
Autophagy in Crotonaldehyde-Induced Endothelial Toxicity

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
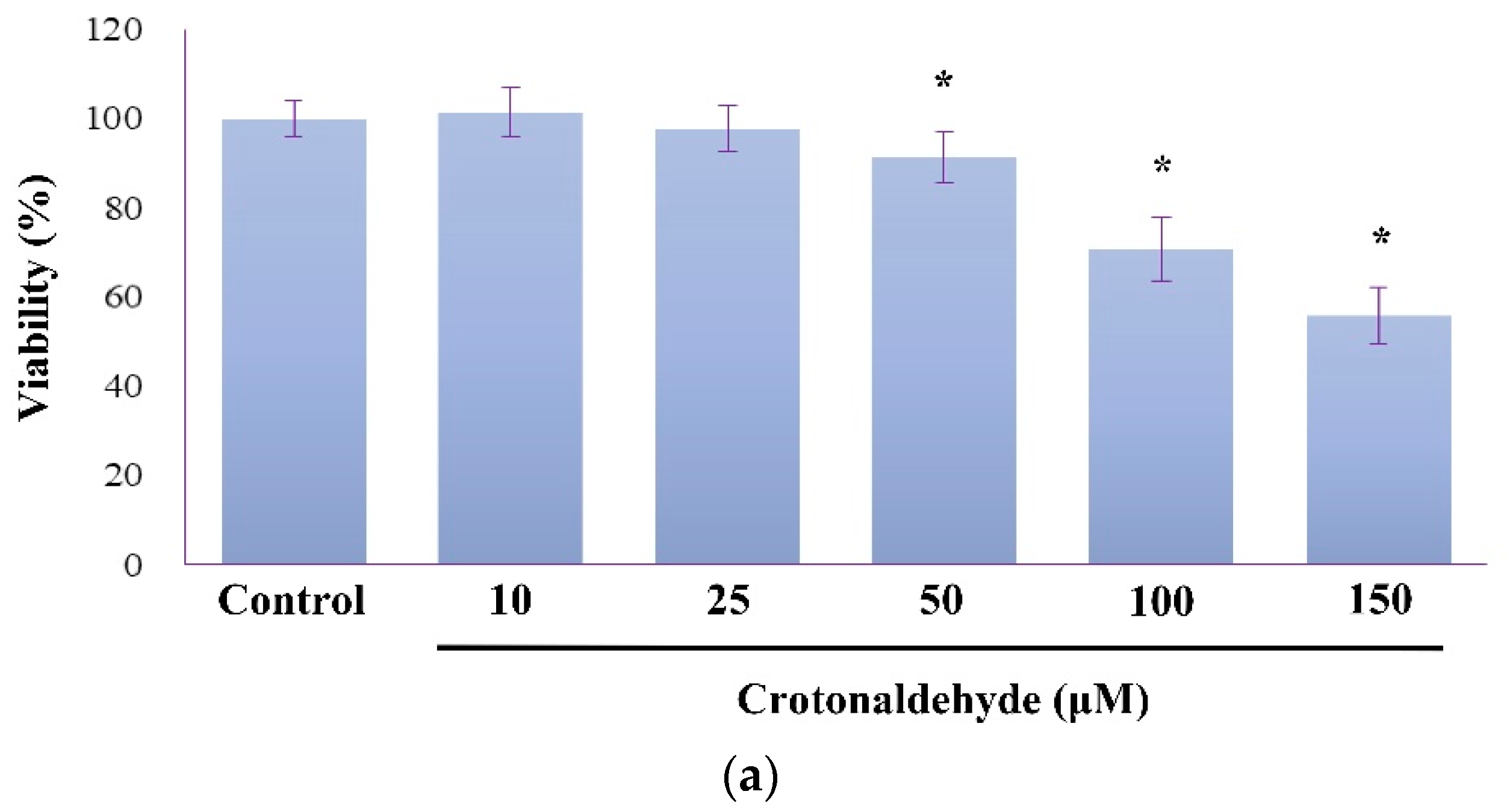
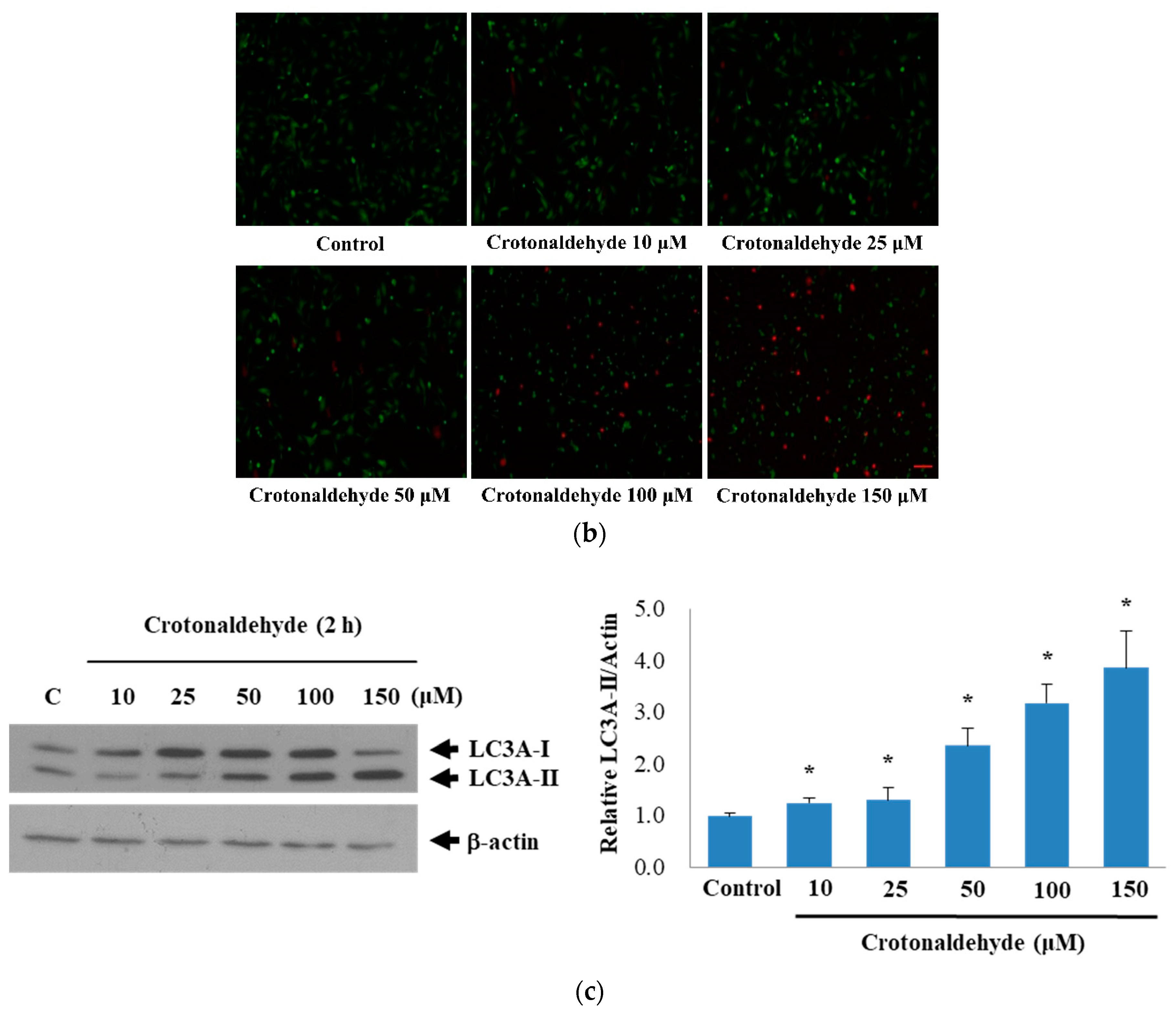
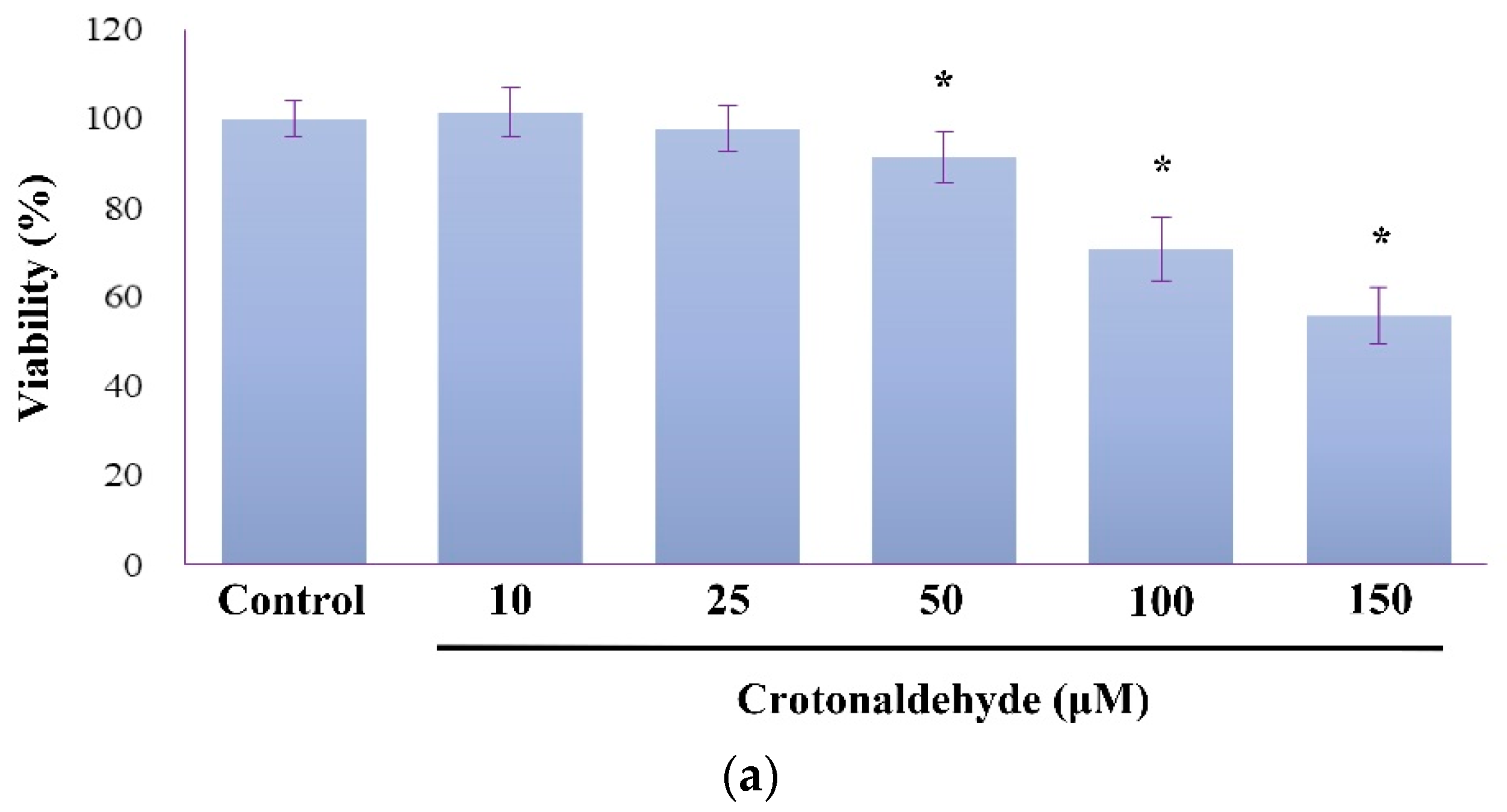
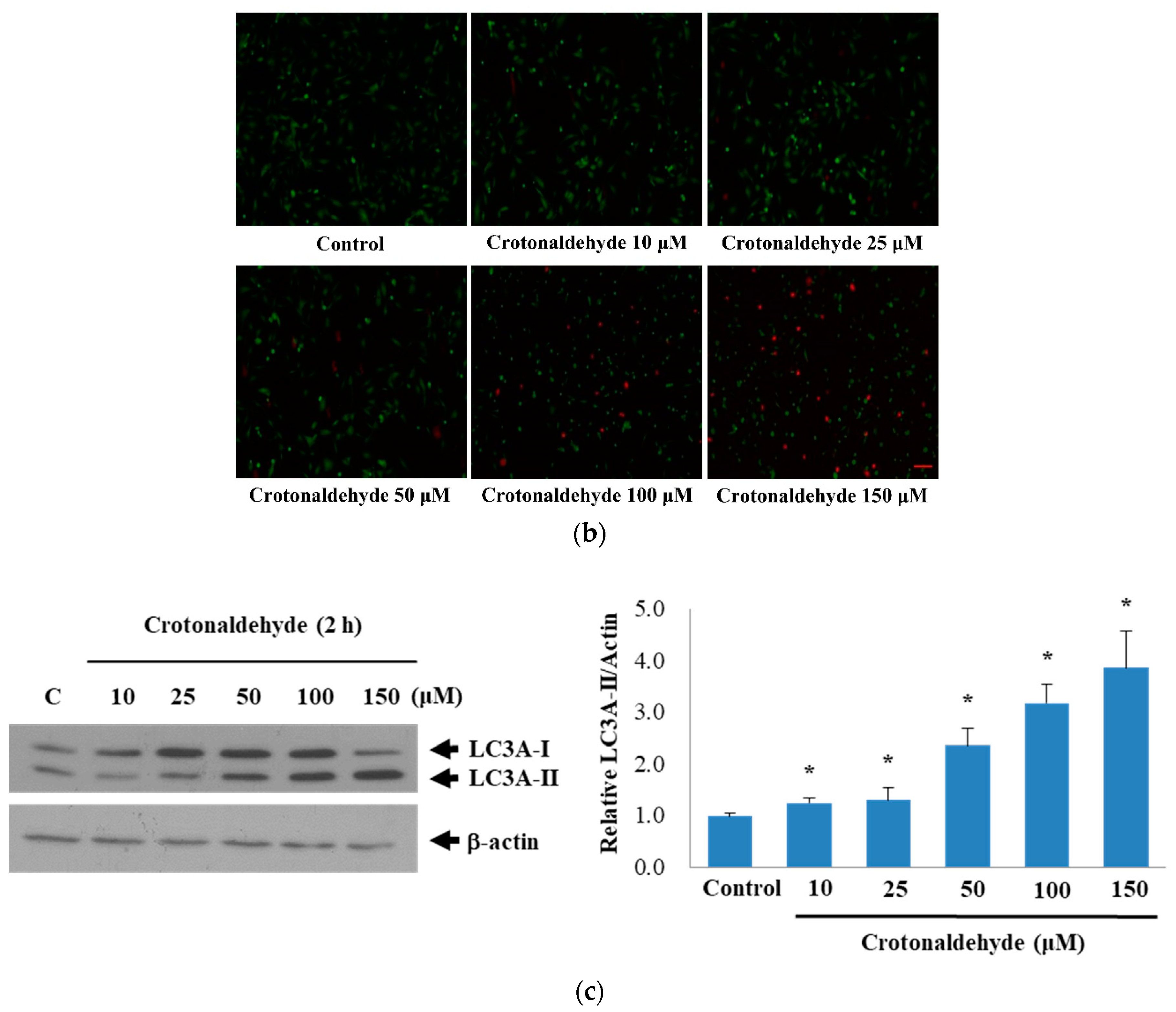
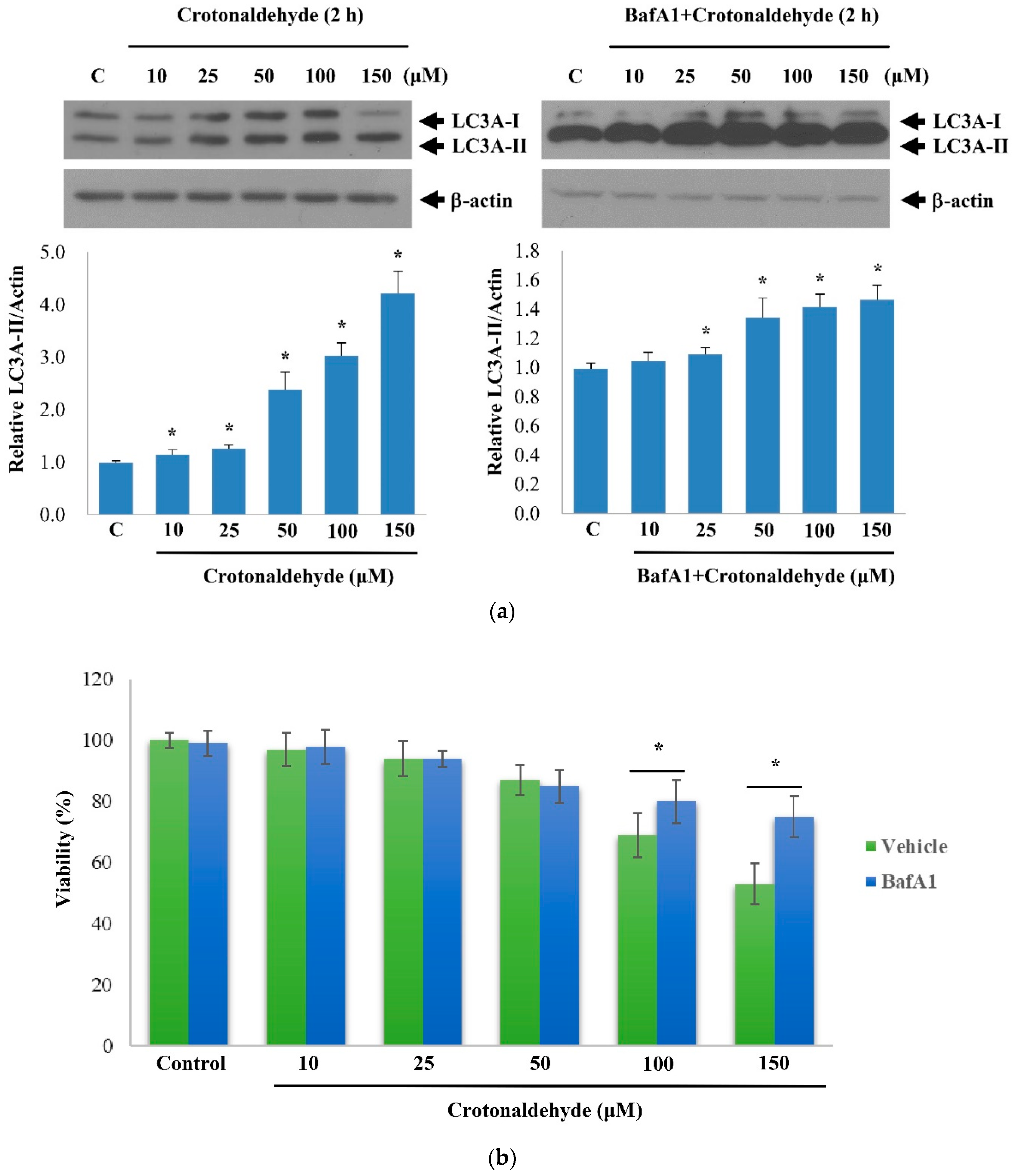
2.1. Induction of Cell Death and Autophagy In Endothelial Cells by Crotonaldehyde
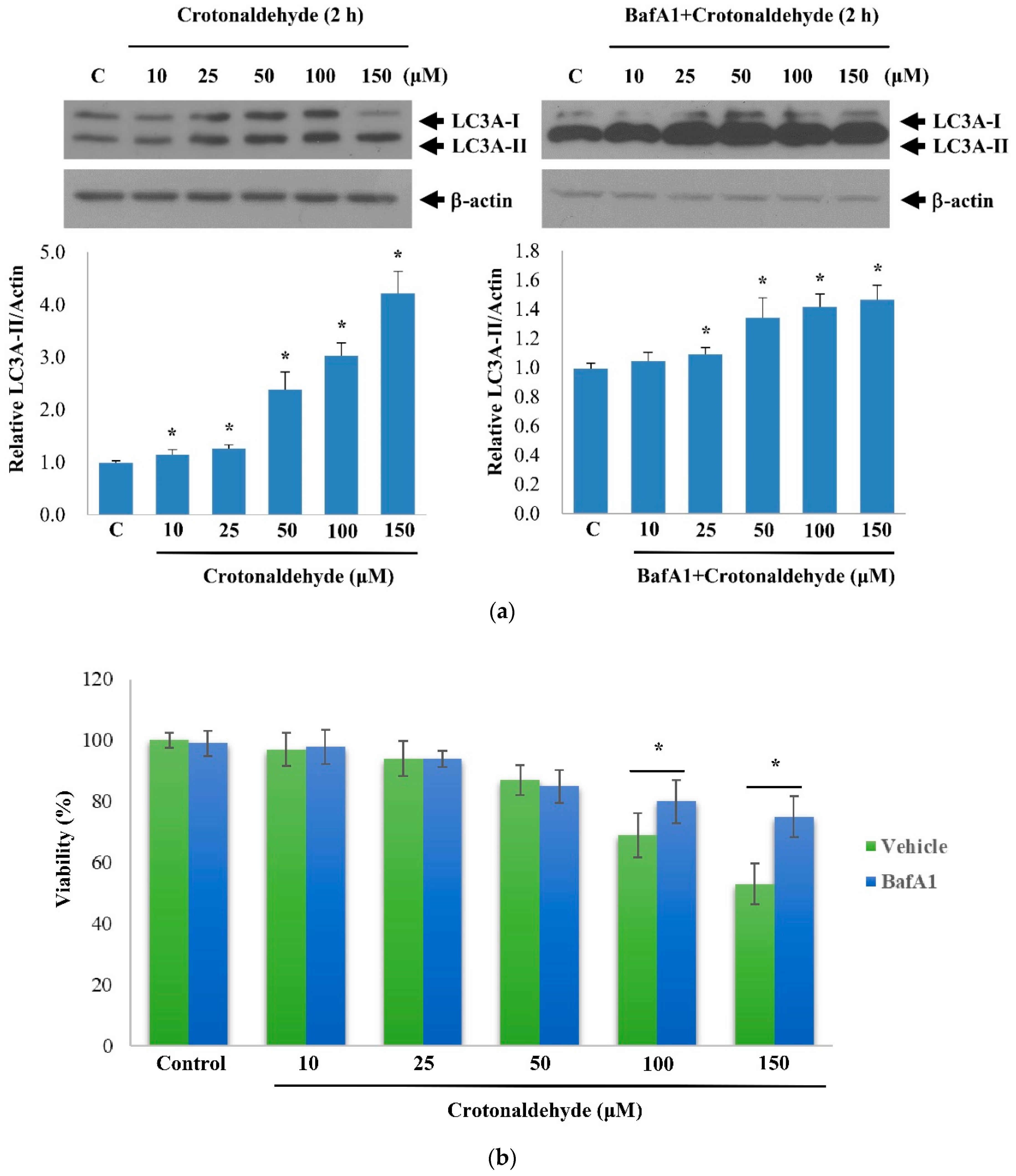
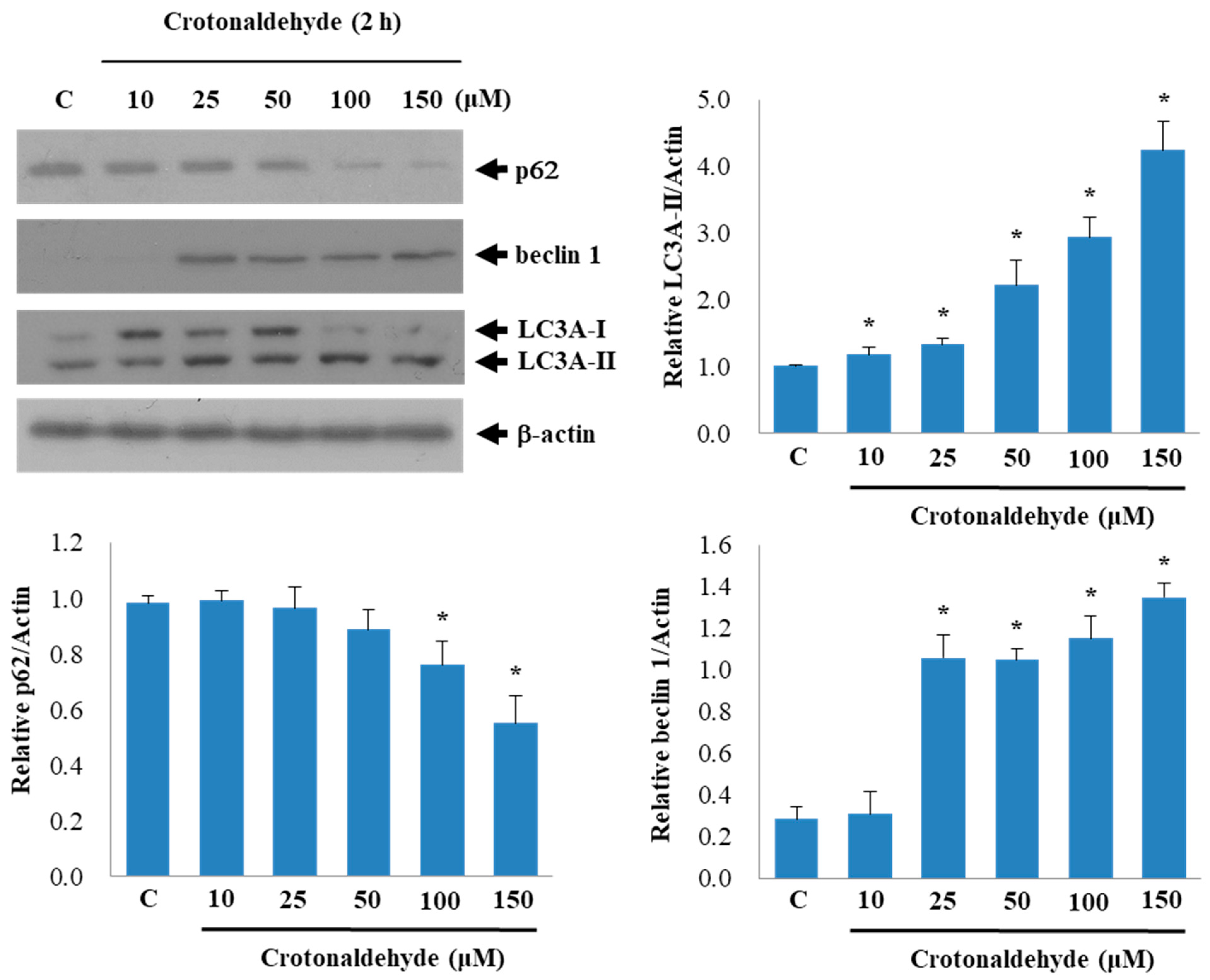
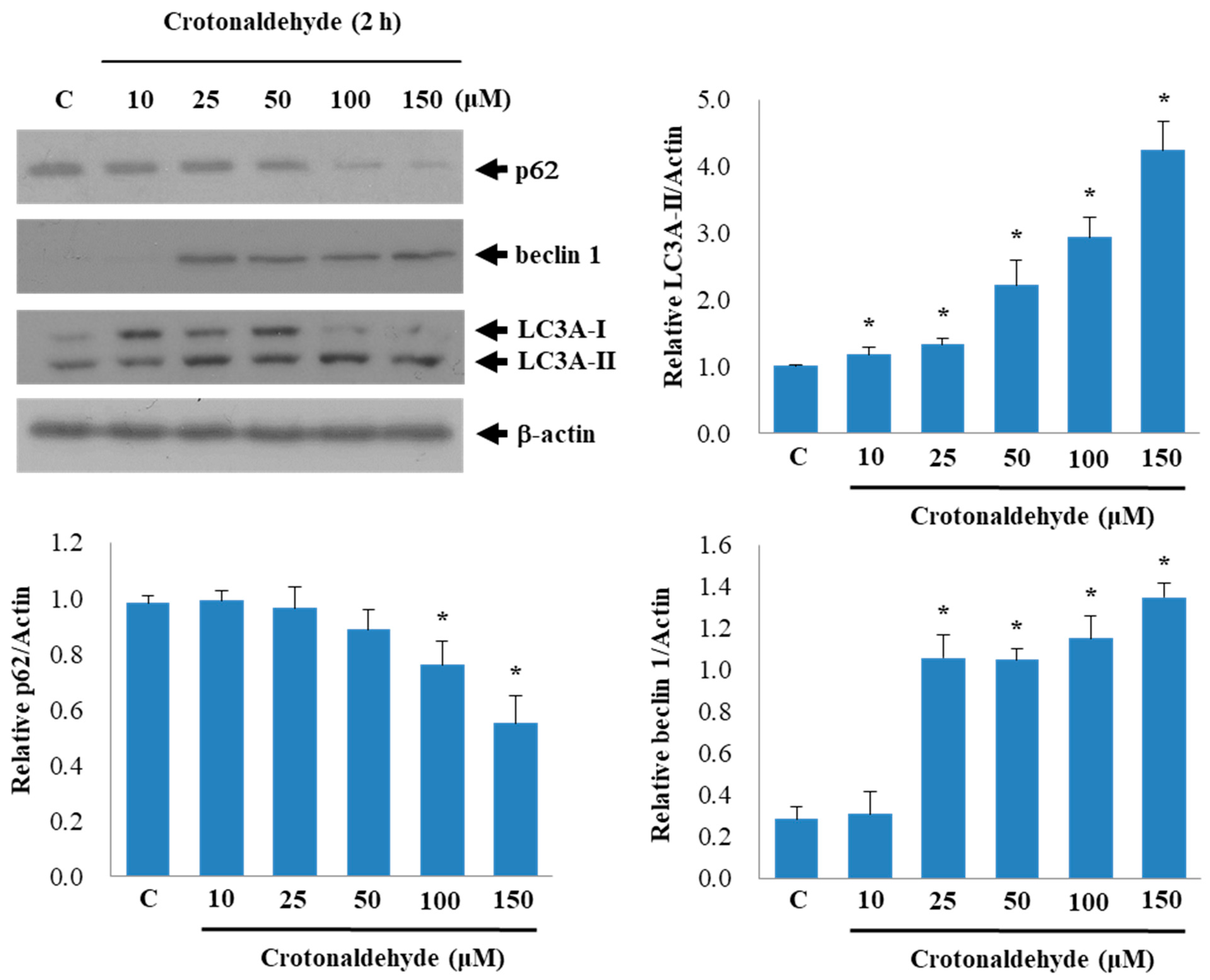
2.2. Effect of Crotonaldehyde on Autophagic Flux
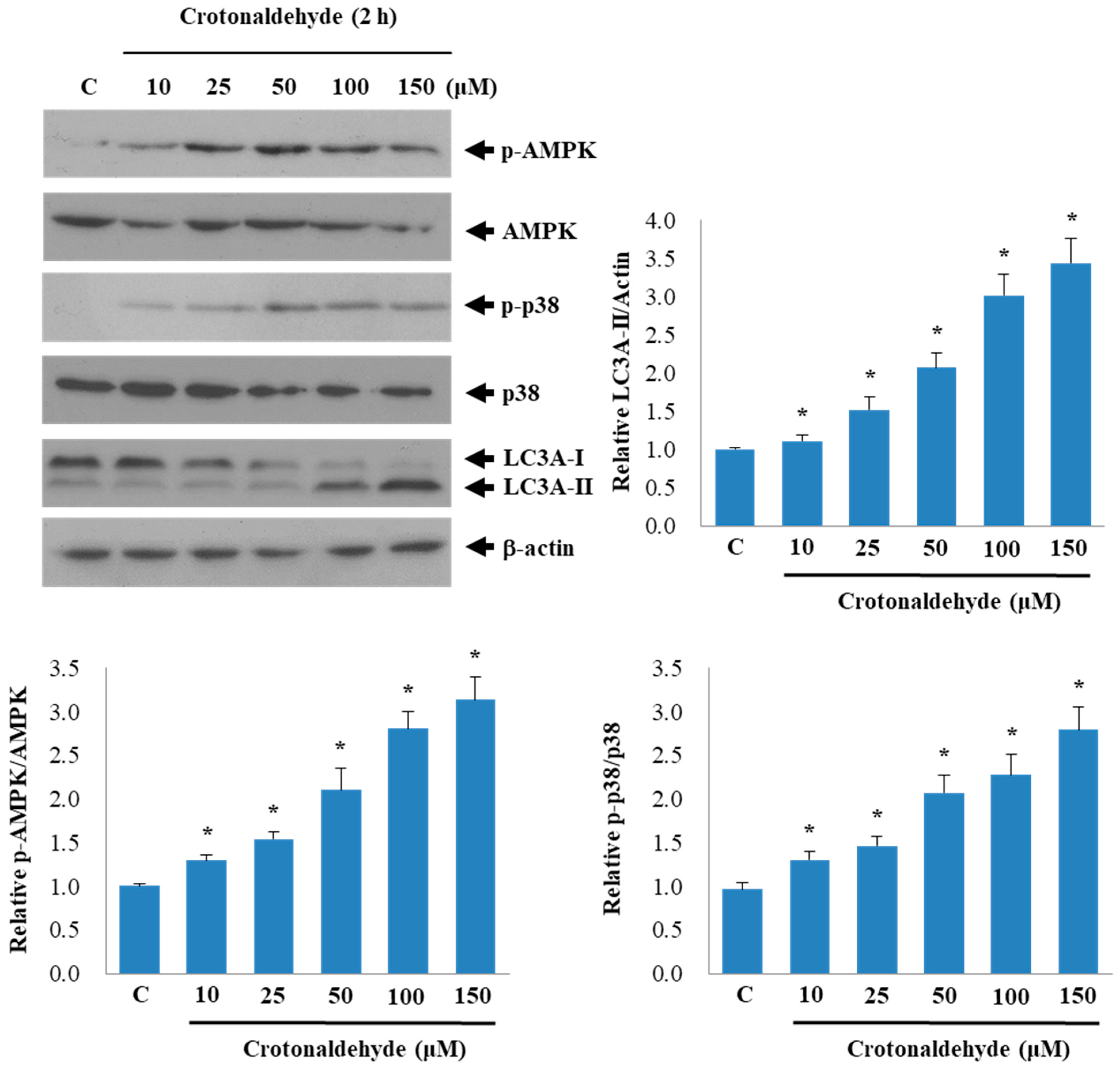
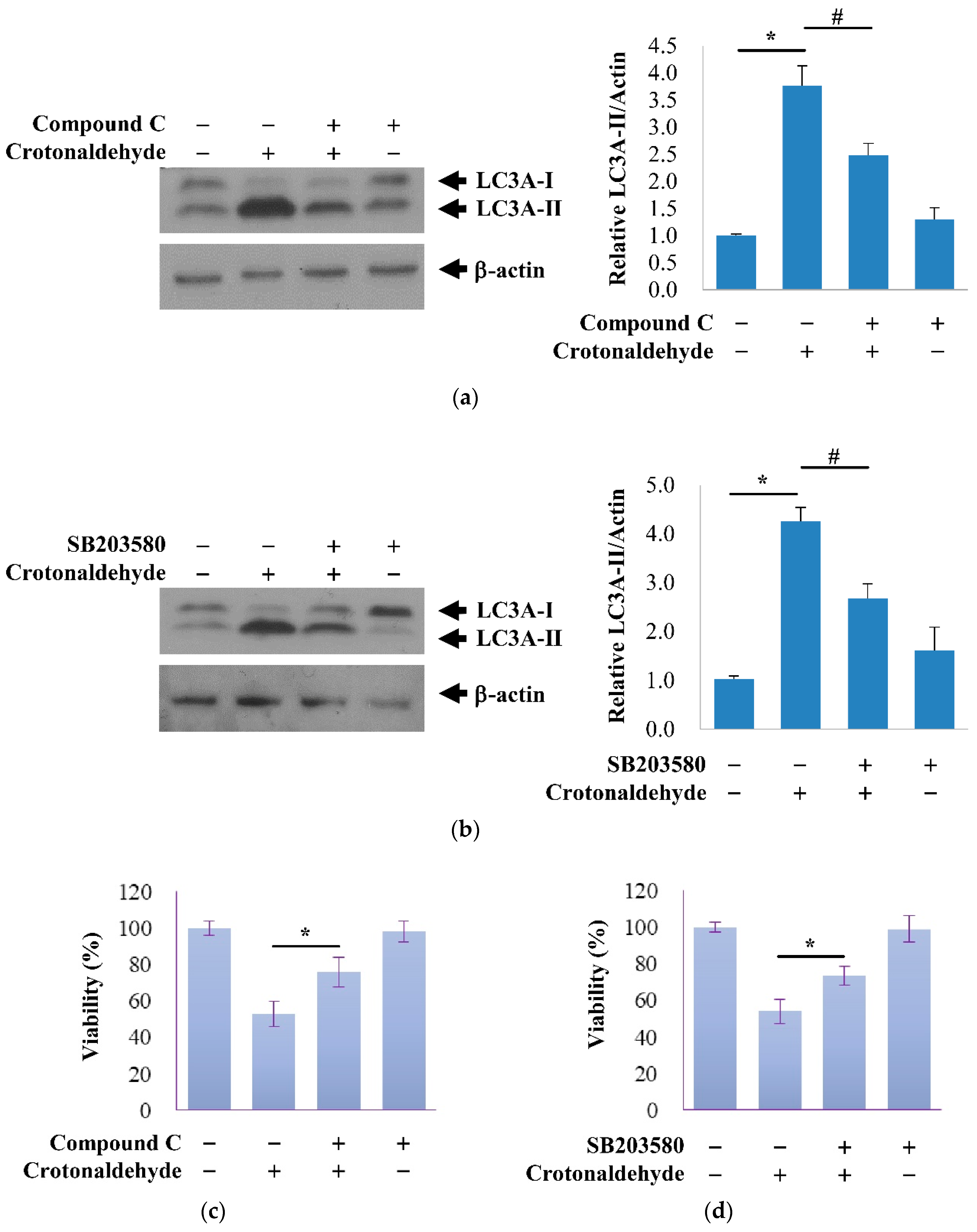
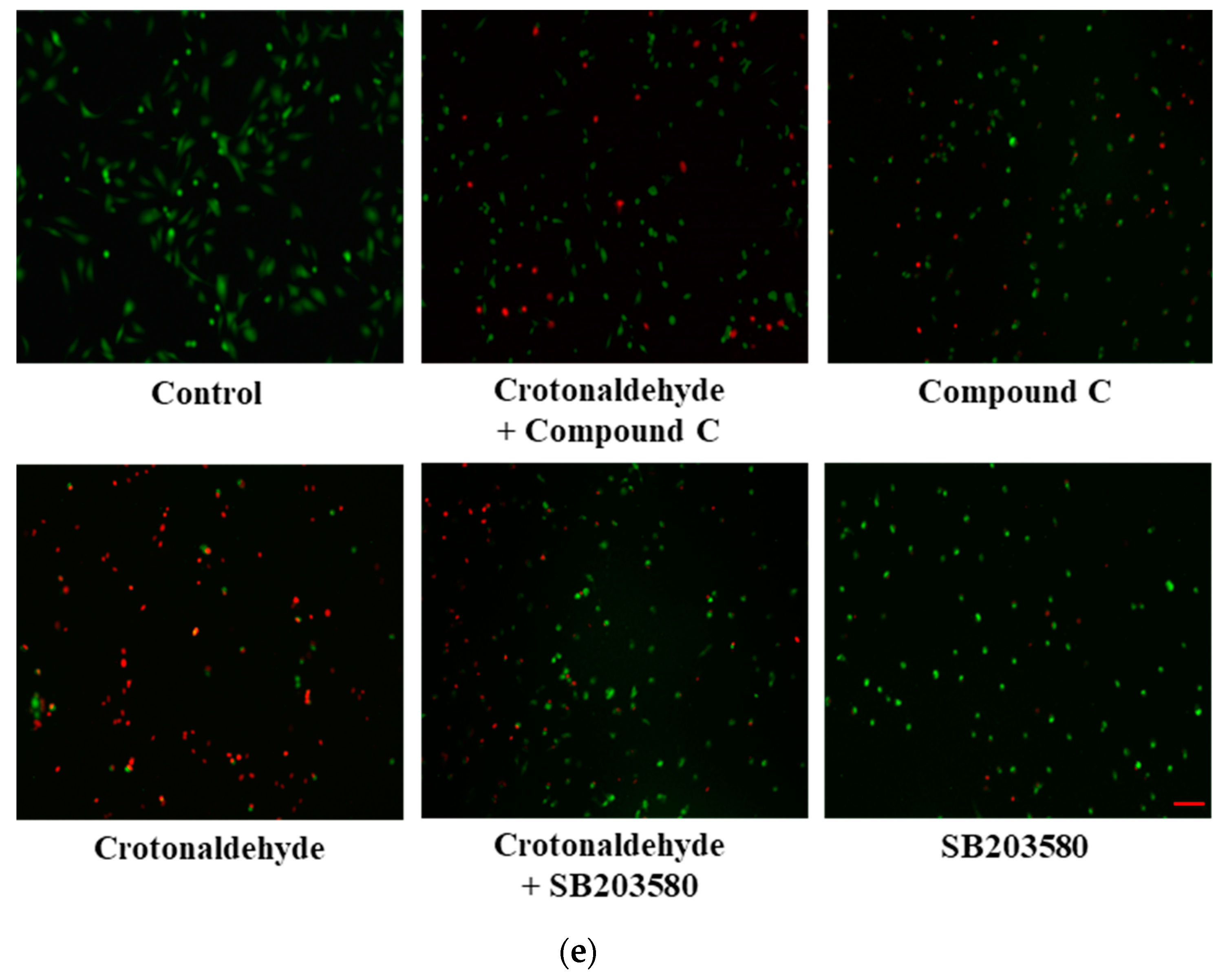
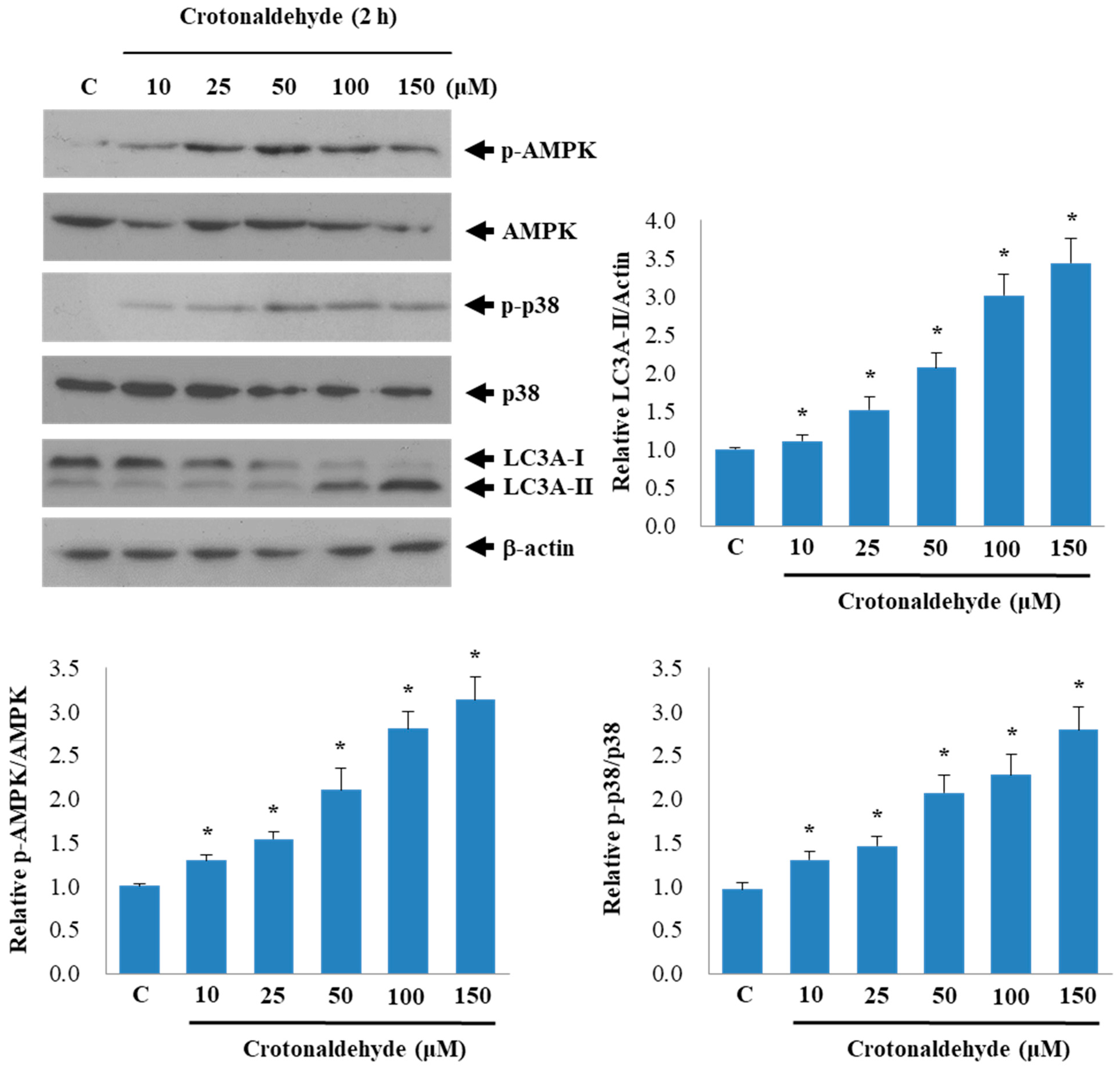
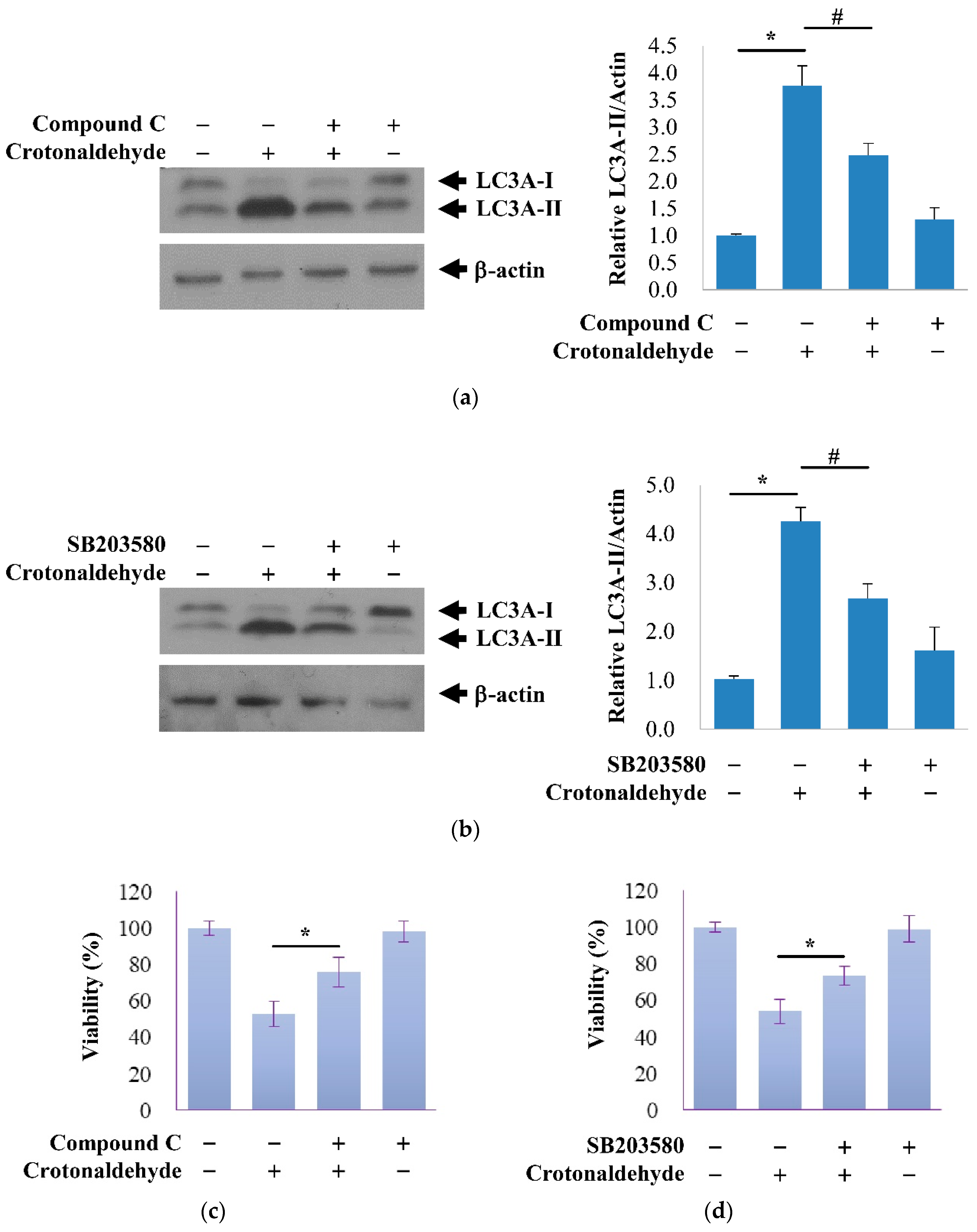
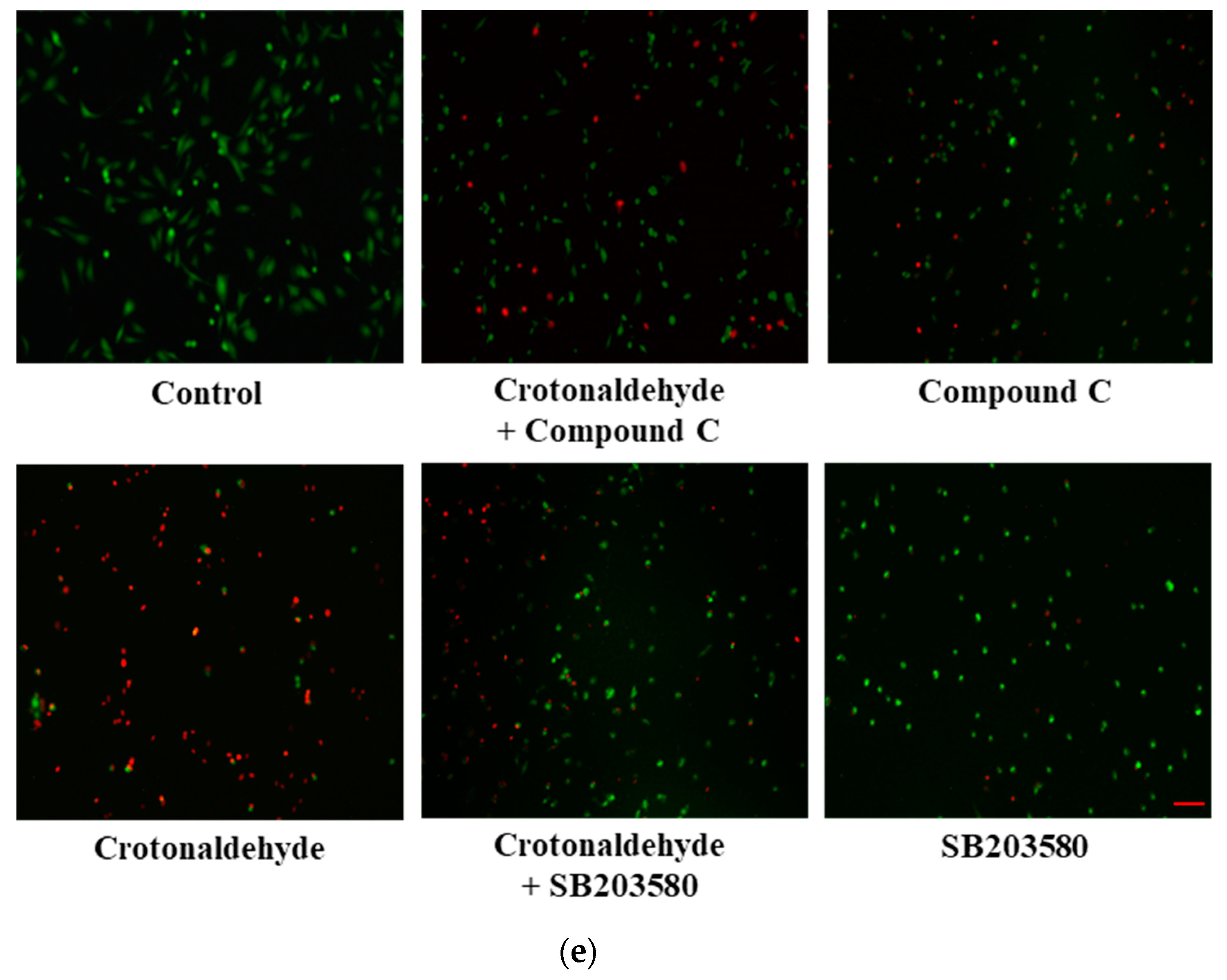
2.3. Involvement of AMPK and p38 MAPK in Crotonaldehyde-Induced Autophagy
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Viability Assay
4.4. Live/Dead Assay
4.5. Western Blot Analysis
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schroeder, S.A. New Evidence That Cigarette Smoking Remains the Most Important Health Hazard. N. Engl. J. Med. 2013, 368, 389–390. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, J.A.; Barua, R.S. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J. Am. Coll. Cardiol. 2004, 43, 1731–1737. [Google Scholar] [CrossRef]
- Streppel, M.T.; Boshuizen, H.C.; Ocke, M.C.; Kok, F.J.; Kromhout, D. Mortality and life expectancy in relation to long-term cigarette, cigar and pipe smoking: the Zutphen Study. Tob. Control 2007, 16, 107–113. [Google Scholar] [CrossRef]
- Katsiki, N.; Papadopoulou, S.K.; Fachantidou, A.I.; Mikhailidis, D.P. Smoking and vascular risk: Are all forms of smoking harmful to all types of vascular disease? Pub. Health 2013, 127, 435–441. [Google Scholar] [CrossRef]
- Branton, P.J.; McAdam, K.G.; Winter, D.B.; Liu, C.; Duke, M.G.; Proctor, C.J. Reduction of aldehydes and hydrogen cyanide yields in mainstream cigarette smoke using an amine functionalised ion exchange resin. Chem. Central J. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Pazo, D.Y.; Moliere, F.; Sampson, M.M.; Reese, C.M.; Agnew-Heard, K.A.; Walters, M.J.; Holman, M.R.; Blount, B.C.; Watson, C.H.; Chambers, D.M. Mainstream Smoke Levels of Volatile Organic Compounds in 50 U.S. Domestic Cigarette Brands Smoked With the ISO and Canadian Intense Protocols. Nicotine Tob. Res. 2016, 18, 1886–1894. [Google Scholar] [CrossRef]
- Bagchi, P.; Geldner, N.; de Castro, B.; De Jesus, V.R.; Park, S.K.; Blount, B.C. Crotonaldehyde exposure in U.S. tobacco smokers and nonsmokers: NHANES 2005–2006 and 2011–2012. Environ. Res. 2018, 163, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Voulgaridou, G.P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. 2011, 711, 13–27. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Dry Cleaning, Some Chlorinated Solvents and Other Industrial Chemicals, 1st ed.; World Health Organization: Geneva, Switzerland, 1995. [Google Scholar]
- Lee, S.E.; Park, Y.S. Role of Lipid Peroxidation-Derived alpha, beta-Unsaturated Aldehydes in Vascular Dysfunction. Oxidat. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef]
- Yang, B.C.; Yang, Z.H.; Pan, X.J.; Xiao, F.J.; Liu, X.Y.; Zhu, M.X.; Xie, J.P. Crotonaldehyde-exposed macrophages induce IL-8 release from airway epithelial cells through NF-kappa B and AP-1 pathways. Toxicol. Lett. 2013, 219, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.C.; Yang, Z.H.; Pan, X.J.; Liu, X.Y.; Zhu, M.X.; Xie, J.P. Crotonaldehyde induces apoptosis and immunosuppression in alveolar macrophages. Toxicol. Vitro 2013, 27, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Moretto, N.; Facchinetti, F.; Southworth, T.; Civelli, M.; Singh, D.; Patacchini, R. α,β-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L839–L848. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Park, H.R.; Kim, H.; Choi, Y.; Jin, Y.H.; Park, C.S.; Ahn, H.J.; Cho, J.J.; Park, Y.S. Effect of crotonaldehyde on the induction of COX-2 expression in human endothelial cells. Mol. Cell. Toxicol. 2017, 13, 345–350. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Yang, Z.; Pan, X.; Liu, X.; Zhu, M.; Xie, J. Crotonaldehyde induces autophagy-mediated cytotoxicity in human bronchial epithelial cells via PI3K, AMPK and MAPK pathways. Environ. Pollut. 2017, 228, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, M.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Lee, S.J.; Smith, A.; Choi, A.M. Autophagy in vascular disease. Proc. Am. Thorac. Soc. 2010, 7, 40–47. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X.Y. Autophagy and oxidative stress in cardiovascular diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in cardiovascular biology. J. Clin. Invest. 2015, 125, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Fougeray, S.; Pallet, N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat. Rev. Nephrol. 2015, 11, 34–45. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Dis. 2012, 11, U709–U784. [Google Scholar] [CrossRef]
- Csordas, A.; Kreutmayer, S.; Ploner, C.; Braun, P.R.; Karlas, A.; Backovic, A.; Wick, G.; Bernhard, D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc. Res. 2011, 92, 141–148. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.H. 2-Deoxy-d-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef]
- Maejima, Y.; Isobe, M.; Sadoshima, J. Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell. Cardiol. 2016, 95, 19–25. [Google Scholar] [CrossRef]
- Nezis, I.P.; Stenmark, H. p62 at the Interface of Autophagy, Oxidative Stress Signaling, and Cancer. Antiox. Redox Signal. 2012, 17, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, U132–U171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Kawauchi, J.; Adachi, M.T.; Hashimoto, Y.; Oshiro, S.; Aso, T.; Kitajima, S. Activation of JNK and transcriptional repressor ATF3/LRF1 through the IRE1/TRAF2 pathway is implicated in human vascular endothelial cell death by homocysteine. Biochem. Biophys. Res. Commun. 2001, 289, 718–724. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Global Report on Mortality Attributable to Tobacco. Available online: https://www.who.int/tobacco/publications/surveillance/rep_mortality_attributable/en/ (accessed on 5 February 2019).
- Shah, R.H.; Cole, J.W. Smoking and stroke: The more you smoke the more you stroke. Expert Rev. Cardiovasc. Ther. 2010, 8, 917–932. [Google Scholar] [CrossRef]
- Lee, K.; Hong, S.; Seong, G.J.; Kim, C.Y. Cigarette Smoke Extract Causes Injury in Primary Retinal Ganglion Cells via Apoptosis and Autophagy. Cur. Eye Res. 2016, 41, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.K.; Ounpuu, S.; Hawken, S.; Pandey, M.R.; Valentin, V.; Hunt, D.; Diaz, R.; Rashed, W.; Freeman, R.; Jiang, L.X.; et al. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: A case-control study. Lancet 2006, 368, 647–658. [Google Scholar] [CrossRef]
- Lekakis, J.; Papamichael, C.; Vemmos, C.; Stamatelopoulos, K.; Voutsas, A.; Stamatelopoulos, S. Effects of acute cigarette smoking on endothelium-dependent arterial dilatation in normal subjects. Am. J. Cardiol. 1998, 81, 1225–1228. [Google Scholar] [CrossRef]
- Mercado, C.; Jaimes, E.A. Cigarette smoking as a risk factor for atherosclerosis and renal disease: Novel pathogenic insights. Curr. Hyperten. Rep. 2007, 9, 66–72. [Google Scholar] [CrossRef]
- Kuper, H.; Adami, H.O.; Boffetta, P. Tobacco use, cancer causation and public health impact. J. Int. Med. 2002, 251, 455–466. [Google Scholar] [CrossRef]
- Park, H.E.; Lee, S.E.; Son, G.W.; Yun, H.D.; Park, C.S.; Ahn, H.J.; Cho, J.J.; Lee, J.; Park, Y.S. Profiling of gene expression using microarray in acrolein-treated human pulmonary fibroblasts. Mol. Cell. Toxicol. 2017, 13, 49–58. [Google Scholar] [CrossRef]
- Colombo, G.; Aldini, G.; Orioli, M.; Giustarini, D.; Gornati, R.; Rossi, R.; Colombo, R.; Carini, M.; Milzani, A.; Dalle-Donne, I. Water-Soluble alpha,beta-Unsaturated Aldehydes of Cigarette Smoke Induce Carbonylation of Human Serum Albumin. Antiox. Redox Signal. 2010, 12, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Carmella, S.G.; Chen, M.; Patel, Y.; Stram, D.O.; Haiman, D.A.; Le Marchand, A.; Hecht, S.S. Mercapturic Acids Derived from the Toxicants Acrolein and Crotonaldehyde in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer. PLoS ONE 2015, 10, e0124841. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.S.; Yang, H.; Lee, S.E.; Park, C.S.; Jin, Y.S.; Park, Y.S. Crotonaldehyde induces heat shock protein 72 expression that mediates anti-apoptotic effects in human endothelial cells. Toxicol. Lett. 2013, 223, 116–123. [Google Scholar] [CrossRef]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Ademi, C.; Bertram, S.; Schmid, K.W.; Baba, H.A. Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J. Surg. Oncol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef]
- Weikel, K.J.; Cacicedo, J.M.; Ruderman, N.B.; Ido, Y. Knockdown of GSK3 beta increases basal autophagy and AMPK signalling in nutrient-laden human aortic endothelial cells. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Jung, Y.K.; Oh, S.H. Autophagy Induction by Capsaicin in Malignant Human Breast Cells Is Modulated by p38 and Extracellular Signal-Regulated Mitogen-Activated Protein Kinases and Retards Cell Death by Suppressing Endoplasmic Reticulum Stress-Mediated Apoptosis. Mol. Pharmacol. 2010, 78, 114–125. [Google Scholar] [CrossRef]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS … Thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef]
- Lee, S.E.; Jeong, S.I.; Kim, G.D.; Yang, H.; Park, C.S.; Jin, Y.H.; Park, Y.S. Upregulation of heme oxygenase-1 as an adaptive mechanism for protection against crotonaldehyde in human umbilical vein endothelial cells. Toxicol. Lett. 2011, 201, 240–248. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.E.; Park, H.R.; Park, C.-S.; Ahn, H.-J.; Cho, J.-J.; Lee, J.; Park, Y.S. Autophagy in Crotonaldehyde-Induced Endothelial Toxicity. Molecules 2019, 24, 1137. https://doi.org/10.3390/molecules24061137
Lee SE, Park HR, Park C-S, Ahn H-J, Cho J-J, Lee J, Park YS. Autophagy in Crotonaldehyde-Induced Endothelial Toxicity. Molecules. 2019; 24(6):1137. https://doi.org/10.3390/molecules24061137
Chicago/Turabian StyleLee, Seung Eun, Hye Rim Park, Cheung-Seog Park, Hyun-Jong Ahn, Jeong-Je Cho, Jongsung Lee, and Yong Seek Park. 2019. "Autophagy in Crotonaldehyde-Induced Endothelial Toxicity" Molecules 24, no. 6: 1137. https://doi.org/10.3390/molecules24061137
APA StyleLee, S. E., Park, H. R., Park, C.-S., Ahn, H.-J., Cho, J.-J., Lee, J., & Park, Y. S. (2019). Autophagy in Crotonaldehyde-Induced Endothelial Toxicity. Molecules, 24(6), 1137. https://doi.org/10.3390/molecules24061137