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Lippincott Williams and Wilkins, Wolters Kluwer: Baltimore, MD, USA, 2013; p. 1126. [Google Scholar]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization, Department of Control of Neglected Tropical Diseases. Working to Overcome the Global Impact of Neglected Tropical Diseases: First WHO Report on Neglected Tropical Diseases; WHO Press: Geneva, Switzerland, 2010; ISBN 978 92 4 1564090. Available online: https://apps.who.int/iris/bitstream/handle/10665/44440/9789241564090_eng.pdf;jsessionid=016AB1F2B46847F532878327511591A8?sequence=1 (accessed on 25 September 2017).
- Chandler, J.A.; James, P.M. Discovery of Trypanosomatid Parasites in Globally Distributed Drosophila Species. PLoS ONE 2013, 8, e61937. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Dawson, A.; Poohner, I.; Costa, D.M.; Sa, M.S.; Cordeiro-da-Silva, A.; Luciani, R.; Gul, S.; Witt, G.; Ellinger, B.; et al. Exploiting the 2-amino-1,3,4-thiadiazole scaffold to inhibit Trypanosoma brucei pteridine reductase in support of early-stage drug discovery. ACS Omega 2017, 2, 5666–5683. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Neglected Tropical Diseases. Available online: http://www.who.int/neglected_diseases/diseases/en/ (accessed on 21 July 2018).
- Tahghighi, A.; Emami, S.; Razmi, S.; Marznaki, F.R.; Ardestani, S.K.; Dastmalchi, S.; Kobarfard, F.; Shafiee, A.; Foroumadi, A. New 5-(nitroheteroaryl)-1,3,4-thiadiazoles containing acyclic amines at C-2: Synthesis and SAR study for their antileishmanial activity. J. Enzyme Inhib. Med. Chem. 2013, 28, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Poorrajab, F.; Ardestani, S.K.; Emami, S.; Behrouzi-Fardmoghadam, M.; Shafiee, A.; Foroumadi, A. Nitroimidazolyl-1,3,4-thiadiazole-based anti-leishmanial agents: Synthesis and in vitro biological evaluation. Eur. J. Med. Chem. 2009, 44, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Tiuman, T.S.; Santos, A.O.; Ueda-Nakamura, T.; Dias Filho, B.P.; Nakamura, C.V. Recent advances in leishmaniasis treatment. Int. J. Infect. Dis. 2011, 15, e525–e532. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. Available online: http://www.who.int/mediacentre/factsheets /fs375/en/Leishmaniasis (accessed on 25 September 2017).
- World Health Organization, Department of Control of Neglected Tropical Diseases. Leishmaniasis in high-burden countries: An epidemiological update based on data reported in 2014. Wkly. Epidemiol. Rec. 2016, 91, 285–296. Available online: https://www.who.int/neglected diseases/ resources /who_wer9122/en/ (accessed on 25 September 2017).
- Al-Qahtani, A.; Siddiqui, Y.M.; Bekhit, A.A.; El-Sayed, O.A.; Aboul-Enein, H.Y.; Al-Ahdal, M.N. Inhibition of growth of Leishmania donovani promastigotes by newly synthesized 1,3,4-thiadiazole analogs. Saudi Pharm. J. 2009, 17, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef]
- World Health Organization. Leishmaniasis. 14 March 2018. Available online: http://www.who.int/news- room/fact-sheets/detail/leishmaniasis (accessed on 10 July 2018).
- Tahghighi, A.; Babalouei, F. Thiadiazoles: The appropriate pharmacological scaffolds with leishmanicidal and antimalarial activities: A review. Iran. J. Basic Med. Sci. 2017, 20, 613–622. [Google Scholar]
- Rodrigues, R.F.; da Silva, E.F.; Echevarria, A.; Fajardo-Bonin, R.; Amaral, V.F.; Leon, L.L.; Canto-Cavalheiro, M. A comparative study of mesoionic compounds in Leishmania sp. and toxicity evaluation. Eur. J. Med. Chem. 2007, 42, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Cobo, F. Leishmaniasis. In Imported Infectious Diseases: TheImpact inDeveloped Countries, 1st ed.; Woodhead Publishing Elsevier: Cambridge, UK, 2014; pp. 234–236. ISBN 978-1-907568-57-2. [Google Scholar]
- Passero, L.F.D.; Laurenti, M.D.; Santos-Gomes, G.; Soares Campos, B.L.; Sartorelli, P.; Lago, J.H.G. In vivo antileishmanial activity of plant-based secondary metabolites. In Fighting Multidrug Resistance with Herbal Extracts, Essential Oils and Their Components, 1st ed.; Rai, M., Kon, K., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2013; pp. 96–97. [Google Scholar]
- Frezard, F.; Demicheli, C.; Da Silva, S.M.; Azevedo, E.G.; Ribeiro, R.R. Nanostructures for improved antimonial therapy of leishmaniasis. In Nano- and Microscale Drug Delivery Systems: Design and Fabrication, 1st ed.; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; p. 421. [Google Scholar]
- Pund, S.; Joshi, A. Nanoarchitectures for neglected tropical diseases: Challenges and state of the art. In Nano- and Microscale Drug Delivery Systems: Design andFabrication, 1st ed.; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; p. 449. [Google Scholar]
- Tahghighi, A.; Marznaki, F.R.; Kobarfard, F.; Dastmalchi, S.; Mojarrad, J.S.; Razmi, S.; Ardestani, S.K.; Emami, S.; Shafiee, A.; Foroumadi, A. Synthesis and antileishmanial activity of novel 5-(5-nitrofuran-2-yl)-1,3,4-thiadiazoles with piperazinyl-linked benzamidine substituents. Eur. J. Med. Chem. 2011, 46, 2602–2608. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.F.; Canto-Cavalheiro, M.M.; Braz, V.R.; Cysne-Finkelstein, L.; Leon, L.L.; Echevarria, A. Synthesis and biological evaluation of new 1,3,4-thiadiazolium-2-phenylamine derivatives against Leishmania amazonensis promastigotes and amastigotes. Eur. J. Med. Chem. 2002, 37, 979–984. [Google Scholar] [CrossRef]
- Ferrari, S.; Morandi, F.; Motiejunas, D.; Nerini, E.; Henrich, S.; Luciani, R.; Venturelli, A.; Lazzari, S.; Calo, S.; Gupta, S.; et al. Virtual screening identification of nonfolate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J. Med. Chem. 2011, 54, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Tahghighi, A.; Razmi, S.; Mahdavi, M.; Foroumadi, P.; Ardestani, S.K.; Emami, S.; Kobarfard, F.; Dastmalchi, S.; Shafiee, A.; Foroumadi, A. Synthesis and antileishmanial activity of 5-(5-nitrofuran-2-yl)-1,3,4-thiadiazol-2-amines containing-N-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]moieties. Eur. J. Med. Chem. 2012, 50, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.F.; Charret, K.S.; da Silva, E.F.; Echevarria, A.; Amaral, V.F.; Leon, L.L.; Canto-Cavalheiro, M. Antileishmanial activity of 1,3,4-thiadiazolium-2-aminide in mice infected with Leishmania amazonensis. Antimicrob. Agents Chemother. 2009, 53, 839–842. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pourrajab, F.; Forouzannia, S.K.; Tabatabaee, S.A. Novel immunomodulatory function of 1,3,4-thiadiazole derivatives with leishmanicidal activity. J. Antimicrob. Chemother. 2012, 67, 1968–1978. [Google Scholar] [CrossRef]
- Liew, F.Y.; Wei, X.Q.; Proudfoot, L. Cytokines and nitric oxide as effector molecules against parasitic infections. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 1311–1315. [Google Scholar] [CrossRef]
- Rodrigues, R.F.; Castro-Pinto, D.; Echevarria, A.; dos Reis, C.M.; Del Cistia, C.N.; Sant’Anna, C.M.R.; Teixeira, F.; Castro, H.; Canto-Cavalheiro, M.; Leon, L.L.; et al. Investigation of trypanothione reductase inhibitory activity by 1,3,4-thiadiazolium-2-aminide derivatives and molecular docking studies. Bioorg. Med. Chem. 2012, 20, 1760–1766. [Google Scholar] [CrossRef]
- Soares-Bezerra, R.J.; da Silva, E.F.; Echevarria, A.; Gomes-da-Silva, L.; Cysne-Finkelstein, L.; Monteiro, F.P.; Leon, L.L.; Genestra, M. Effect of mesoionic 4-phenyl-5-(cinnamoyl)-1,3,4-thiadiazolium-2-phenylamine chloride derivative salts on the activities of the nitric oxide synthase and arginase of Leishmaniaamazonensis. J. Enzyme Inhib. Med. Chem. 2008, 23, 328–333. [Google Scholar] [CrossRef][Green Version]
- Kavoosi, G.; Ardestani, S.K.; Kariminia, A.Z.; Tavakoli, Z. Production of nitric oxide by murine macrophages induced by lipophosphoglycan of Leishmania major. Korean J. Parasitol. 2006, 44, 35–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lemos de Souza, V.; Souza, J.A.; Silva, T.M.C.; Veras, P.S.T. Different Leishmania species determine distinct profiles of immune and histopathological responses in CBA mice. Microbes Infect. 2000, 2, 1807–1815. [Google Scholar] [CrossRef]
- Liew, F.Y.; Li, Y.; Moss, D.; Parkinson, C.; Rogers, M.V.; Moncada, S. Resistance to Leishmania majorinfection correlates with the inductionof nitric oxide synthase in murine macrophages. Eur. J. Immunol. 1991, 21, 3009–3014. [Google Scholar] [CrossRef]
- Nashleanas, M.; Scott, P. Activated T cells induce macrophages to produce NO and controle Leishmania major in the absence of tumor necrosis factor receptor p55. Infect Immun. 2000, 68, 1428–1434. [Google Scholar] [CrossRef]
- Soares-Bezerra, R.J.; Leon, L.L.; Echevarria, A.; Reis, C.M.; Gomes-Silva, L.; Agostinho, C.G.; Fernandes, R.A.; Canto-Cavalheiro, M.M.; Genestra, M.S. In vitro evaluation of 4-phenyl-5-(4′-X-phenyl)-1,3,4-thiadiazolium-2-phenylaminide chlorides and 3[N-4′-X-phenyl]-1,2,3-oxadiazolium-5-olate derivatives on nitric oxide synthase and arginase activities of Leishmania amazonensis. Exp. Parasitol. 2013. [Google Scholar] [CrossRef]
- Colotti, G.; Baiocco, P.; Fiorillo, A.; Boffi, A.; Poser, E.; Di Chiaro, F.; Ilari, A. Structural insights into the enzymes of the trypanothione pathway: Targets forantileishmaniasis drugs. Future Med. Chem. 2013, 5, 1861–1875. [Google Scholar] [CrossRef]
- Khan, M.O.F. Trypanothione reductase: A viable chemotherapeutic target for antitrypanosomal and antileishmanial drug design. Drug Target Insights 2007, 2, 129–146. [Google Scholar] [CrossRef]
- Benitez, D.; Medeiros, A.; Fiestas, L.; Panozzo-Zenere, E.A.; Maiwald, F.; Prousis, K.C.; Roussaki, M.; Calogeropoulou, T.; Detsi, A.; Jaeger, T.; et al. Identification of novel chemical scaffolds inhibiting trypanothione synthetase from pathogenic trypanosomatids. PLoS Negl. Trop. Dis. 2016, 10, e0004617. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Fiorillo, A.; Genovese, I.; Colotti, G. An update on structural insights into the enzymes of the polyamine-trypanothione pathway: Targets for new drugs against leishmaniasis. Future Med. Chem. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Fairlamb, A.H.; Blackburn, P.; Ulrich, P.; Chait, B.T.; Cerami, A. Trypanothione: A novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science 1985, 227, 1485–1487. [Google Scholar] [CrossRef]
- Olin-Sandoval, V.; Gonzalez-Chavez, Z.; Berzunza-Cruz, M.; Martinez, I.; Jasso-Chavez, R.; Becker, I.; Espinoza, B.; Moreno-Sanchez, R.; Saavedra, E. Drug target validation of the trypanothione pathway enzymes through metabolic modeling. FEBS J. 2012, 279, 1811–1833. [Google Scholar] [CrossRef] [PubMed]
- Spinks, D.; Torrie, L.S.; Thompson, S.; Harrison, J.R.; Frearson, J.A.; Read, K.D.; Fairlamb, A.H.; Wyatt, P.G.; Gilbert, I.H. Design, synthesis and biological evaluation of Trypanosoma brucei trypanothione synthetase inhibitors. Chem. Med. Chem. 2012, 7, 95–106. [Google Scholar] [CrossRef]
- Sousa, A.F.; Gomes-Alves, A.G.; Benitez, D.; Comini, M.A.; Flohe, L.; Jaeger, T.; Passos, J.; Stuhlmann, F.; Tomas, A.M.; Castro, H. Genetic and chemical analyses reveal that trypanothione synthetase but not glutathionylspermidine synthetase is essential for Leishmania infantum. Free Radic. Biol. Med. 2014, 73, 229–238. [Google Scholar] [CrossRef]
- Fyfe, P.K.; Oza, S.L.; Fairlamb, A.H.; Hunter, W.N. Leishmania trypanothione synthetase-amidase structure reveals a basis for regulation of conflicting synthetic and hydrolytic activities. J. Biol. Chem. 2008, 283, 17672–17680. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef]
- Sienkiewicz, N.; Ong, H.B.; Fairlamb, A.H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol. Microbiol. 2010, 77, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.B.; Sienkiewicz, N.; Wyllie, S.; Fairlamb, A.H. Dissecting the Metabolic Roles of Pteridine Reductase 1 in Trypanosoma brucei and Leishmania major. J. Biol. Chem. 2011, 286, 10429–10438. [Google Scholar] [CrossRef] [PubMed]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New approaches to Leishmania chemotherapy: Pteridine reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitology 1997, 114, S101–S110. [Google Scholar]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | IC50 (μM) | CC50 (μM) | R | IC50 (μM) | CC50 (μM) | ||
|---|---|---|---|---|---|---|---|
| 4 | Me | 54 ± 0.17 | - | 16 | Me | 64 ± 0.32 | - |
| 5 | Et | 50 ± 0.8 | - | 17 | Et | 98.4 ± 0.12 | - |
| 6 | c-Pr | 55.5 ± 0.66 | - | 18 | c-Pr | 97.3 ± 0.98 | - |
| 7 | (CH2)2OH | 21 ± 0.65 | 122.11 | 19 | (CH2)3OH | 3 ± 0.41 | 42.16 |
| 8 | (CH2)3OH | 18 ± 0.2 | 62.33 | 20 | (CH2)3OMe | 3 ± 0.5 | 37.80 |
| 9 | CH2CH(OMe)2 | 13 ± 0.53 | 62.46 | 21 | CH2CH(OMe)2 | 56 ± 0.74 | - |
| 10 | (CH2)2N(Et)2 | 44 ± 0.72 | 83.29 | 22 | (CH2)2N(Et)2 | 36 ± 0.24 | 43.80 |
| 11 | (CH2)3N(Et)2 | 13 ± 0.43 | 104.60 | 23 | (CH2)3N(Me)2 | 25 ± 0.17 | 87.35 |
| 12 | (CH2)2N(i-Pr)2 | 13 ± 0.23 | 42.18 | 24 | (CH2)2O(CH2)2OH | 42 ± 0.45 | - |
| 13 | CH(Et)CH2OH | 10 ± 0.66 | 83.29 | 25 | CH2CH=CH2 | 34.3 ± 0.5 | 129.32 |
| 14 | (CH2)2O(CH2)2OH | 116 ± 0.76 | - | ||||
| 15 | CH(Me)(CH2)3N(Et)2 | 33 ± 0.76 | 367.65 |
| R | IC50 (μM) | |
|---|---|---|
| 26 | H | 19.6 ± 0.56 |
| 27 | 2-F | 19.0 ± 0.35 |
| 28 | 4-F | 21.4 ± 1.86 |
| 29 | 2-Cl | 18.9 ± 0.12 |
| 30 | 4-Cl | 88.8 ± 0.9 |
| 31 | 2-Me | 25.2 ± 0.54 |
| 32 | 4-Me | 12.2 ± 0.66 |
| 33 | 2,4-Cl2 | 19.9 ± 0.84 |
| 34 | 2-F-6-Cl | 17.4 ± 0.76 |
| X | Y | L. amazonensis | L. braziliensis ED50 (μM) | L. chagasi ED50 (μM) | TD50 (μM) | ||
|---|---|---|---|---|---|---|---|
| Promastigotes IC50 (μM) | Amastigotes IC50 (μM) | ||||||
| 37 | H | H | 0.47 ± 0.03 | 104.54 ± 11.95 | 49.61 | 22.76 | 3.06 |
| 38 | OMe | H | 0.17 ± 0.01 | 23.93 ± 4.88 | 46.20 | 8.31 | 1.13 |
| 39 | F | H | 0.92 ± 0.06 | 5.37 ± 0.28 | 5.1 | 3.42 | 5.82 |
| 40 | Cl | H | 1.51 ± 0.22 | 186.34 ± 18.11 | 6.23 | 13.17 | 11.98 |
| 41 | Br | H | 0.87 ± 0.1 | 33.26 ± 2.38 | 2.93 | 9.93 | 6.59 |
| 42 | H | OMe | 0.04 ± 0.01 | 41.88 ± 2.83 | 30.64 | 4.75 | 0.61 |
| 43 | H | Cl | 0.48 ± 0.05 | 178.11 ± 15.39 | 1.72 | 5.17 | 6.23 |
| 44 | H | Br | 0.52 ± 0.05 | 5.48 ± 0.04 | 25.95 | 144.68 | 2.47 |
| 45 | NO2 | H | 1.00 ± 0.12 | 52.92 ± 5.92 | 70.77 | 22.83 | 5.49 |
| Pentamidine | 0.46 ± 0.08 | 118.00 ± 7.31 | - | - | - | ||
| Pentamidine (Filaxis Lab) | 23.64/ED50 | - | 64.18 | 27.5 | 2.21 | ||
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serban, G. Future Prospects in the Treatment of Parasitic Diseases: 2-Amino-1,3,4-Thiadiazoles in Leishmaniasis. Molecules 2019, 24, 1557. https://doi.org/10.3390/molecules24081557
Serban G. Future Prospects in the Treatment of Parasitic Diseases: 2-Amino-1,3,4-Thiadiazoles in Leishmaniasis. Molecules. 2019; 24(8):1557. https://doi.org/10.3390/molecules24081557
Chicago/Turabian StyleSerban, Georgeta. 2019. "Future Prospects in the Treatment of Parasitic Diseases: 2-Amino-1,3,4-Thiadiazoles in Leishmaniasis" Molecules 24, no. 8: 1557. https://doi.org/10.3390/molecules24081557
APA StyleSerban, G. (2019). Future Prospects in the Treatment of Parasitic Diseases: 2-Amino-1,3,4-Thiadiazoles in Leishmaniasis. Molecules, 24(8), 1557. https://doi.org/10.3390/molecules24081557