Novel C-2 Symmetric Molecules as α-Glucosidase and α-Amylase Inhibitors: Design, Synthesis, Kinetic Evaluation, Molecular Docking and Pharmacokinetics

 , , ,
, , ,  ,
,  , and
, and 
Abstract
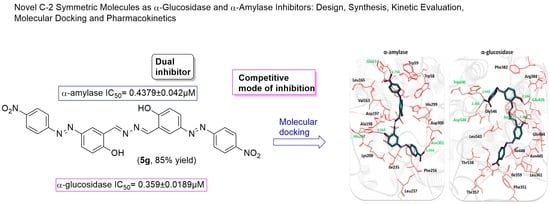
:
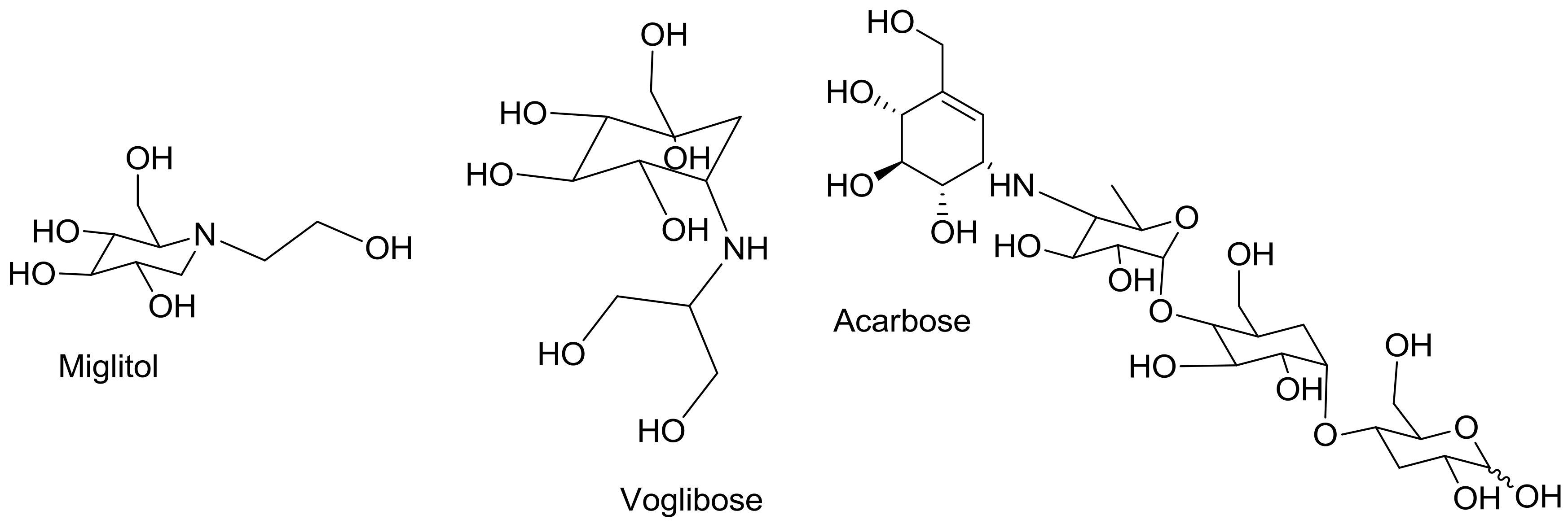
1. Introduction
2. Results and Discussion
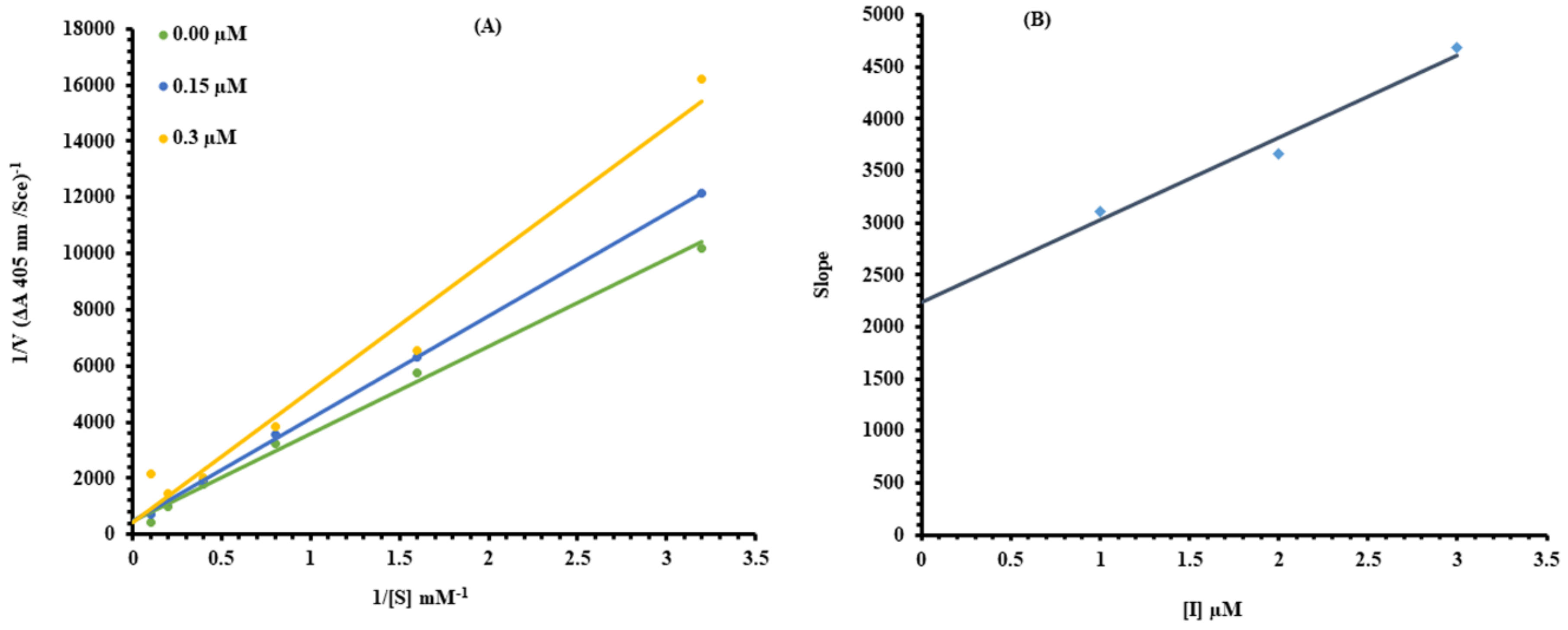
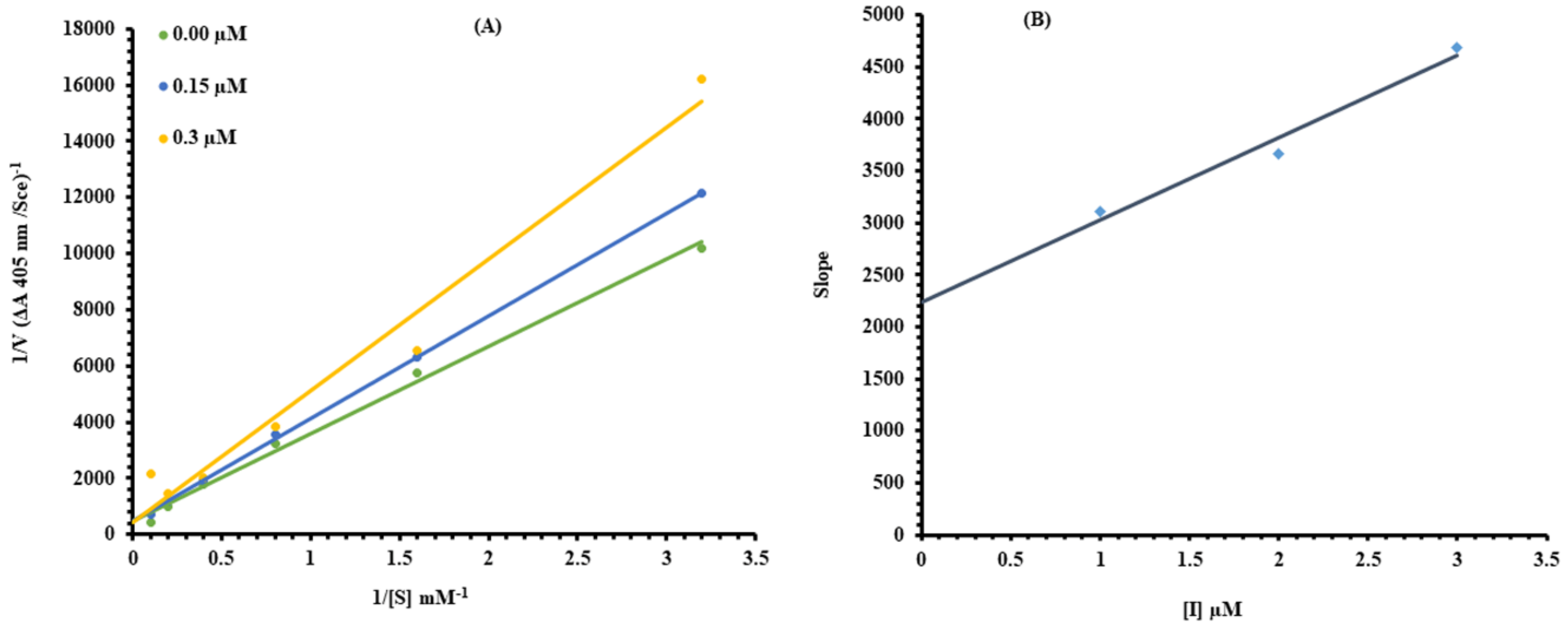
2.1. Kinetic Analysis
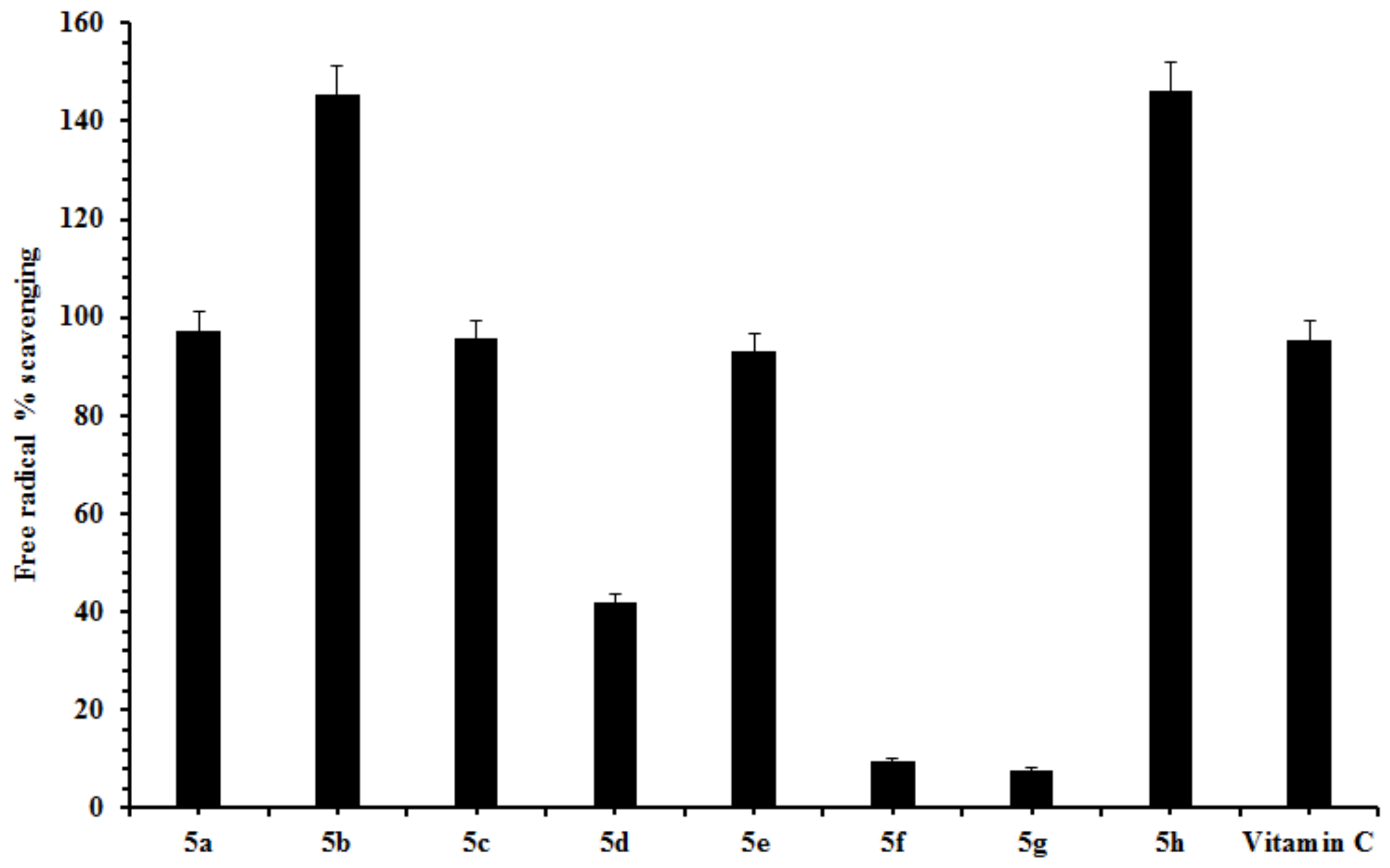
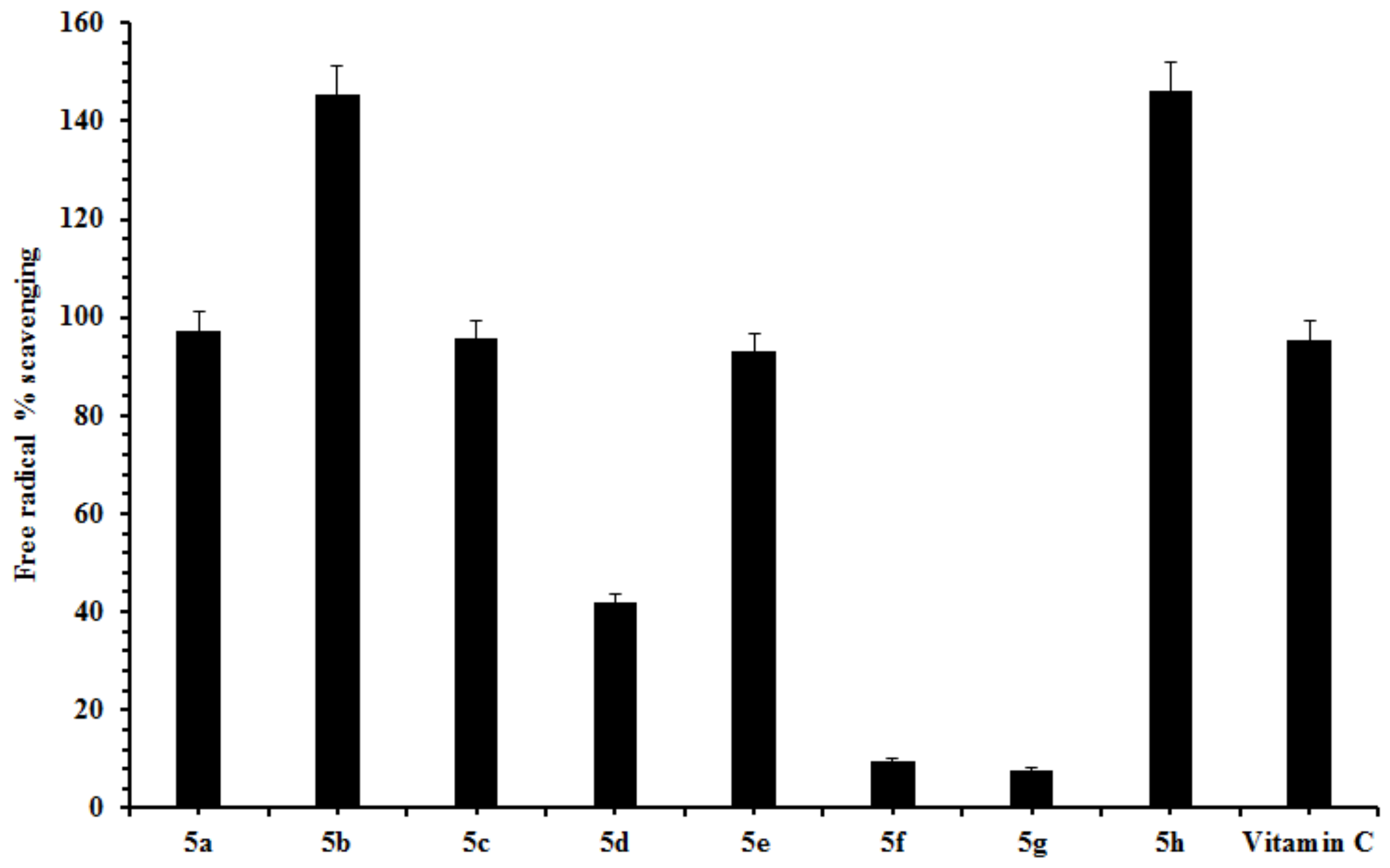
2.2. Free Radical Scavenging
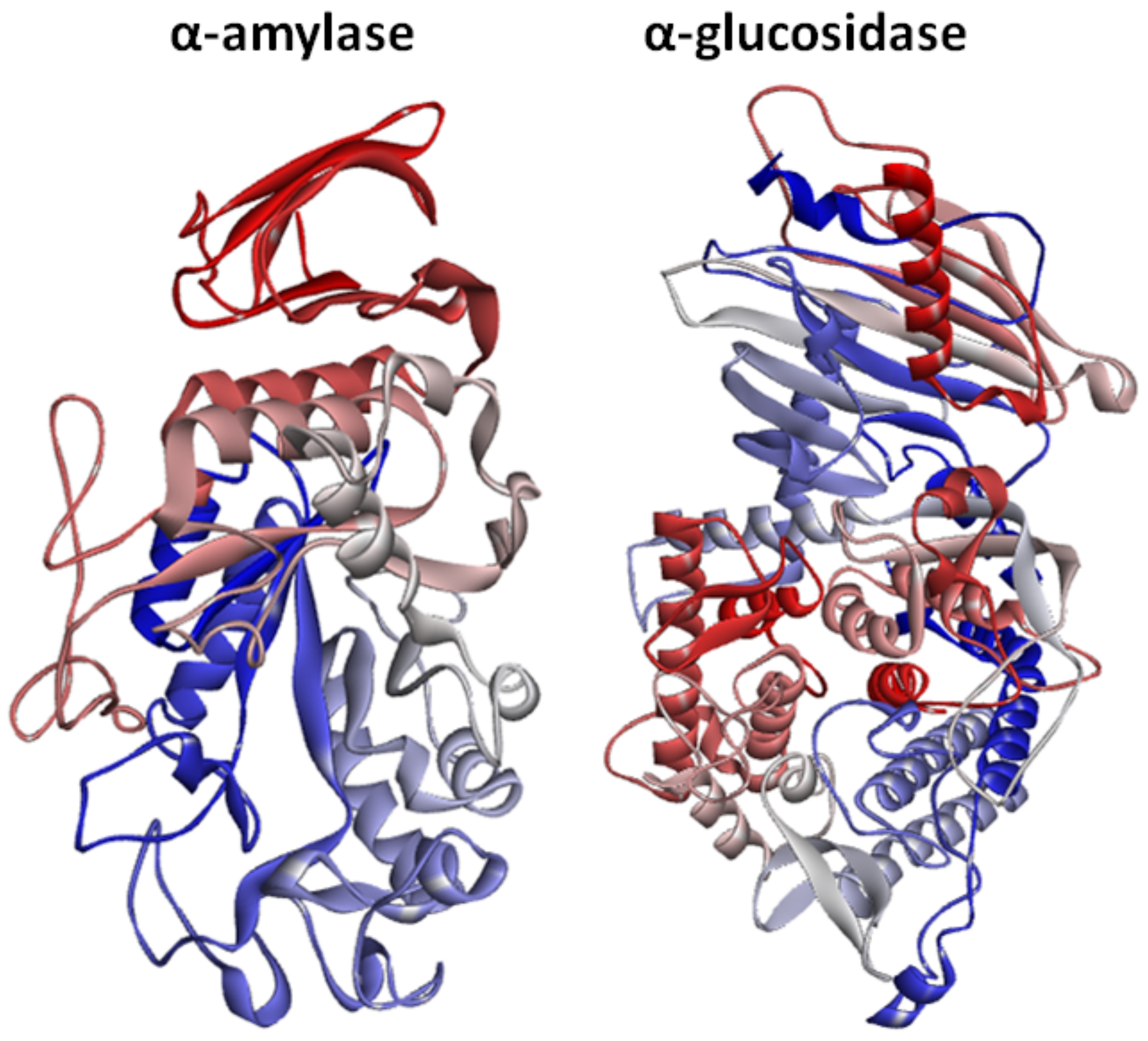
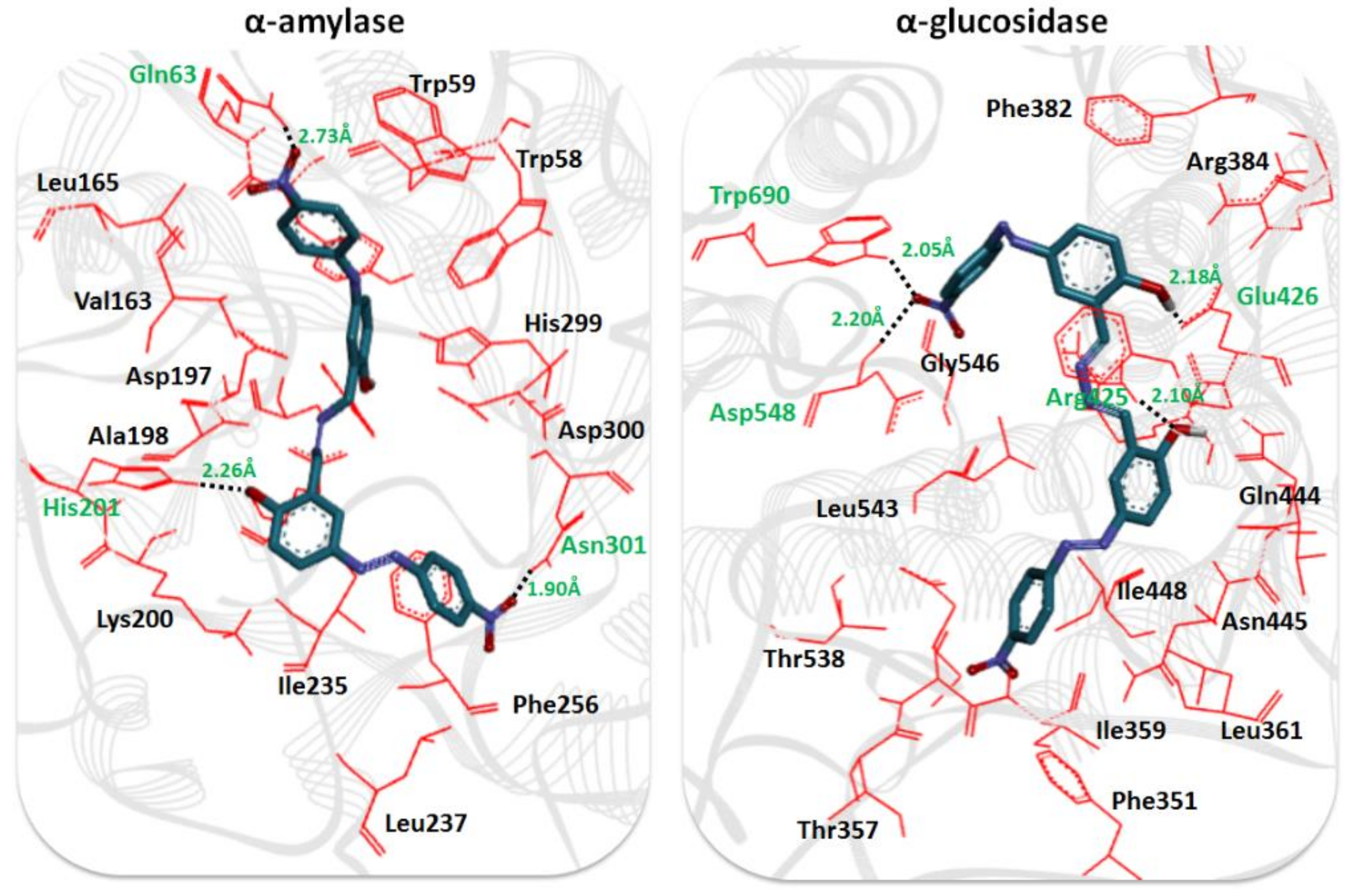
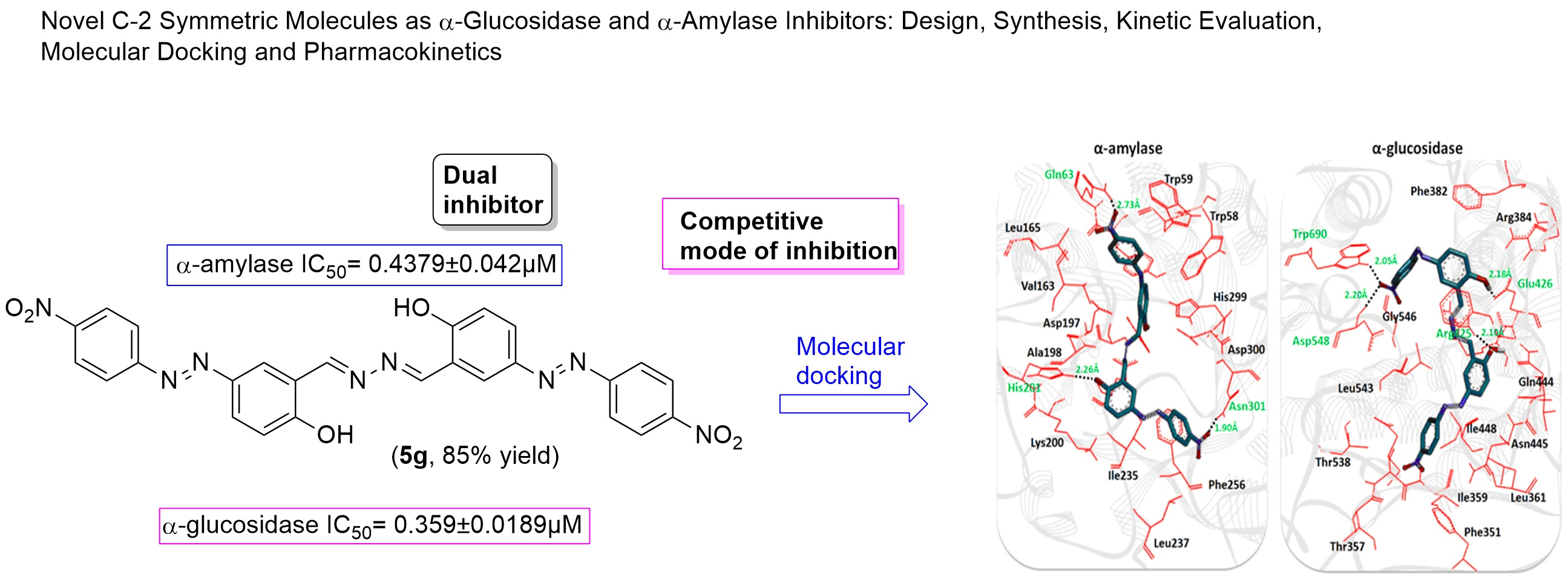
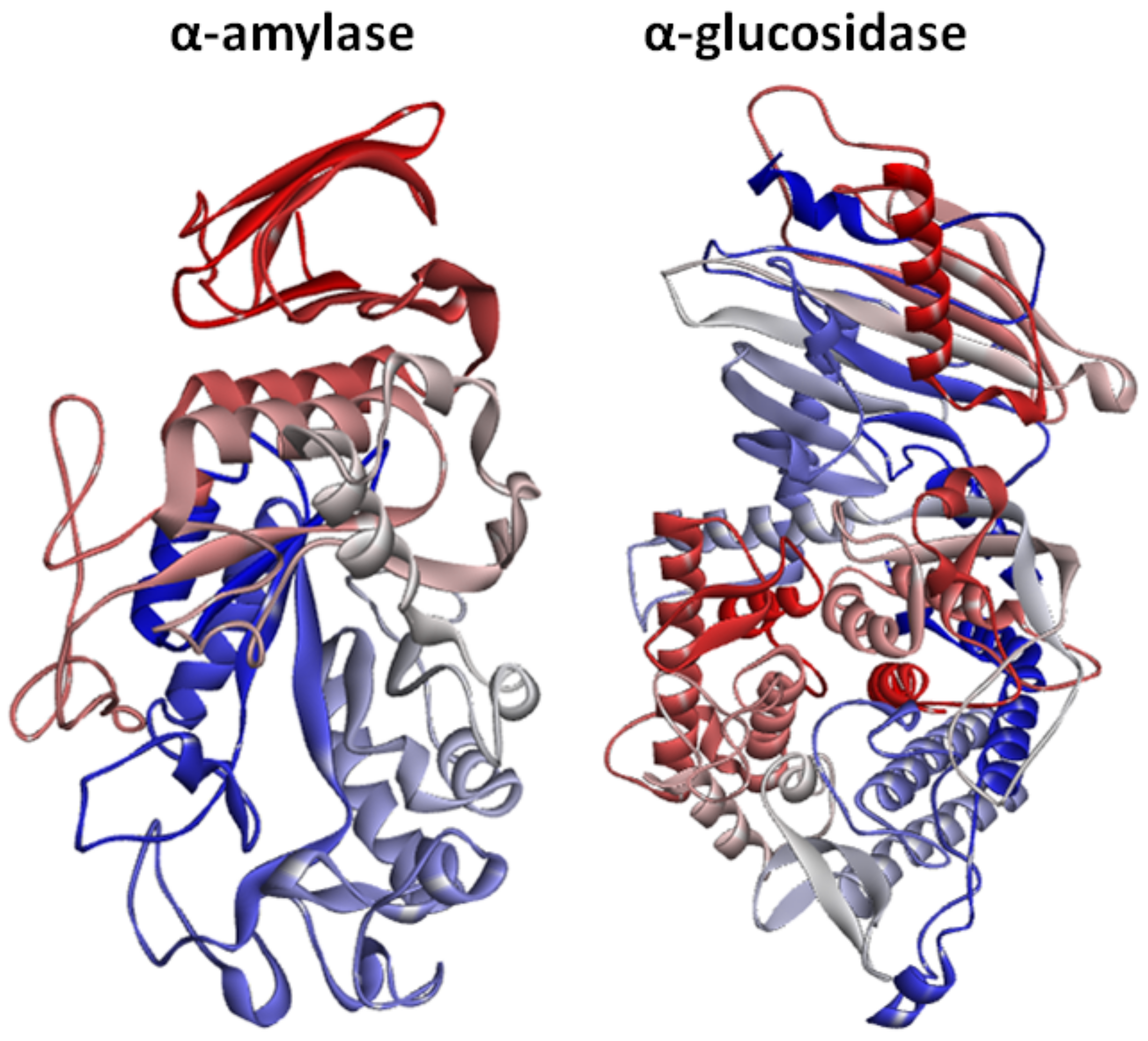
2.3. Structural Assessment of α-Amylase and α-Glucosidase
2.4. Chemo-Informatics and Lipinski’s Rule of Five (RO5) Assessments of the Ligands
2.5. Lead Optimization and Lipophilicity Values
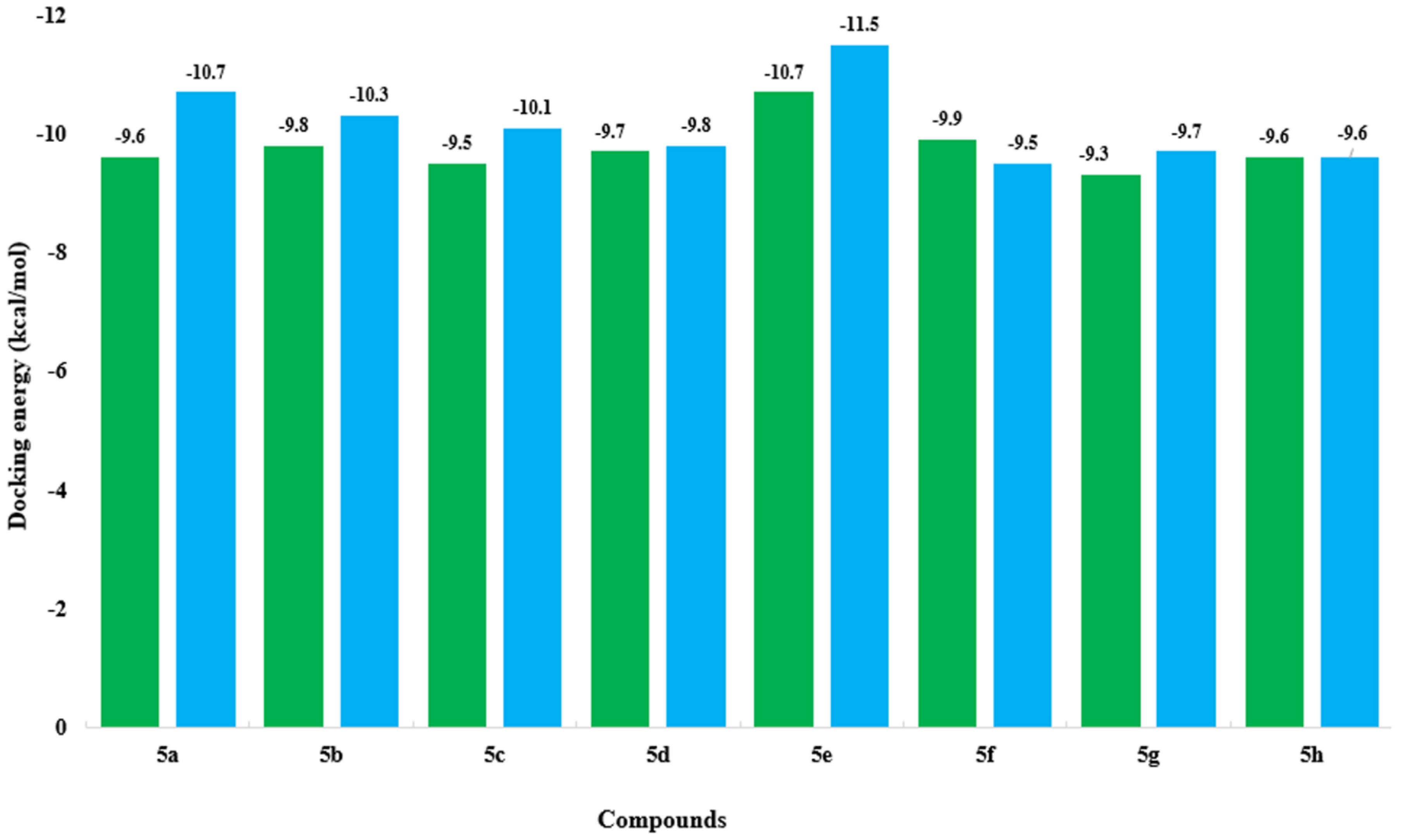
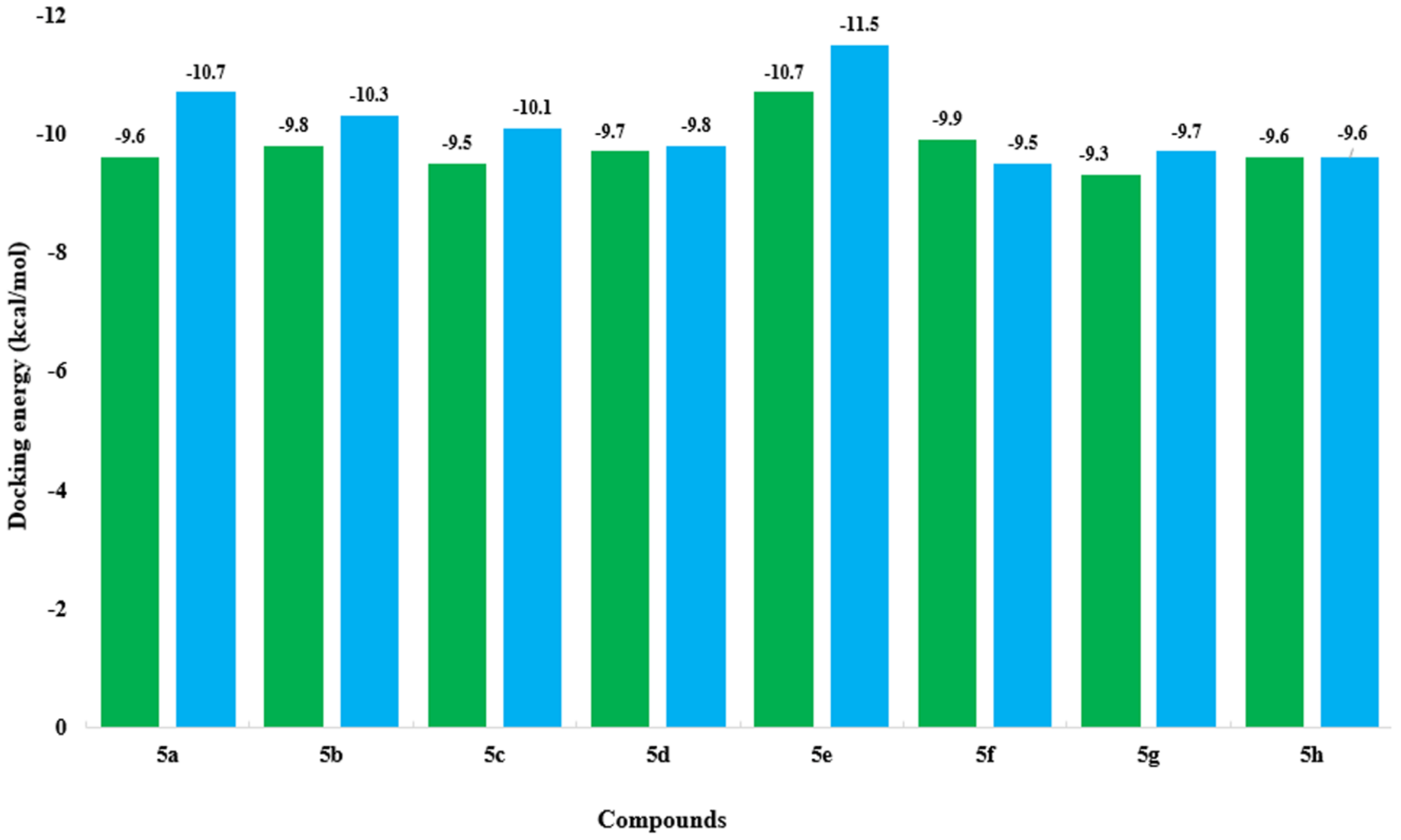
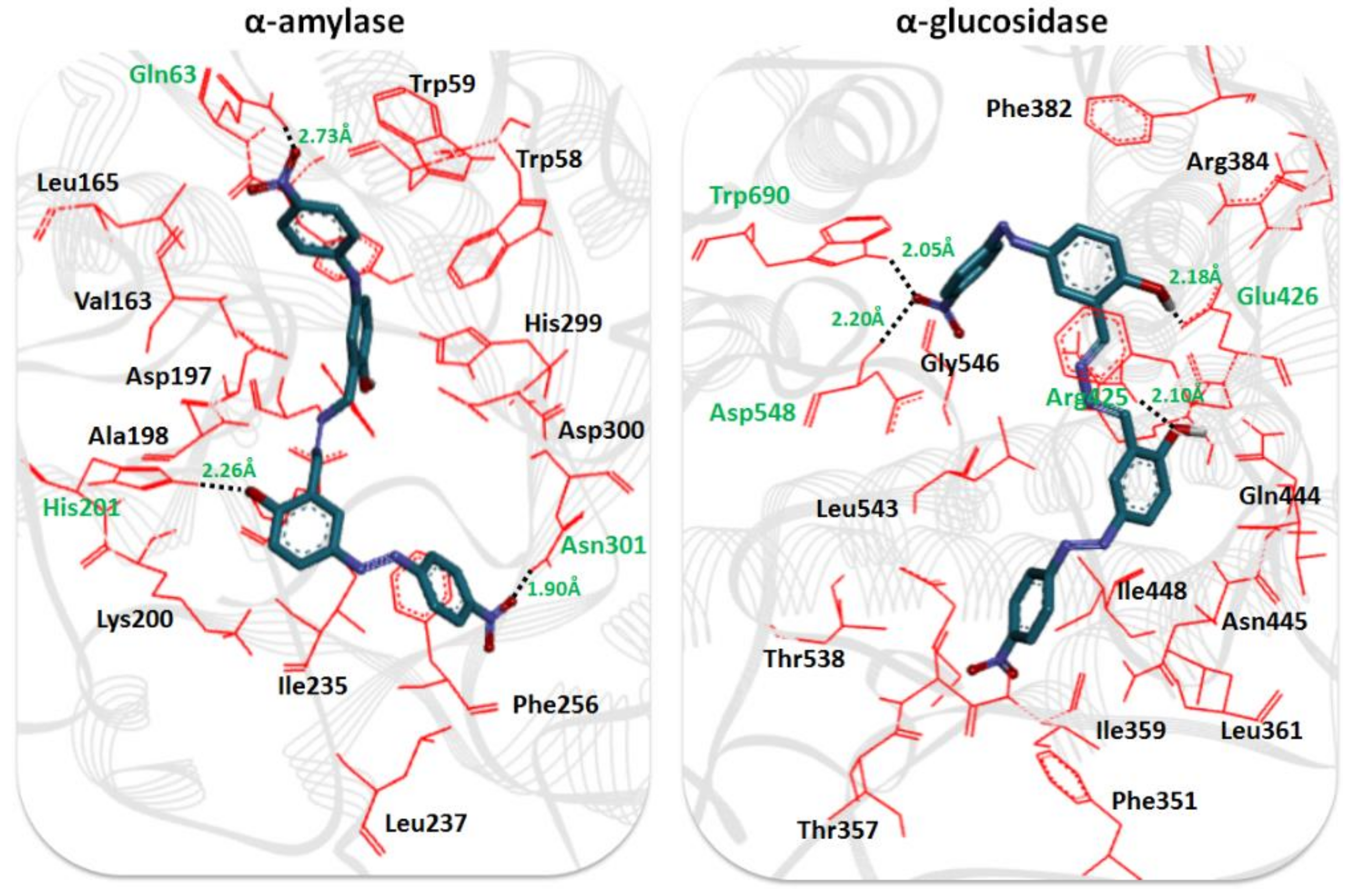
2.6. Molecular Docking Analysis
2.7. Structure-Activity Relationship (SAR) Analysis
3. Experimental
3.1. Methods and Materials
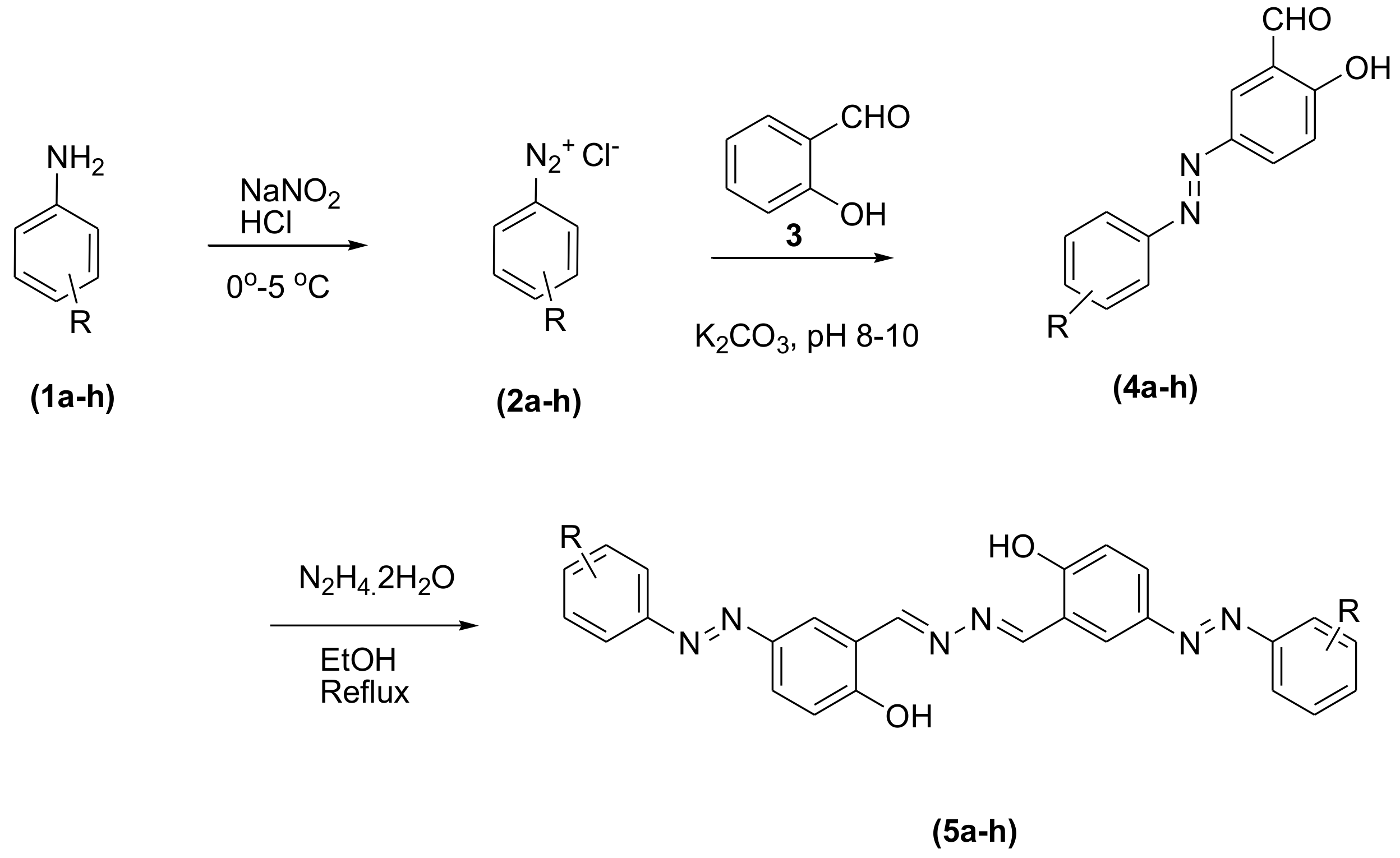
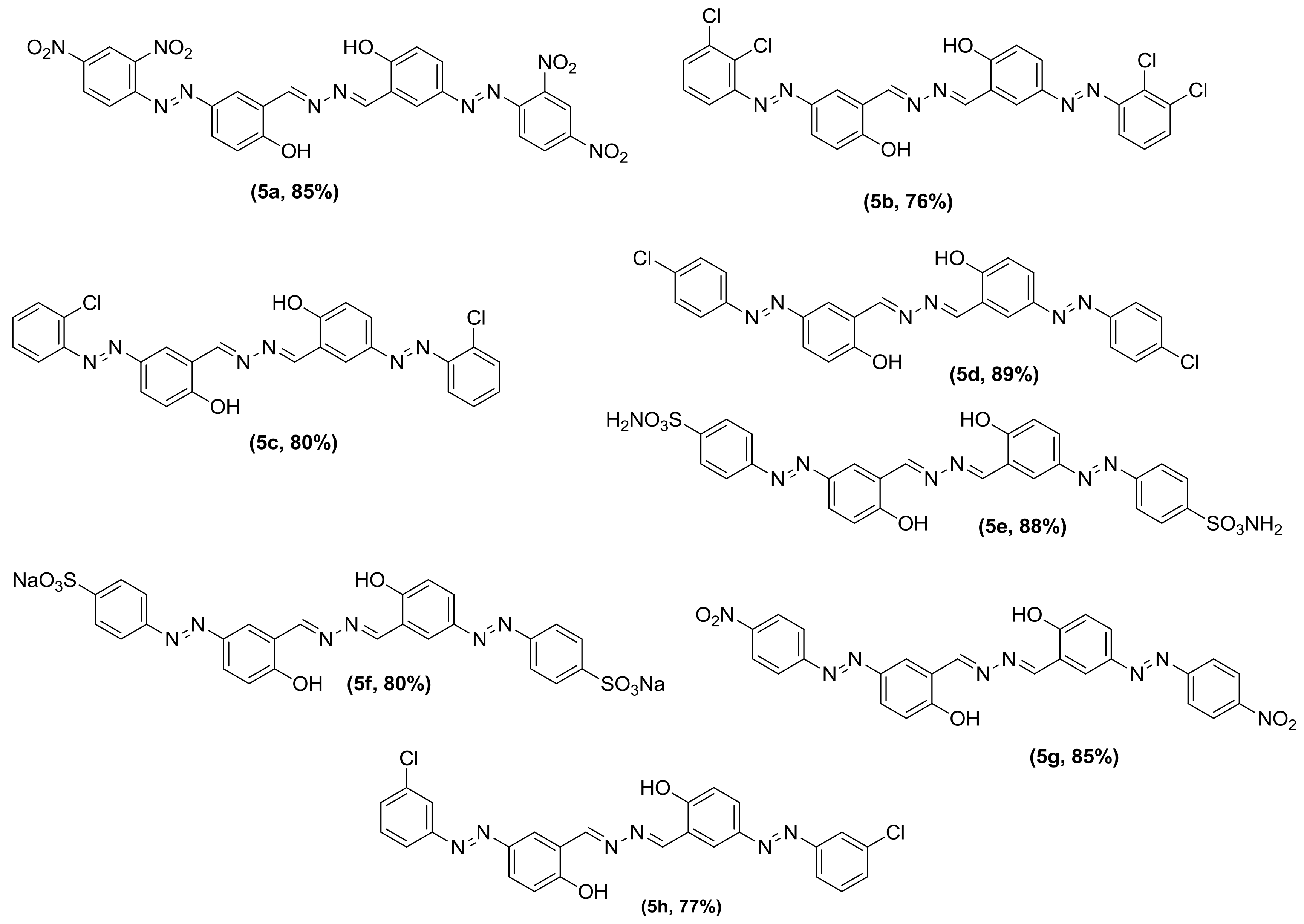
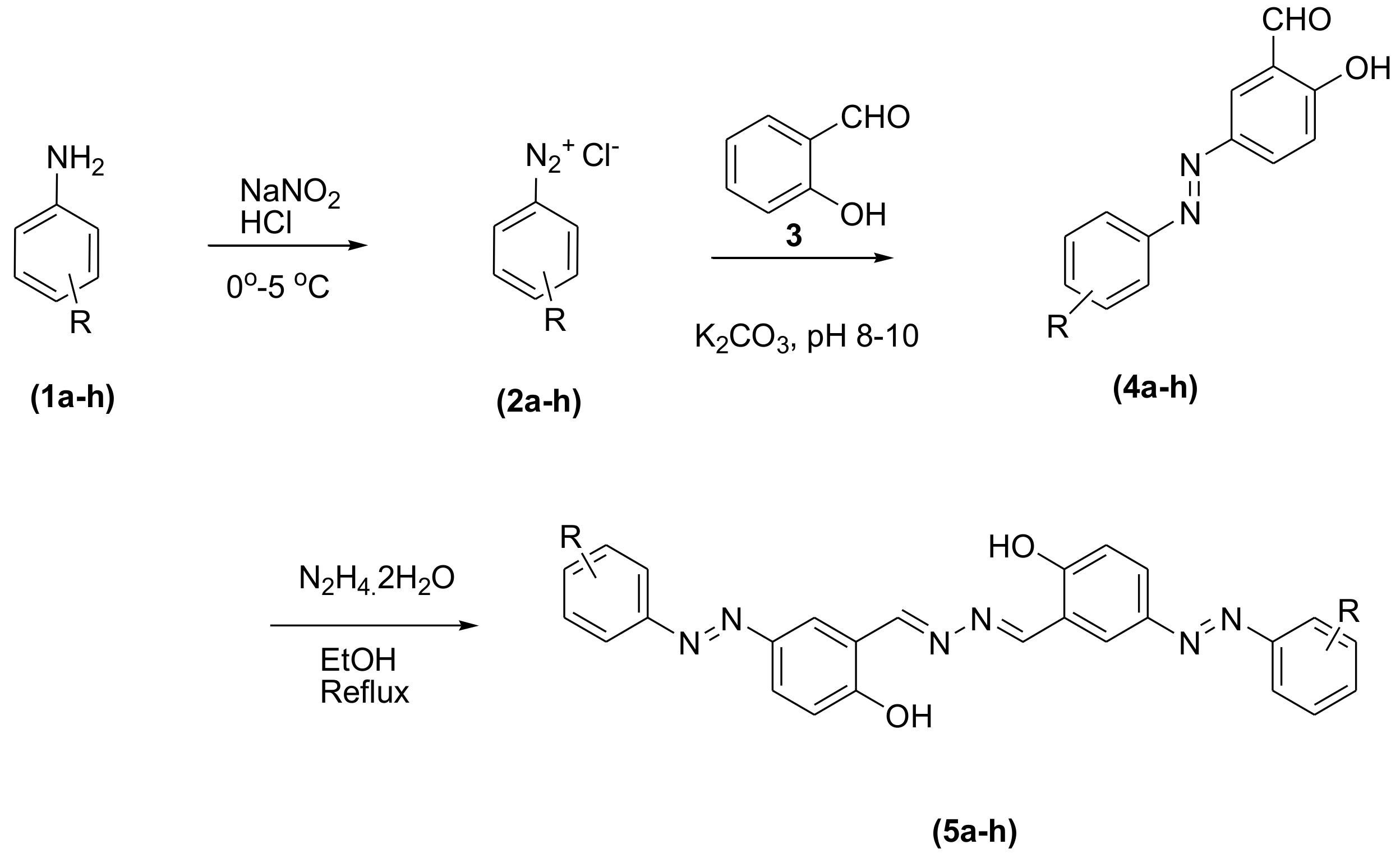
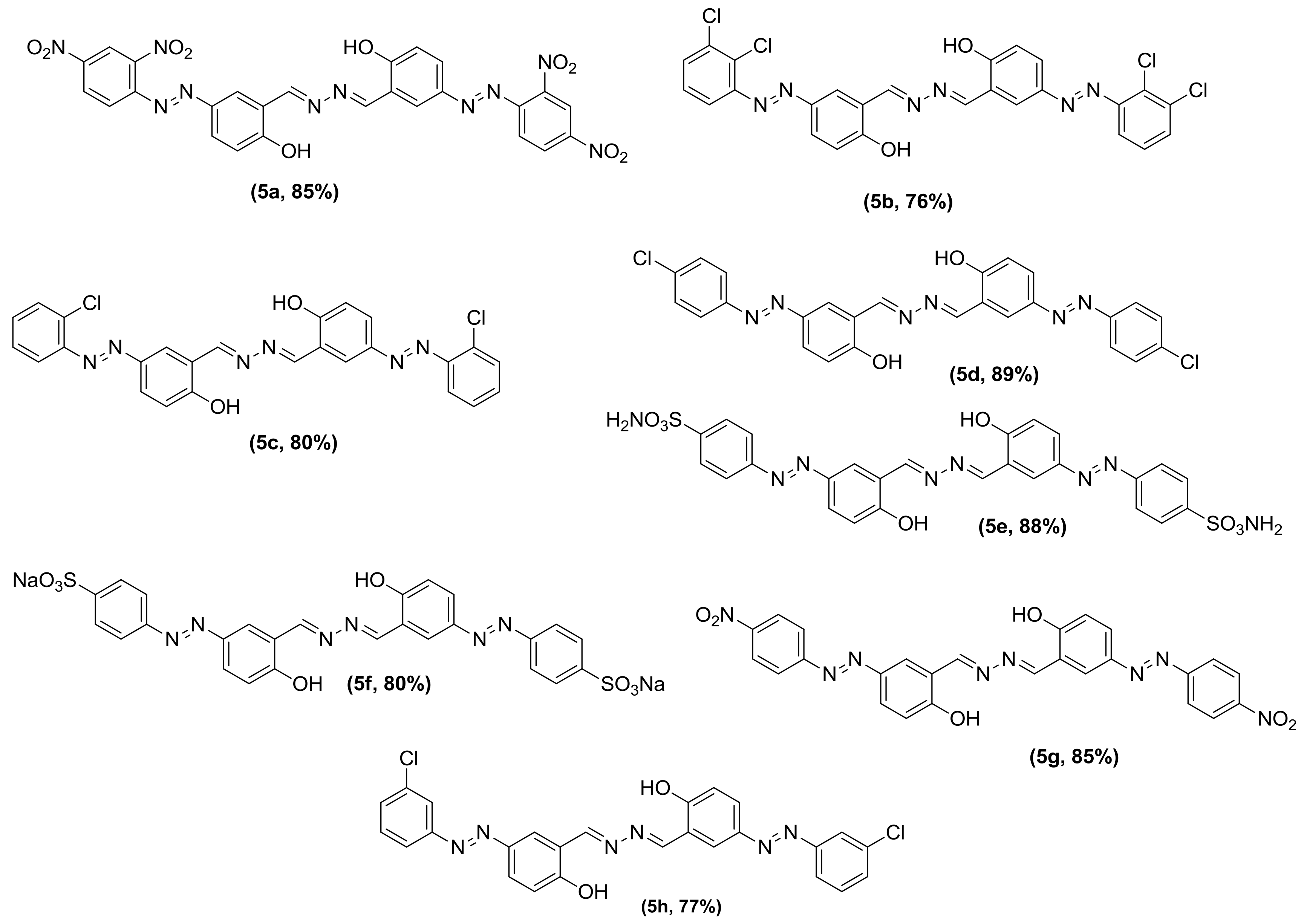
3.2. General Procedure for the Synthesis of Phenolic Azo Dyes (4a–4j) and Their Condensation with Hydrazine Dihydrate (5a–5j)
3.3. Biological Activity Methods
3.3.1. α-Glucosidase Inhibition Assay
3.3.2. α-Amylase Inhibition Assay
3.3.3. Free radical Scavenging Assay
3.3.4. Kinetic Study of α-Glucosidase
3.4. Computational Methodology
3.4.1. Selection of Target Proteins from the PDB
3.4.2. In-Silico Design of Ligand Structures
3.4.3. Ligand-Based Docking Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, G.; Chen, M.; Wang, J.; Peng, Y.; Li, L.; Xie, Z.; Deng, B.; Chen, S.; Li, W. Synthesis, biological evaluation and molecular docking studies of chromone hydrazone derivatives as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2957–2961. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.J.; Dutta, S.; Gajbhiye, R.L.; Jaisankar, P.; Sen, A.K. Synthesis, in vitro evaluation and molecular docking studies of novel amide linked triazolyl glycoconjugates as new inhibitors of α-glucosidase. Bioorg. Chem. 2017, 72, 11–20. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Xie, Z.; Chen, M.; Li, L.; Peng, Y.; Chen, S.; Li, W.; Deng, B. Discovery of 3, 3-di (indolyl) indolin-2-one as a novel scaffold for α-glucosidase inhibitors: In silico studies and SAR predictions. Bioorg. Chem. 2017, 72, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Choudhry, S.; Huma, R.; Ashraf, M.; Al-Rashida, M.; Munir, R.; Sohail, R.; Jahan, B.; Munawar, M.A.; Khan, M.A. Hetarylcoumarins: synthesis and biological evaluation as potent α-glucosidase inhibitors. Bioorg. Chem. 2017, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kasturi, S.; Surarapu, S.; Uppalanchi, S.; Anireddy, J.S.; Dwivedi, S.; Anantaraju, H.S.; Perumal, Y.; Sigalapalli, D.K.; Babu, B.N.; Ethiraj, K.S. Synthesis and α-glucosidase inhibition activity of dihydroxy pyrrolidines. Bioorg. Med. Chem. Lett. 2017, 27, 2818–2823. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Channar, P.A.; Larik, F.A.; Jabeen, F.; Muqadar, U.; Saeed, S.; Flörke, U.; Ismail, H.; Dilshad, E.; Mirza, B. Design, synthesis, molecular docking studies of organotin-drug derivatives as multi-target agents against antibacterial, antifungal, α-amylase, α-glucosidase and butyrylcholinesterase. Inorg. Chim. Acta 2017, 464, 204–213. [Google Scholar] [CrossRef]
- Muraoka, O.; Ying, S.; Yoshikai, K.; Matsuura, Y.; Yamada, E.; Minematsu, T.; Tanabe, G.; Matsuda, H.; Yoshikawa, M. Synthesis of a nitrogen analogue of salacinol and its α-glucosidase inhibitory activity. Chem. Pharma Bull. 2001, 49, 1503–1505. [Google Scholar] [CrossRef]
- Arshad, N.; Perveen, F.; Saeed, A.; Channar, P.A.; Farooqi, S.I.; Larik, F.A.; Ismail, H.; Mirza, B. Spectroscopic, molecular docking and structural activity studies of (E)-N′-(substituted benzylidene/methylene) isonicotinohydrazide derivatives for DNA binding and their biological screening. J. Mol. Struct. 2017, 1139, 371–380. [Google Scholar] [CrossRef]
- Cai, C.Y.; Rao, L.; Rao, Y.; Guo, J.X.; Xiao, Z.Z.; Cao, J.Y.; Huang, Z.S.; Wang, B. Analogues of xanthones—Chalcones and bis-chalcones as α-glucosidase inhibitors and anti-diabetes candidates. Eur. J. Med. Chem. 2017, 130, 51–59. [Google Scholar] [CrossRef]
- Chaudhry, F.; Naureen, S.; Huma, R.; Shaukat, A.; Al-Rashida, M.; Asif, N.; Ashraf, M.; Munawar, M.A.; Khan, M.A. In search of new α-glucosidase inhibitors: Imidazolylpyrazole derivatives. Bioorg. Chem. 2017, 71, 102–109. [Google Scholar] [CrossRef]
- Noreen, T.; Taha, M.; Imran, S.; Chigurupati, S.; Rahim, F.; Selvaraj, M.; Ismail, N.H.; Mohammad, J.I.; Ullah, H.; Nawaz, F.; et al. Synthesis of alpha amylase inhibitors based on privileged indole scaffold. Bioorg. Chem. 2017, 72, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Balan, K.; Perumal, P.; Sundarabaalaji, N.; Palvannan, T. Synthesis, molecular modeling and biological evaluation of novel 2-allyl amino 4-methyl sulfanyl butyric acid as α-amylase and α-glucosidase inhibitor. J. Mol. Struct. 2015, 1081, 62–68. [Google Scholar] [CrossRef]
- Saeed, A.; Bosch, A.; Bettiol, M.; Nossa González, D.L.; Erben, M.F.; Lamberti, Y. Novel Guanidine Compound against Multidrug-Resistant Cystic Fibrosis-Associated Bacterial Species. Molecules 2018, 23, 1158. [Google Scholar] [CrossRef]
- Saeed, A.; Shahzad, D.; Larik, F.A.; Channar, P.A.; Mehfooz, H.; Abbas, Q.; Hassan, M.; Raza, H.; Seo, S.Y.; Shabbir, G. Synthesis of 4-aryl-2, 6-dimethyl-3, 5-bis-N-(aryl)-carbamoyl-1, 4-dihydropyridines as novel skin protecting and anti-aging agents. Bangladesh J. Pharmacol. 2017, 12, 210–215. [Google Scholar] [CrossRef]
- Kato, E.; Iwano, N.; Yamada, A.; Kawabata, J. Synthesis and α-amylase inhibitory activity of glucose–deoxynojirimycin conjugates. Tetrahedron 2011, 67, 7692–7702. [Google Scholar] [CrossRef]
- Patel, B.D.; Bhadada, S.V.; Ghate, M.D. synthesis and anti-diabetic activity of triazolotriazine derivatives as dipeptidyl peptidase-4 (DPP-4) inhibitors. Bioorg. Chem. 2017, 72, 345–358. [Google Scholar] [CrossRef]
- Shahidpour, S.; Panahi, F.; Yousefi, R.; Nourisefat, M.; Nabipoor, M.; Ali Khalafi-Nezhad, A. Design and synthesis of new antidiabetic α-glucosidase and α-amylase inhibitors based on pyrimidine-fused heterocycles. Med. Chem. Res. 2015, 24, 3086–3096. [Google Scholar] [CrossRef]
- Aslam, M.A.S.; Mahmood, S.U.; Shahid, M.; Saeed, A.; Iqbal, J. Synthesis, biological assay in vitro and molecular docking studies of new Schiff base derivatives as potential urease inhibitors. Eur. J. Med. Chem. 2011, 46, 5473–5479. [Google Scholar] [CrossRef]
- Vigato, P.A.; Tamburini, S. The challenge of cyclic and acyclic Schiff bases and related derivatives. Coord. Chem. Rev. 2004, 248, 1717–2128. [Google Scholar] [CrossRef]
- Karthikeyan, M.S.; Prasad, D.J.; Poojary, B.; Bhat, K.S.; Holla, B.S.; Kumari, N.S. Synthesis and biological activity of Schiff and Mannich bases bearing 2, 4-dichloro-5-fluorophenyl moiety. Bioorg. Med. Chem. 2006, 14, 7482–7489. [Google Scholar] [CrossRef]
- Ren, S.; Wang, R.; Komatsu, K.; Bonaz-Krause, P.; Zyrianov, Y.; McKenna, C.E.; Csipke, C.; Tokes, Z.A.; Lien, E.J. Synthesis, biological evaluation, and quantitative structure− activity relationship analysis of new Schiff bases of hydroxysemicarbazide as potential antitumor agents. J. Med. Chem. 2002, 45, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.M.; Da Silva, D.L.; Modolo, L.V.; Alves, R.B.; De Resende, M.A.; Martins, C.V.; De Fátima, Â. Schiff bases: A short review of their antimicrobial activities. J. Adv. Res. 2011, 2, 1–8. [Google Scholar] [CrossRef]
- Vicini, P.; Geronikaki, A.; Incerti, M.; Busonera, B.; Poni, G.; Cabras, C.A.; La Colla, P. Synthesis and biological evaluation of benzo [d] isothiazole, benzothiazole and thiazole Schiff bases. Bioorg. Med. Chem. 2003, 11, 4785–4789. [Google Scholar] [CrossRef]
- Borisova, N.E.; Reshetova, M.D.; Ustynyuk, Y.A. Metal-free methods in the synthesis of macrocyclic Schiff bases. Chem. Rev. 2007, 107, 46–79. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Ge, H.M.; Tan, S.H.; Li, H.Q.; Song, Y.C.; Zhu, H.L.; Tan, R.X. Synthesis and antimicrobial activities of Schiff bases derived from 5-chloro-salicylaldehyde. Eur. J. Med. Chem. 2007, 42, 558–564. [Google Scholar] [CrossRef]
- Dinçalp, H.; Toker, F.; Durucasu, I.; Avcıbaşı, N.; Icli, S. New thiophene-based azo ligands containing azo methine group in the main chain for the determination of copper (II) ions. Dyes Pigments 2007, 75, 11–24. [Google Scholar] [CrossRef]
- Kurtoglu, G.; Avar, B.; Zengin, H.; Kose, M.; Sayin, K.; Kurtoglu, M. A novel azo-azomethine based fluorescent dye and its Co (II) and Cu (II) metal chelates. J. Mol. Liq. 2014, 200, 105–114. [Google Scholar] [CrossRef]
- Nejati, K.; Rezvani, Z.; Seyedahmadian, M. The synthesis, characterization, thermal and optical properties of copper, nickel, and vanadyl complexes derived from azo dyes. Dyes Pigments 2009, 83, 304–311. [Google Scholar] [CrossRef]
- Refat, M.S.; El-Deen, I.M.; Anwer, Z.M.; El-Ghol, S. Spectroscopic studies and biological evaluation of some transition metal complexes of Schiff-base ligands derived from 5-arylazo-salicylaldehyde and thiosemicarbazide. J. Coord. Chem. 2009, 62, 1709–1718. [Google Scholar] [CrossRef]
- Hallas, G.; Towns, A.D. Synthesis of some nitro-substituted thiophene-based azo disperse dyes. Dyes Pigments 1997, 33, 319–336. [Google Scholar] [CrossRef]
- Hallas, G.; Choi, J.H. Synthesis and spectral properties of azo dyes derived from 2-aminothiophenes and 2-aminothiazoles. Dyes Pigments 1999, 42, 249–265. [Google Scholar] [CrossRef]
- Singh, H.; Sindhu, J.; Khurana, J.M.; Sharma, C.; Aneja, K.R. Syntheses, biological evaluation and photophysical studies of novel 1, 2, 3-triazole linked azo dyes. RSC Adv. 2014, 4, 5915–5926. [Google Scholar] [CrossRef]
- Garjani, A.; Davaran, S.; Rashidi, M.; Malek, N. Protective effects of some azo derivatives of 5-aminosalicylic acid and their pegylated prodrugs on acetic acid-induced rat colitis. Daru J. Pharm. Sci. 2004, 12, 24–30. [Google Scholar]
- Ono, M.; Wada, Y.; Wu, Y.; Nemori, R.; Jinbo, Y.; Wang, H.; Lo, K.M.; Yamaguchi, N.; Brunkhorst, B.; Otomo, H.; et al. FP-21399 blocks HIV envelope protein-mediated membrane fusion and concentrates in lymph nodes. Nat. Biotechnol. 1997, 15, 343. [Google Scholar] [CrossRef] [PubMed]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Ismail, H.; Dilshad, E.; Mirza, B. New 1-octanoyl-3-aryl thiourea derivatives: Solvent-free synthesis, characterization and multi-target biological activities. Bangladesh J. Pharmacol. 2016, 11, 894–902. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.-Y.; Liou, J.-J. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.A.; Yar, M.S.; Abdel-Hamid, S.G.; Al-Qasoumi, S.I.; Samad, A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5862–5869. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Prerana, B.; Jadhav, A.; Yadav, R.; Megha, G.G. Concept of drug likeness in pharmaceutical research. Int. J. Pharm. Bio Sci. 2015, 6, 142–154. [Google Scholar]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C. Predicting selectivity and druggability in drug discovery. Annu. Rep. Comput. Chem. 2008, 4, 23–37. [Google Scholar]
- Litterman, N.K.; Lipinski, C.A.; Bunin, B.A.; Ekins, S. Computational prediction and validation of an expert’s evaluation of chemical probes. J. Chem. Inf. Model. 2014, 54, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Abbas, Q.; Ashraf, Z.; Moustafa, A.A.; Seo, S.Y. Pharmacoinformatics exploration of polyphenol oxidases leading to novel inhibitors by virtual screening and molecular dynamic simulation study. Comput. Biol. Chem. 2017, 68, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Rangan, L. Molecular docking and inhibition studies of α-amylase activity by labdane diterpenes from Alpinia nigra seeds. Med. Chem. Res. 2014, 23, 4836–4852. [Google Scholar] [CrossRef]
- Abbas, Q.; Hassan, M.; Raza, H.; Kim, S.J.; Chung, K.W.; Kim, G.H.; Seo, S.Y. In vitro, in vivo and in silico anti-hyperglycemic inhibition by sinigrin. Asian Pac. J. Trop. Med. 2017, 10, 372–379. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | α-Glucosidase (µM) | α-Amylase (µM) |
|---|---|---|
| 5a | 0.547 ± 0.0289 | 8.541 ± 0.653 |
| 5b | 5.345 ± 0.2826 | 28.373 ± 2.171 |
| 5c | 8.061 ± 0.4263 | 9.5482 ± 0.730 |
| 5d | 19.521 ± 1.0321 | 9.7183 ± 0.863 |
| 5e | 0.367 ± 0.01941 | 28.4828 ± 2.081 |
| 5f | 1.841 ± 0.09738 | 4.2861 ± 0.328 |
| 5g | 0.359 ± 0.0189 | 0.4379 ± 0.042 |
| 5h | 0.400 ± 0.0211 | 0.5902 ± 0.012 |
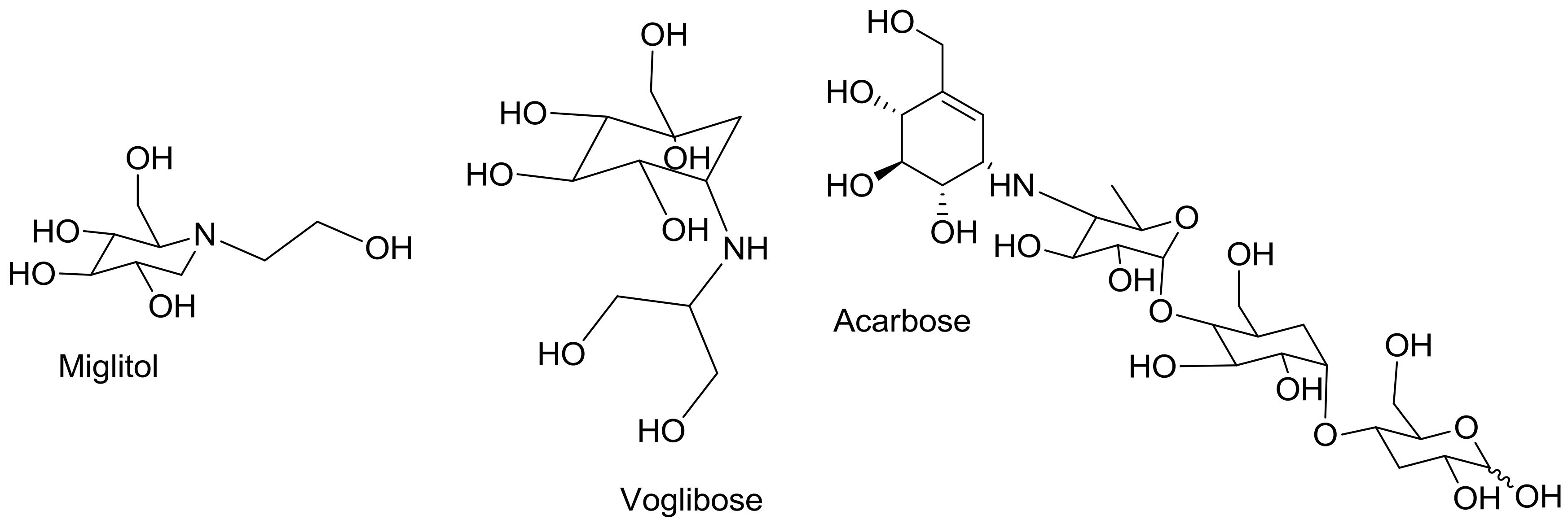
| Acarbose | 6.109 ± 0.329 | 33.178 ± 2.392 |
| Properties | 5a | 5b | 5c | 5d | 5e | 5f | 5g | 5h |
|---|---|---|---|---|---|---|---|---|
| Mol. Weight (g/mol) | 632.14 | 584.01 | 516.09 | 516.09 | 638.10 | 652.04 | 540.15 | 516.09 |
| No. HBA | 16 | 8 | 8 | 8 | 16 | 14 | 12 | 8 |
| No. HBD | 6 | 2 | 2 | 2 | 6 | 2 | 4 | 2 |
| Mol. LogP | 6.77 | 11.01 | 9.82 | 10.06 | 4.42 | 6.26 | 7.82 | 10.06 |
| No of SC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mol. Vol (A3) | 536.68 | 477.50 | 446.16 | 448.00 | 510.16 | 515.19 | 474.01 | 448.15 |
| Molar Refractivity | 154.84 | 150.61 | 141.40 | 141.40 | 158.32 | N/A | 143.52 | 141.40 |
| Density | 1.65 | 1.47 | 1.36 | 1.36 | 1.57 | N/A | 1.45 | 1.36 |
| Polarizability | 61.38 | 59.70 | 56.05 | 56.05 | 62.76 | N/A | 56.89 | 56.05 |
| Drug Likeness Score | −1.49 | −0.51 | −0.67 | −0.54 | −1.22 | −1.33 | −1.16 | −0.73 |
| Ligands | cLogP | LE | LLE | LELP | Mutagenic | Tumorigenic | Irritant |
|---|---|---|---|---|---|---|---|
| 5a | 4.8122 | 0.24806 | 3.5055 | 19.399 | high | high | none |
| 5b | 10.923 | 0.28743 | −2.960 | 38.001 | high | high | none |
| 5c | 9.7106 | 0.30535 | −1.697 | 31.802 | high | high | none |
| 5d | 9.7106 | 0.30535 | −1.697 | 31.802 | high | high | none |
| 5e | 7.027 | 0.25421 | 1.1262 | 27.642 | high | high | high |
| 5f | 1.5448 | 0.28781 | 7.2633 | 5.3675 | high | high | none |
| 5g | 6.6554 | 0.28044 | 1.5214 | 23.732 | high | high | none |
| 5h | 9.7106 | 0.30535 | −1.697 | 31.802 | high | high | None |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahzad, D.; Saeed, A.; Larik, F.A.; Channar, P.A.; Abbas, Q.; Alajmi, M.F.; Arshad, M.I.; Erben, M.F.; Hassan, M.; Raza, H.; et al. Novel C-2 Symmetric Molecules as α-Glucosidase and α-Amylase Inhibitors: Design, Synthesis, Kinetic Evaluation, Molecular Docking and Pharmacokinetics. Molecules 2019, 24, 1511. https://doi.org/10.3390/molecules24081511
Shahzad D, Saeed A, Larik FA, Channar PA, Abbas Q, Alajmi MF, Arshad MI, Erben MF, Hassan M, Raza H, et al. Novel C-2 Symmetric Molecules as α-Glucosidase and α-Amylase Inhibitors: Design, Synthesis, Kinetic Evaluation, Molecular Docking and Pharmacokinetics. Molecules. 2019; 24(8):1511. https://doi.org/10.3390/molecules24081511
Chicago/Turabian StyleShahzad, Danish, Aamer Saeed, Fayaz Ali Larik, Pervaiz Ali Channar, Qamar Abbas, Mohamed F. Alajmi, M. Ifzan Arshad, Mauricio F. Erben, Mubashir Hassan, Hussain Raza, and et al. 2019. "Novel C-2 Symmetric Molecules as α-Glucosidase and α-Amylase Inhibitors: Design, Synthesis, Kinetic Evaluation, Molecular Docking and Pharmacokinetics" Molecules 24, no. 8: 1511. https://doi.org/10.3390/molecules24081511
APA StyleShahzad, D., Saeed, A., Larik, F. A., Channar, P. A., Abbas, Q., Alajmi, M. F., Arshad, M. I., Erben, M. F., Hassan, M., Raza, H., Seo, S.-Y., & El-Seedi, H. R. (2019). Novel C-2 Symmetric Molecules as α-Glucosidase and α-Amylase Inhibitors: Design, Synthesis, Kinetic Evaluation, Molecular Docking and Pharmacokinetics. Molecules, 24(8), 1511. https://doi.org/10.3390/molecules24081511