Using Self-Assembling Peptides to Integrate Biomolecules into Functional Supramolecular Biomaterials


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Peptide Assemblies as Scaffolds for Receptor-Binding Ligands
3. Moving Beyond Peptides as the Functional Ligand
3.1. Glycosylated Nanomaterials Fabricated from Carbohydrate-Modified Self-Assembling Peptides
3.2. Incorporating Folded Proteins into Supramolecular Nanomaterials
4. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Grzelczak, M.; Vermant, J.; Furst, E.M.; Liz-Marzan, L.M. Directed self-assembly of nanoparticles. ACS Nano 2010, 4, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Bong, D.T.; Clark, T.D.; Granja, J.R.; Ghadiri, M.R. Self-assembling organic nanotubes. Angew. Chem. Int. Ed. 2001, 40, 988–1011. [Google Scholar] [CrossRef]
- Kunitake, T. Synthetic Bilayer Membranes: Molecular Design, Self-Organization, and Application. Angew. Chem. Int. Ed. 1992, 31, 709–726. [Google Scholar] [CrossRef]
- Gazit, E. Self-assembled peptide nanostructures: The design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 2007, 36, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Seroski, D.T.; Hudalla, G.A. Self-Assembled Peptide and Protein Nanofibers for Biomedical Applications. In Biomedical Applications of Functionalized Nanomaterials; Elsevier: Amsterdam, The Netherlands, 2018; pp. 569–598. [Google Scholar]
- Webber, M.J.; Appel, E.A.; Meijer, E.; Langer, R. Supramolecular biomaterials. Nat. Mater. 2016, 15, 13–26. [Google Scholar] [CrossRef]
- Stupp, S.I.; Zha, R.H.; Palmer, L.C.; Cui, H.; Bitton, R. Self-assembly of biomolecular soft matter. Faraday Discuss. 2013, 166, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.C.; Velichko, Y.S.; Olvera de La Cruz, M.; Stupp, S.I. Supramolecular self-assembly codes for functional structures. Philos. Trans. R. Soc. A 2007, 365, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.A.; Czeisler, C.; Niece, K.L.; Beniash, E.; Harrington, D.A.; Kessler, J.A.; Stupp, S.I. Selective differentiation of neural progenitor cells by high-epitope density nanofibers. Science 2004, 303, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; Shelby, S.A.; Choi, P.H.; Marciel, A.B.; Chen, R.; Tan, L.; Chu, T.K.; Mesch, R.A.; Lee, B.-C.; Connolly, M.D. Free-floating ultrathin two-dimensional crystals from sequence-specific peptoid polymers. Nat. Mater. 2010, 9, 454–460. [Google Scholar] [CrossRef]
- Leclere, P.; Surin, M.; Viville, P.; Lazzaroni, R.; Kilbinger, A.; Henze, O.; Feast, W.; Cavallini, M.; Biscarini, F.; Schenning, A. About oligothiophene self-assembly: From aggregation in solution to solid-state nanostructures. Chem. Mater. 2004, 16, 4452–4466. [Google Scholar] [CrossRef]
- Figg, C.A.; Simula, A.; Gebre, K.A.; Tucker, B.S.; Haddleton, D.M.; Sumerlin, B.S. Polymerization-induced thermal self-assembly (PITSA). Chem. Sci. 2015, 6, 1230–1236. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Thermo-sensitive polymeric micelles based on poly (N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, Z.L.; Shen, Y.; Radosz, M. Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog. Polym. Sci. 2010, 35, 1128–1143. [Google Scholar] [CrossRef]
- Jakab, K.; Norotte, C.; Marga, F.; Murphy, K.; Vunjak-Novakovic, G.; Forgacs, G. Tissue engineering by self-assembly and bio-printing of living cells. Biofabrication 2010, 2, 022001. [Google Scholar] [CrossRef] [PubMed]
- Yemini, M.; Reches, M.; Rishpon, J.; Gazit, E. Novel electrochemical biosensing platform using self-assembled peptide nanotubes. Nano Lett. 2005, 5, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Pochan, D.J.; Ozbas, B.; Rajagopal, K.; Pakstis, L.; Kretsinger, J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [Google Scholar] [CrossRef]
- Parry, A.L.; Clemson, N.A.; Ellis, J.; Bernhard, S.S.; Davis, B.G.; Cameron, N.R. ‘Multicopy multivalent’ glycopolymer-stabilized gold nanoparticles as potential synthetic cancer vaccines. J. Am. Chem. Soc. 2013, 135, 9362–9365. [Google Scholar] [CrossRef]
- Habibi, N.; Kamaly, N.; Memic, A.; Shafiee, H. Self-assembled peptide-based nanostructures: Smart nanomaterials toward targeted drug delivery. Nano Today 2016, 11, 41–60. [Google Scholar] [CrossRef]
- Mandal, D.; Shirazi, A.N.; Parang, K. Self-assembly of peptides to nanostructures. Org. Biomol. Chem. 2014, 12, 3544–3561. [Google Scholar] [CrossRef]
- Saiani, A.; Mohammed, A.; Frielinghaus, H.; Collins, R.; Hodson, N.; Kielty, C.; Sherratt, M.; Miller, A. Self-assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter 2009, 5, 193–202. [Google Scholar] [CrossRef]
- Bowerman, C.J.; Liyanage, W.; Federation, A.J.; Nilsson, B.L. Tuning β-sheet peptide self-assembly and hydrogelation behavior by modification of sequence hydrophobicity and aromaticity. Biomacromolecules 2011, 12, 2735–2745. [Google Scholar] [CrossRef]
- Klok, H.A. Protein-inspired materials: Synthetic concepts and potential applications. Angew. Chem. Int. Ed. 2002, 41, 1509–1513. [Google Scholar] [CrossRef]
- Ramakers, B.; Van Hest, J.; Löwik, D. Molecular tools for the construction of peptide-based materials. Chem. Soc. Rev. 2014, 43, 2743–2756. [Google Scholar] [CrossRef]
- Sun, L.; Zheng, C.; Webster, T.J. Self-assembled peptide nanomaterials for biomedical applications: Promises and pitfalls. Int. J. Nanomed. 2017, 12, 73–86. [Google Scholar] [CrossRef]
- De Santis, E.; Ryadnov, M.G. Peptide self-assembly for nanomaterials: The old new kid on the block. Chem. Soc. Rev. 2015, 44, 8288–8300. [Google Scholar] [CrossRef]
- Raymond, D.M.; Nilsson, B.L. Multicomponent peptide assemblies. Chem. Soc. Rev. 2018, 47, 3659–3720. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Mándity, I.M.; Monsignori, A.; Fülöp, L.; Forró, E.; Fülöp, F. Exploiting Aromatic Interactions for β-Peptide Foldamer Helix Stabilization: A Significant Design Element. Chem. Eur. J. 2014, 20, 4591–4597. [Google Scholar] [CrossRef]
- Martinek, T.A.; Hetényi, A.; Fülöp, L.; Mándity, I.M.; Tóth, G.K.; Dékány, I.; Fülöp, F. Secondary Structure Dependent Self-Assembly of β-Peptides into Nanosized Fibrils and Membranes. Angew. Chem. Int. Ed. 2006, 45, 2396–2400. [Google Scholar] [CrossRef]
- Inaba, H.; Matsuura, K. Peptide Nanomaterials Designed from Natural Supramolecular Systems. Chem. Rec. 2018. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Controlled patterning of aligned self-assembled peptide nanotubes. Nat. Nanotechnol. 2006, 1, 195. [Google Scholar] [CrossRef]
- Mahler, A.; Reches, M.; Rechter, M.; Cohen, S.; Gazit, E. Rigid, Self-Assembled Hydrogel Composed of a Modified Aromatic Dipeptide. Adv. Mater. 2006, 18, 1365–1370. [Google Scholar] [CrossRef]
- Kol, N.; Adler-Abramovich, L.; Barlam, D.; Shneck, R.Z.; Gazit, E.; Rousso, I. Self-Assembled Peptide Nanotubes Are Uniquely Rigid Bioinspired Supramolecular Structures. Nano Lett. 2005, 5, 1343–1346. [Google Scholar] [CrossRef]
- Matsuura, K. Synthetic approaches to construct viral capsid-like spherical nanomaterials. Chem. Commun. 2018, 54, 8944–8959. [Google Scholar] [CrossRef]
- Yu, Z.; Cai, Z.; Chen, Q.; Liu, M.; Ye, L.; Ren, J.; Liao, W.; Liu, S. Engineering β-sheet peptide assemblies for biomedical applications. Biomater. Sci. 2016, 4, 365–374. [Google Scholar] [CrossRef]
- Loo, Y.; Zhang, S.; Hauser, C.A. From short peptides to nanofibers to macromolecular assemblies in biomedicine. Biotechnol. Adv. 2012, 30, 593–603. [Google Scholar] [CrossRef]
- Valéry, C.; Artzner, F.; Paternostre, M. Peptide nanotubes: Molecular organisations, self-assembly mechanisms and applications. Soft Matter 2011, 7, 9583–9594. [Google Scholar] [CrossRef]
- Wu, E.C.; Zhang, S.; Hauser, C.A. Self-assembling peptides as cell-interactive scaffolds. Adv. Funct. Mater. 2012, 22, 456–468. [Google Scholar] [CrossRef]
- Rad-Malekshahi, M.; Lempsink, L.; Amidi, M.; Hennink, W.E.; Mastrobattista, E. Biomedical applications of self-assembling peptides. Bioconjugate Chem. 2015, 27, 3–18. [Google Scholar] [CrossRef]
- Eskandari, S.; Guerin, T.; Toth, I.; Stephenson, R.J. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Adv. Drug Deliv. Rev. 2017, 110, 169–187. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Self-assembling peptide nanofiber hydrogels in tissue engineering and regenerative medicine: Progress, design guidelines, and applications. J. Biomed. Mater. Res. A 2016, 104, 1002–1016. [Google Scholar] [CrossRef]
- Wu, Y.; Collier, J.H. α-Helical coiled-coil peptide materials for biomedical applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1424. [Google Scholar] [CrossRef]
- M Leite, D.; Barbu, E.; Pilkington, G.J.; Lalatsa, A. Peptide self-assemblies for drug delivery. Curr. Top. Med. Chem. 2015, 15, 2277–2289. [Google Scholar] [CrossRef][Green Version]
- Fukunaga, K.; Tsutsumi, H.; Mihara, H. Self-assembling peptides as building blocks of functional materials for biomedical applications. Bull. Chem. Soc. Jpn. 2018, 92, 391–399. [Google Scholar] [CrossRef]
- Yang, F.; Williams, C.G.; Wang, D.-a.; Lee, H.; Manson, P.N.; Elisseeff, J. The effect of incorporating RGD adhesive peptide in polyethylene glycol diacrylate hydrogel on osteogenesis of bone marrow stromal cells. Biomaterials 2005, 26, 5991–5998. [Google Scholar] [CrossRef]
- Sakiyama-Elbert, S.E. Incorporation of heparin into biomaterials. Acta Biomater. 2014, 10, 1581–1587. [Google Scholar] [CrossRef]
- Perlin, L.; MacNeil, S.; Rimmer, S. Production and performance of biomaterials containing RGD peptides. Soft Matter 2008, 4, 2331–2349. [Google Scholar] [CrossRef]
- Kim, T.G.; Park, T.G. Biomimicking extracellular matrix: Cell adhesive RGD peptide modified electrospun poly (D, L-lactic-co-glycolic acid) nanofiber mesh. Tissue Eng. 2006, 12, 221–233. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef]
- Sever, M.; Mammadov, B.; Guler, M.O.; Tekinay, A.B. Tenascin-C mimetic Peptide nanofibers direct stem cell differentiation to osteogenic lineage. Biomacromolecules 2014, 15, 4480–4487. [Google Scholar] [CrossRef]
- Holmes, T.C.; de Lacalle, S.; Su, X.; Liu, G.; Rich, A.; Zhang, S. Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc. Natl. Acad. Sci. USA 2000, 97, 6728–6733. [Google Scholar] [CrossRef]
- Kisiday, J.; Jin, M.; Kurz, B.; Hung, H.; Semino, C.; Zhang, S.; Grodzinsky, A. Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: Implications for cartilage tissue repair. Proc. Natl. Acad. Sci. USA 2002, 99, 9996–10001. [Google Scholar] [CrossRef]
- Jung, J.P.; Nagaraj, A.K.; Fox, E.K.; Rudra, J.S.; Devgun, J.M.; Collier, J.H. Co-assembling peptides as defined matrices for endothelial cells. Biomaterials 2009, 30, 2400–2410. [Google Scholar] [CrossRef]
- Horii, A.; Wang, X.; Gelain, F.; Zhang, S. Biological designer self-assembling peptide nanofiber scaffolds significantly enhance osteoblast proliferation, differentiation and 3-D migration. PLoS ONE 2007, 2, e190. [Google Scholar] [CrossRef]
- Gelain, F.; Unsworth, L.D.; Zhang, S. Slow and sustained release of active cytokines from self-assembling peptide scaffolds. J. Control. Release 2010, 145, 231–239. [Google Scholar] [CrossRef]
- Gribbon, C.; Channon, K.J.; Zhang, W.; Banwell, E.F.; Bromley, E.H.; Chaudhuri, J.B.; Oreffo, R.O.; Woolfson, D.N. MagicWand: A single, designed peptide that assembles to stable, ordered α-helical fibers. Biochemistry 2008, 47, 10365–10371. [Google Scholar] [CrossRef]
- Haines, L.A.; Rajagopal, K.; Ozbas, B.; Salick, D.A.; Pochan, D.J.; Schneider, J.P. Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2005, 127, 17025–17029. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Choo, J.-E.; Choi, Y.-S.; Suh, J.-S.; Lee, S.-J.; Chung, C.-P.; Park, Y.-J. Osteoblastic differentiation of human bone marrow stromal cells in self-assembled BMP-2 receptor-binding peptide-amphiphiles. Biomaterials 2009, 30, 3532–3541. [Google Scholar] [CrossRef]
- Li, I.-C.; Moore, A.N.; Hartgerink, J.D. “Missing tooth” multidomain peptide nanofibers for delivery of small molecule drugs. Biomacromolecules 2016, 17, 2087–2095. [Google Scholar] [CrossRef]
- Loo, Y.; Lakshmanan, A.; Ni, M.; Toh, L.L.; Wang, S.; Hauser, C.A. Peptide bioink: Self-assembling nanofibrous scaffolds for three-dimensional organotypic cultures. Nano Lett. 2015, 15, 6919–6925. [Google Scholar] [CrossRef]
- O’leary, L.E.; Fallas, J.A.; Bakota, E.L.; Kang, M.K.; Hartgerink, J.D. Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat. Chem. 2011, 3, 821. [Google Scholar] [CrossRef]
- Veiga, A.S.; Sinthuvanich, C.; Gaspar, D.; Franquelim, H.G.; Castanho, M.A.; Schneider, J.P. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials 2012, 33, 8907–8916. [Google Scholar] [CrossRef]
- Zheng, Y.; Wen, Y.; George, A.M.; Steinbach, A.M.; Phillips, B.E.; Giannoukakis, N.; Gawalt, E.S.; Meng, W.S. A peptide-based material platform for displaying antibodies to engage T cells. Biomaterials 2011, 32, 249–257. [Google Scholar] [CrossRef]
- Bian, L.; Guvendiren, M.; Mauck, R.L.; Burdick, J.A. Hydrogels that mimic developmentally relevant matrix and N-cadherin interactions enhance MSC chondrogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 10117–10122. [Google Scholar] [CrossRef]
- Li, R.; Xu, J.; Wong, D.S.H.; Li, J.; Zhao, P.; Bian, L. Self-assembled N-cadherin mimetic peptide hydrogels promote the chondrogenesis of mesenchymal stem cells through inhibition of canonical Wnt/β-catenin signaling. Biomaterials 2017, 145, 33–43. [Google Scholar] [CrossRef]
- Edelbrock, A.N.; Àlvarez, Z.; Simkin, D.; Fyrner, T.; Chin, S.M.; Sato, K.; Kiskinis, E.; Stupp, S.I. Supramolecular Nanostructure Activates TrkB Receptor Signaling of Neuronal Cells by Mimicking Brain-Derived Neurotrophic Factor. Nano Lett. 2018, 18, 6237–6247. [Google Scholar] [CrossRef]
- Qi, G.B.; Gao, Y.J.; Wang, L.; Wang, H. Self-Assembled Peptide-Based Nanomaterials for Biomedical Imaging and Therapy. Adv. Mater. 2018, 30, 1703444. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, X.; Li, Y.; Su, Z.; Jandt, K.D.; Wei, G. Protein-mimetic peptide nanofibers: Motif design, self-assembly synthesis, and sequence-specific biomedical applications. Prog. Polym. Sci. 2018, 80, 94–124. [Google Scholar] [CrossRef]
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Pept. Sci. Orig. Res. Biomol. 2010, 94, 1–18. [Google Scholar] [CrossRef]
- Arslan, E.; Garip, I.C.; Gulseren, G.; Tekinay, A.B.; Guler, M.O. Bioactive supramolecular peptide nanofibers for regenerative medicine. Adv. Healthc. Mater. 2014, 3, 1357–1376. [Google Scholar] [CrossRef]
- Weis, W.I.; Drickamer, K. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 1996, 65, 441–473. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Yamaguchi, Y. Proteoglycans as modulators of growth factor activities. Cell 1991, 64, 867–869. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef]
- Moczar, M.; Moczar, E.; Robert, L. Composition of glycopeptides obtained by proteolytic digestion of the media of porcine aorta. Atherosclerosis 1970, 12, 31–40. [Google Scholar] [CrossRef]
- Margolis, R.K.; Margolis, R.U. Sulfated glycopeptides from rat brain glycoproteins. Biochemistry 1970, 9, 4389–4396. [Google Scholar] [CrossRef]
- Matsuura, K. Construction of functional biomaterials by biomolecular self-assembly. Bull. Chem. Soc. Jpn. 2017, 90, 873–884. [Google Scholar] [CrossRef]
- Ladmiral, V.; Melia, E.; Haddleton, D.M. Synthetic glycopolymers: An overview. Eur. Polym. J. 2004, 40, 431–449. [Google Scholar] [CrossRef]
- Spain, S.G.; Cameron, N.R. A spoonful of sugar: The application of glycopolymers in therapeutics. Polym. Chem. 2011, 2, 60–68. [Google Scholar] [CrossRef]
- Yilmaz, G.; Becer, C.R. Precision glycopolymers and their interactions with lectins. Eur. Polym. J. 2013, 49, 3046–3051. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. Methods for anomeric carbon-linked and fused sugar amino acid synthesis: The gateway to artificial glycopeptides. Chem. Rev. 2000, 100, 4395–4422. [Google Scholar] [CrossRef]
- Bojarová, P.; Křen, V. Sugared biomaterial binding lectins: Achievements and perspectives. Biomater. Sci. 2016, 4, 1142–1160. [Google Scholar] [CrossRef]
- Payne, R.J.; Wong, C.-H. Advances in chemical ligation strategies for the synthesis of glycopeptides and glycoproteins. Chem. Commun. 2010, 46, 21–43. [Google Scholar] [CrossRef]
- Gunay, G.; Sardan Ekiz, M.; Ferhati, X.; Richichi, B.; Nativi, C.; Tekinay, A.B.; Guler, M.O. Antigenic GM3 Lactone Mimetic Molecule Integrated Mannosylated Glycopeptide Nanofibers for the Activation and Maturation of Dendritic Cells. ACS Appl. Mater. Interfaces 2017, 9, 16035–16042. [Google Scholar] [CrossRef]
- Ustun Yaylaci, S.; Sardan Ekiz, M.; Arslan, E.; Can, N.; Kilic, E.; Ozkan, H.; Orujalipoor, I.; Ide, S.; Tekinay, A.B.; Guler, M.O. Supramolecular GAG-like self-assembled glycopeptide nanofibers induce chondrogenesis and cartilage regeneration. Biomacromolecules 2016, 17, 679–689. [Google Scholar] [CrossRef]
- Caliskan, O.S.; Sardan Ekiz, M.; Tekinay, A.B.; Guler, M.O. Spatial Organization of Functional Groups on Bioactive Supramolecular Glycopeptide Nanofibers for Differentiation of Mesenchymal Stem Cells (MSCs) to Brown Adipogenesis. Bioconjugate Chem. 2016, 28, 740–750. [Google Scholar] [CrossRef]
- Zick, Y.; Eisenstein, M.; Goren, R.A.; Hadari, Y.R.; Levy, Y.; Ronen, D. Role of galectin-8 as a modulator of cell adhesion and cell growth. Glycoconj. J. 2002, 19, 517–526. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Toscano, M.A. Turning ‘sweet’ on immunity: Galectin–glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 2009, 9, 338. [Google Scholar] [CrossRef]
- Tribulatti, M.V.; Figini, M.G.; Carabelli, J.; Cattaneo, V.; Campetella, O. Redundant and antagonistic functions of galectin-1,-3, and-8 in the elicitation of T cell responses. J. Immunol. 2012, 188, 2991–2999. [Google Scholar] [CrossRef]
- Vasta, G.R. Roles of galectins in infection. Nat. Rev. Microbiol. 2009, 7, 424–438. [Google Scholar] [CrossRef]
- Restuccia, A.; Tian, Y.F.; Collier, J.H.; Hudalla, G.A. Self-assembled glycopeptide nanofibers as modulators of galectin-1 bioactivity. Cell. Mol. Bioeng. 2015, 8, 471–487. [Google Scholar] [CrossRef]
- Restuccia, A.; Fettis, M.M.; Farhadi, S.A.; Molinaro, M.D.; Kane, B.; Hudalla, G.A. Evaluation of Self-Assembled Glycopeptide Nanofibers Modified with N, N′-Diacetyllactosamine for Selective Galectin-3 Recognition and Inhibition. ACS Biomater. Sci. Eng. 2018, 4, 3451–3459. [Google Scholar] [CrossRef]
- Restuccia, A.; Hudalla, G.A. Tuning carbohydrate density enhances protein binding and inhibition by glycosylated β-sheet peptide nanofibers. Biomater. Sci. 2018, 6, 2327–2335. [Google Scholar] [CrossRef]
- Cairo, C.W.; Gestwicki, J.E.; Kanai, M.; Kiessling, L.L. Control of multivalent interactions by binding epitope density. J. Am. Chem. Soc. 2002, 124, 1615–1619. [Google Scholar] [CrossRef]
- Reynolds, M.; Marradi, M.; Imberty, A.; Penadés, S.; Pérez, S. Influence of ligand presentation density on the molecular recognition of mannose-functionalised glyconanoparticles by bacterial lectin BC2L-A. Glycoconj. J. 2013, 30, 747–757. [Google Scholar] [CrossRef]
- Ladmiral, V.; Mantovani, G.; Clarkson, G.J.; Cauet, S.; Irwin, J.L.; Haddleton, D.M. Synthesis of neoglycopolymers by a combination of “click chemistry” and living radical polymerization. J. Am. Chem. Soc. 2006, 128, 4823–4830. [Google Scholar] [CrossRef]
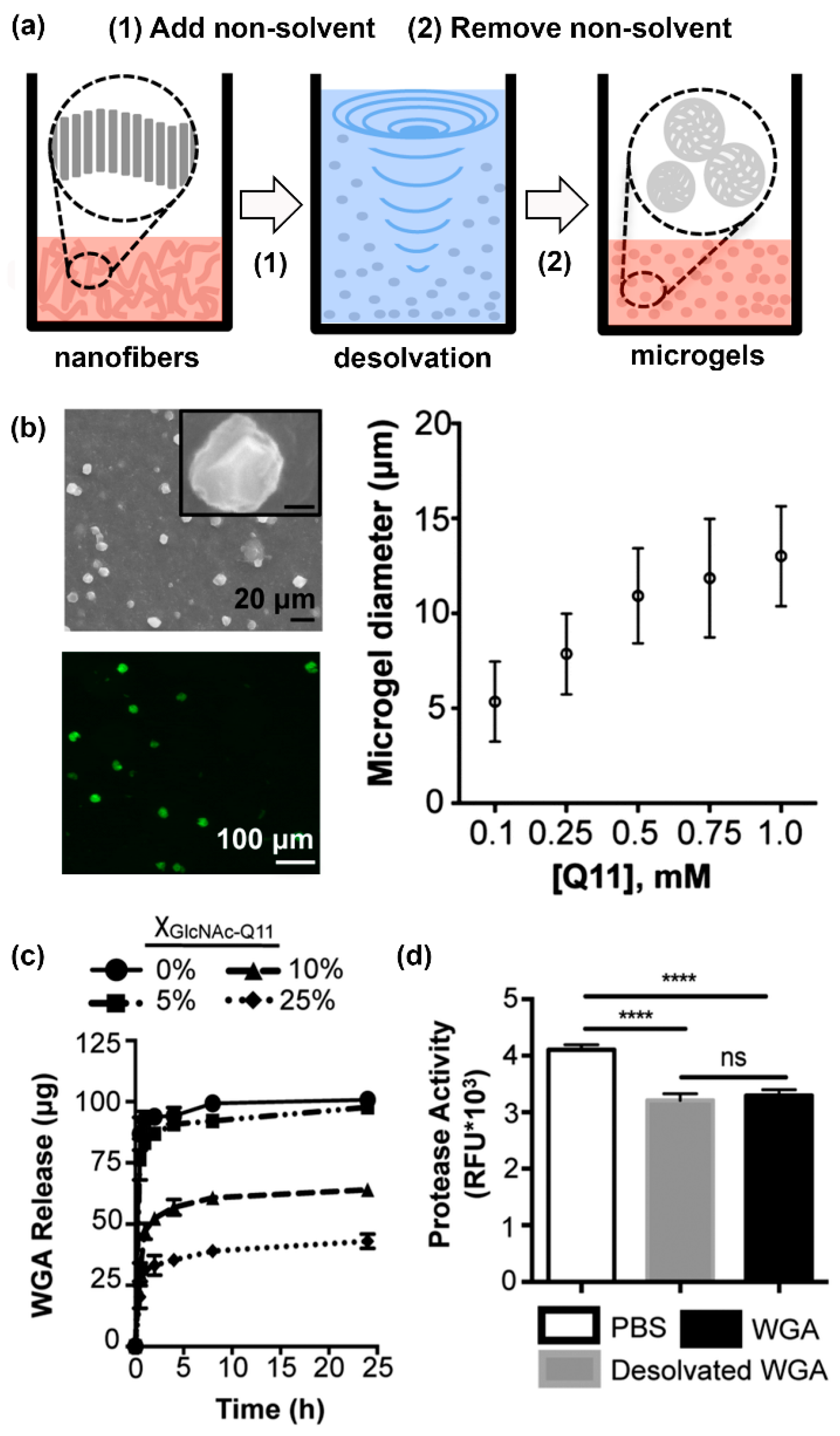
- Fettis, M.M.; Wei, Y.; Restuccia, A.; Kurian, J.J.; Wallet, S.M.; Hudalla, G.A. Microgels with tunable affinity-controlled protein release via desolvation of self-assembled peptide nanofibers. J. Mater. Chem. B 2016, 4, 3054–3064. [Google Scholar] [CrossRef]
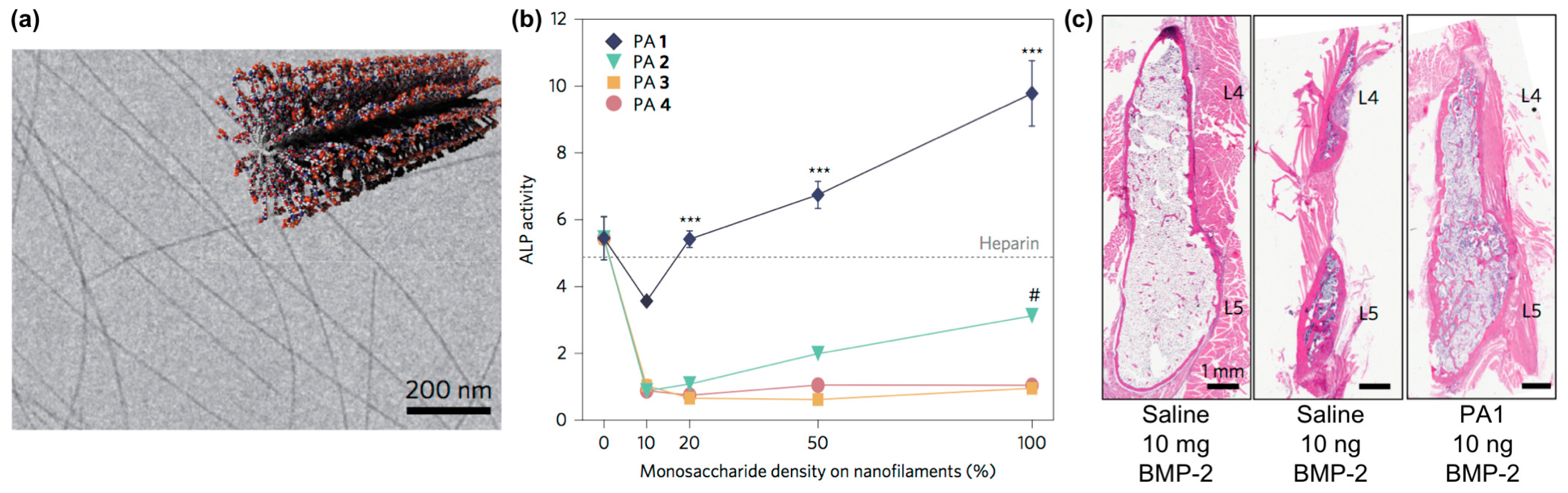
- Lee, S.S.; Fyrner, T.; Chen, F.; Álvarez, Z.; Sleep, E.; Chun, D.S.; Weiner, J.A.; Cook, R.W.; Freshman, R.D.; Schallmo, M.S. Sulfated glycopeptide nanostructures for multipotent protein activation. Nat. Nanotechnol. 2017, 12, 821. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Silver, F.H.; Freeman, J.W.; Seehra, G.P. Collagen self-assembly and the development of tendon mechanical properties. J. Biomech. 2003, 36, 1529–1553. [Google Scholar] [CrossRef]
- Keeley, F.W.; Bellingham, C.M.; Woodhouse, K.A. Elastin as a self–organizing biomaterial: Use of recombinantly expressed human elastin polypeptides as a model for investigations of structure and self–assembly of elastin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2002, 357, 185–189. [Google Scholar] [CrossRef]
- Luo, Q.; Hou, C.; Bai, Y.; Wang, R.; Liu, J. Protein assembly: Versatile approaches to construct highly ordered nanostructures. Chem. Rev. 2016, 116, 13571–13632. [Google Scholar] [CrossRef]
- Wakabayashi, R.; Suehiro, A.; Goto, M.; Kamiya, N. Designer aromatic peptide amphiphiles for self-assembly and enzymatic display of proteins with morphology control. Chem. Commun. 2019, 55, 640–643. [Google Scholar] [CrossRef]
- Rising, A.; Widhe, M.; Johansson, J.; Hedhammar, M. Spider silk proteins: Recent advances in recombinant production, structure–function relationships and biomedical applications. Cell. Mol. Life Sci. 2011, 68, 169–184. [Google Scholar] [CrossRef]
- Kundu, B.; Kurland, N.E.; Bano, S.; Patra, C.; Engel, F.B.; Yadavalli, V.K.; Kundu, S.C. Silk proteins for biomedical applications: Bioengineering perspectives. Prog. Polym. Sci. 2014, 39, 251–267. [Google Scholar] [CrossRef]
- MacEwan, S.R.; Chilkoti, A. Elastin-like polypeptides: Biomedical applications of tunable biopolymers. Pept. Sci. Orig. Res. Biomol. 2010, 94, 60–77. [Google Scholar] [CrossRef]
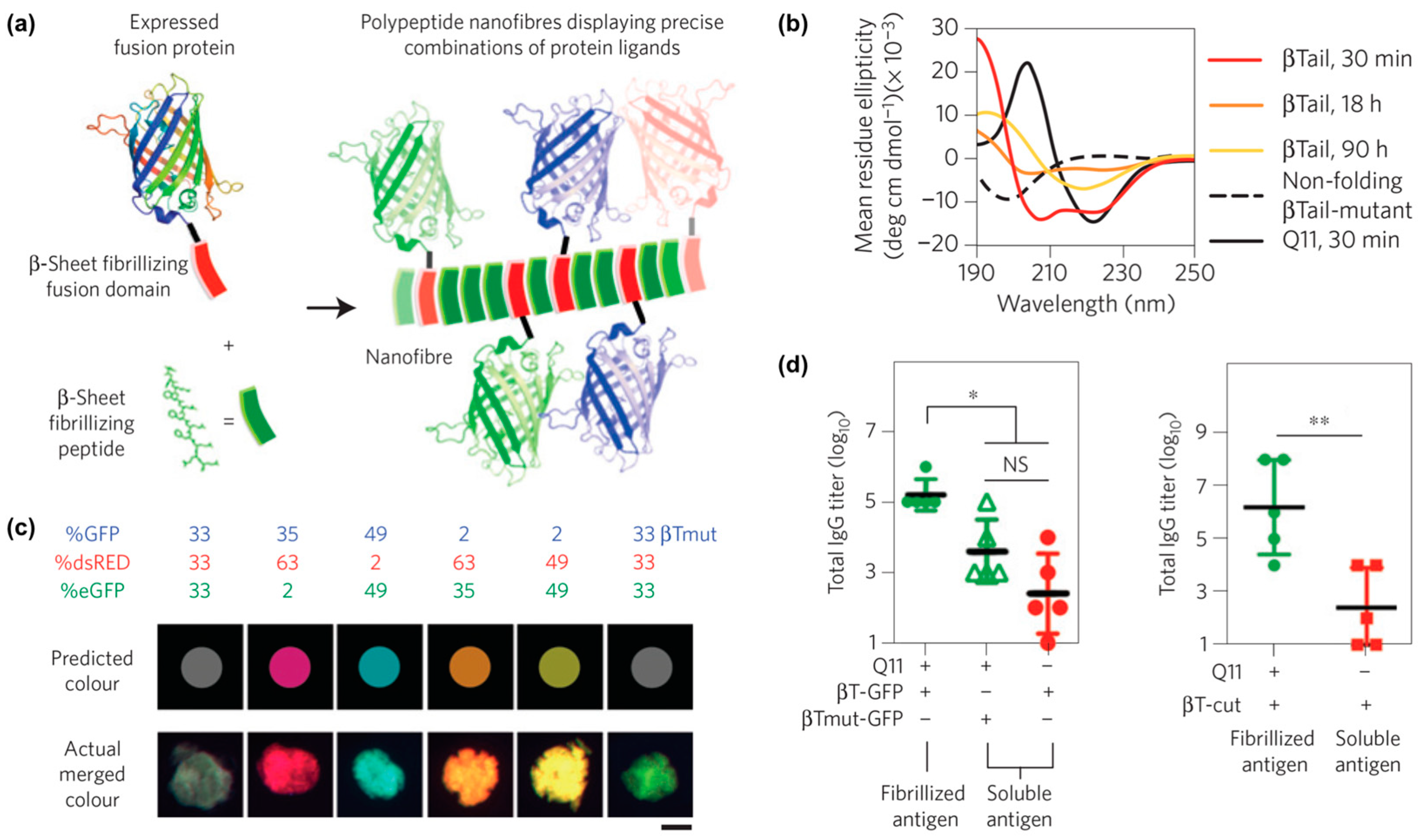
- Hudalla, G.A.; Sun, T.; Gasiorowski, J.Z.; Han, H.; Tian, Y.F.; Chong, A.S.; Collier, J.H. Gradated assembly of multiple proteins into supramolecular nanomaterials. Nat. Mater. 2014, 13, 829–836. [Google Scholar] [CrossRef]
- Ross, J.F.; Bridges, A.; Fletcher, J.M.; Shoemark, D.; Alibhai, D.; Bray, H.E.; Beesley, J.L.; Dawson, W.M.; Hodgson, L.R.; Mantell, J. Decorating Self-Assembled Peptide Cages with Proteins. ACS Nano 2017, 11, 7901–7914. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Harniman, R.L.; Barnes, F.R.; Boyle, A.L.; Collins, A.; Mantell, J.; Sharp, T.H.; Antognozzi, M.; Booth, P.J.; Linden, N. Self-assembling cages from coiled-coil peptide modules. Science 2013, 1226558. [Google Scholar] [CrossRef]
- DiMaio, J.T.; Raymond, D.M.; Nilsson, B.L. Display of functional proteins on supramolecular peptide nanofibrils using a split-protein strategy. Org. Biomol. Chem. 2017, 15, 5279–5283. [Google Scholar] [CrossRef]
- Seroski, D.T.; Restuccia, A.; Sorrentino, A.D.; Knox, K.R.; Hagen, S.J.; Hudalla, G.A. Co-assembly tags based on charge complementarity (CATCH) for installing functional protein ligands into supramolecular biomaterials. Cell. Mol. Bioeng. 2016, 9, 335–350. [Google Scholar] [CrossRef]
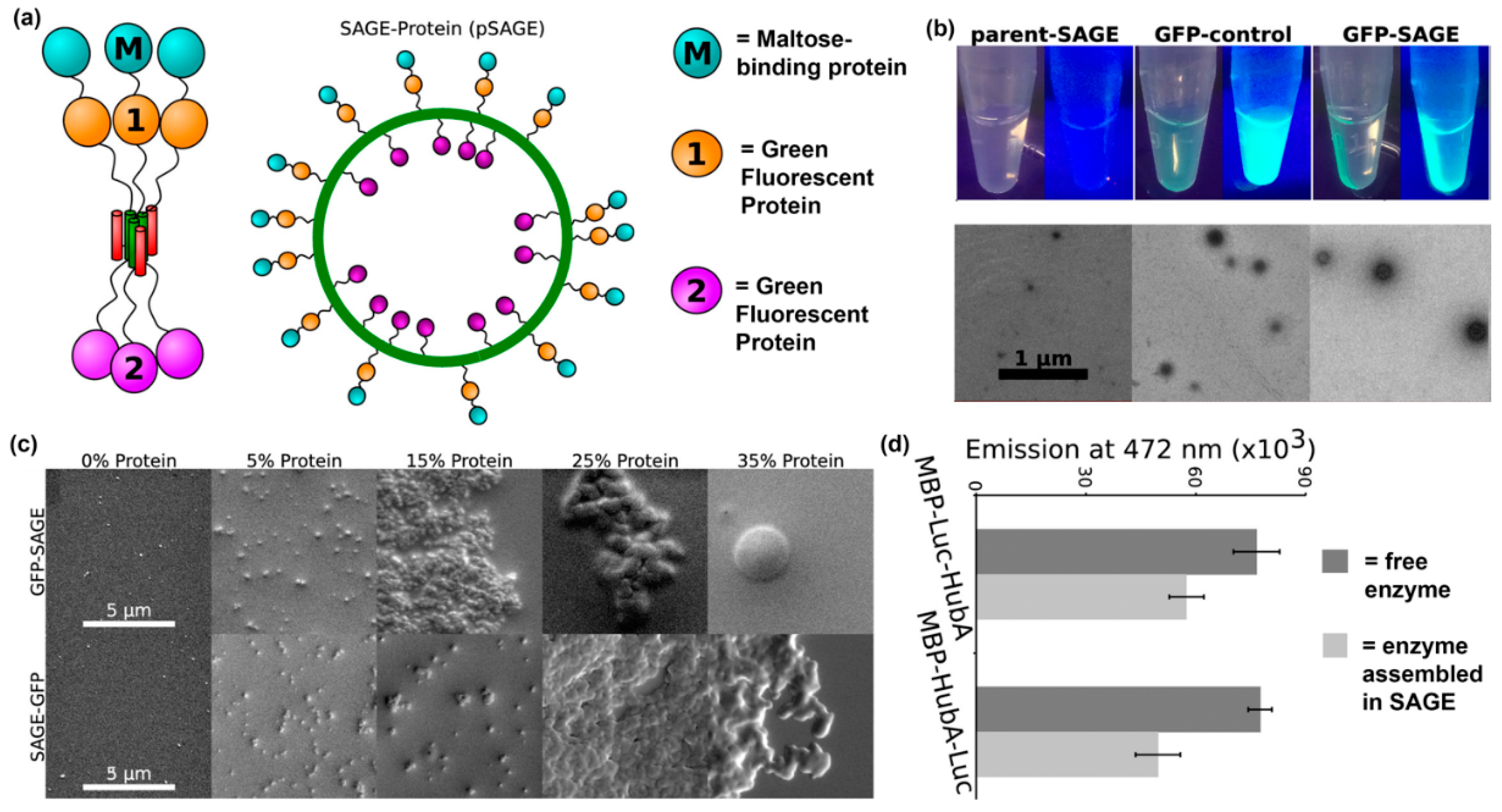
- Farhadi, S.A.; Bracho-Sanchez, E.; Fettis, M.M.; Seroski, D.T.; Freeman, S.L.; Restuccia, A.; Keselowsky, B.G.; Hudalla, G.A. Locally anchoring enzymes to tissues via extracellular glycan recognition. Nat. Commun. 2018, 9, 4943. [Google Scholar] [CrossRef]
- Ahmad, N.; Gabius, H.-J.; Sabesan, S.; Oscarson, S.; Brewer, C.F. Thermodynamic binding studies of bivalent oligosaccharides to galectin-1, galectin-3, and the carbohydrate recognition domain of galectin-3. Glycobiology 2004, 14, 817–825. [Google Scholar] [CrossRef]
- Talaga, M.L.; Fan, N.; Fueri, A.L.; Brown, R.K.; Bandyopadhyay, P.; Dam, T.K. Multitasking human lectin galectin-3 interacts with sulfated glycosaminoglycans and chondroitin sulfate proteoglycans. Biochemistry 2016, 55, 4541–4551. [Google Scholar] [CrossRef]
Sample Availability: Not available. |












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Hudalla, G.A. Using Self-Assembling Peptides to Integrate Biomolecules into Functional Supramolecular Biomaterials. Molecules 2019, 24, 1450. https://doi.org/10.3390/molecules24081450
Liu R, Hudalla GA. Using Self-Assembling Peptides to Integrate Biomolecules into Functional Supramolecular Biomaterials. Molecules. 2019; 24(8):1450. https://doi.org/10.3390/molecules24081450
Chicago/Turabian StyleLiu, Renjie, and Gregory A. Hudalla. 2019. "Using Self-Assembling Peptides to Integrate Biomolecules into Functional Supramolecular Biomaterials" Molecules 24, no. 8: 1450. https://doi.org/10.3390/molecules24081450
APA StyleLiu, R., & Hudalla, G. A. (2019). Using Self-Assembling Peptides to Integrate Biomolecules into Functional Supramolecular Biomaterials. Molecules, 24(8), 1450. https://doi.org/10.3390/molecules24081450