Cohesive Energy Densities Versus Internal Pressures of Near and Supercritical Fluids


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
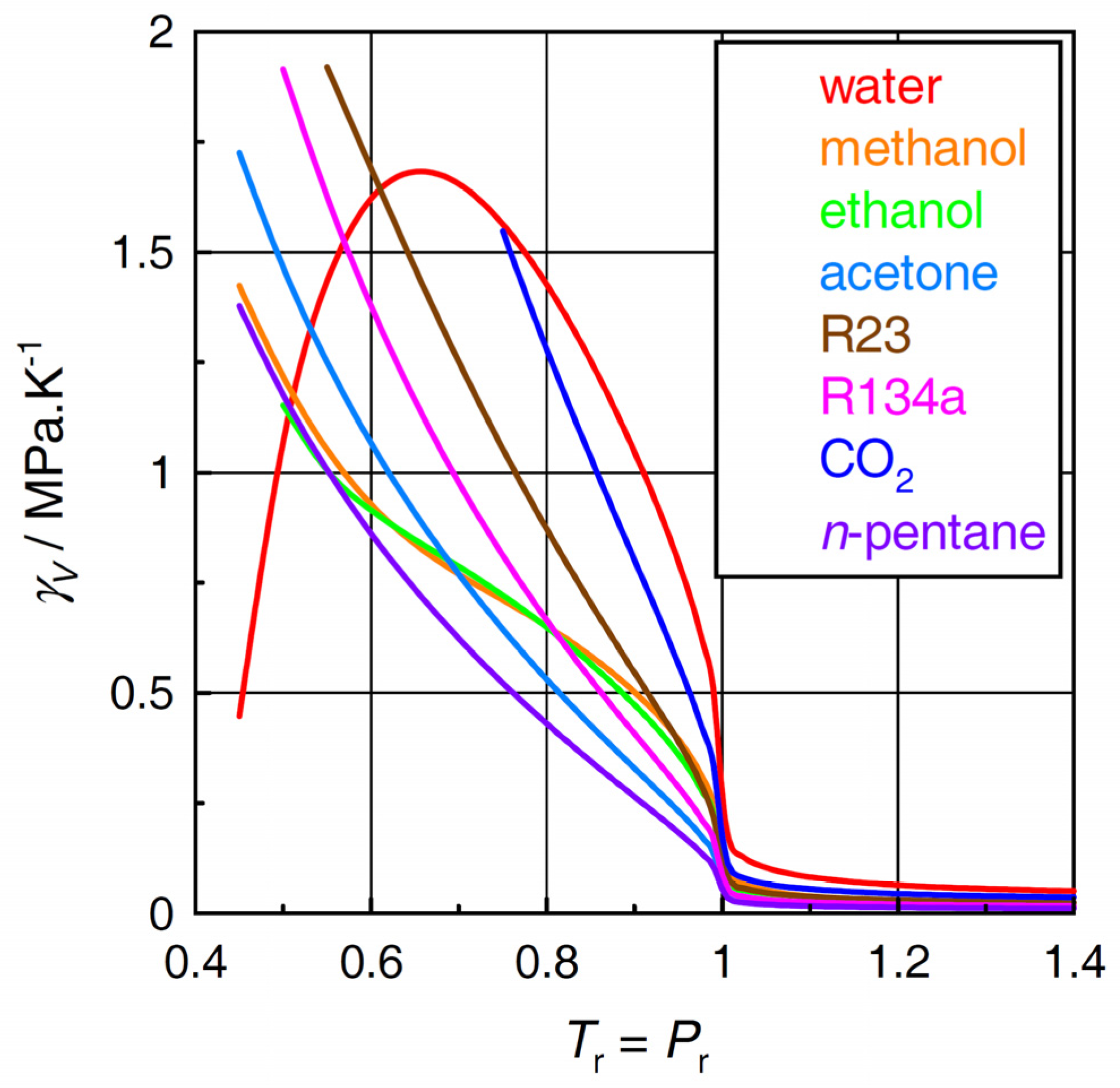
2.1. Thermal Pressure Coefficient
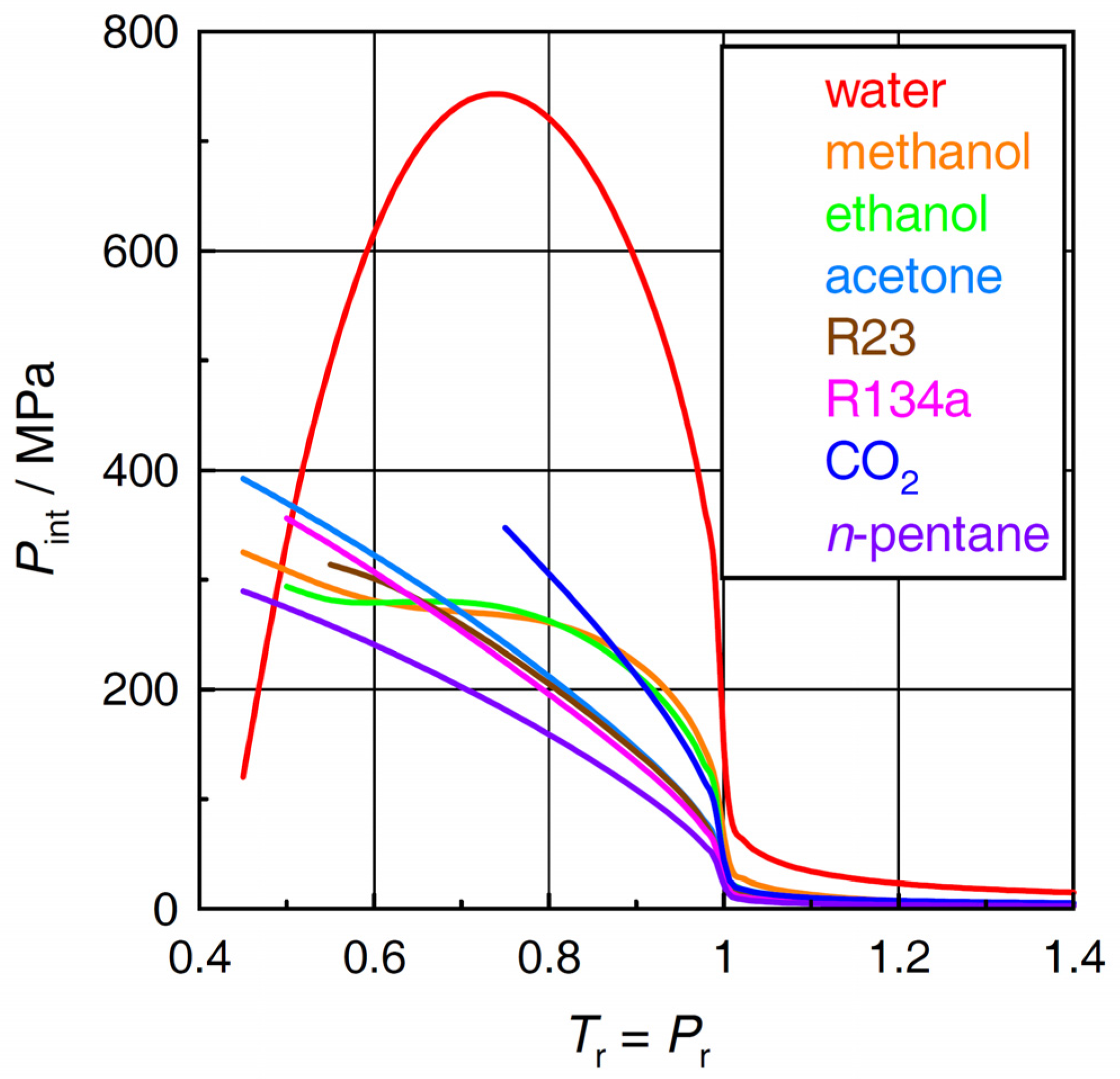
2.2. Internal Pressure
2.3. Cohesive Energy Density
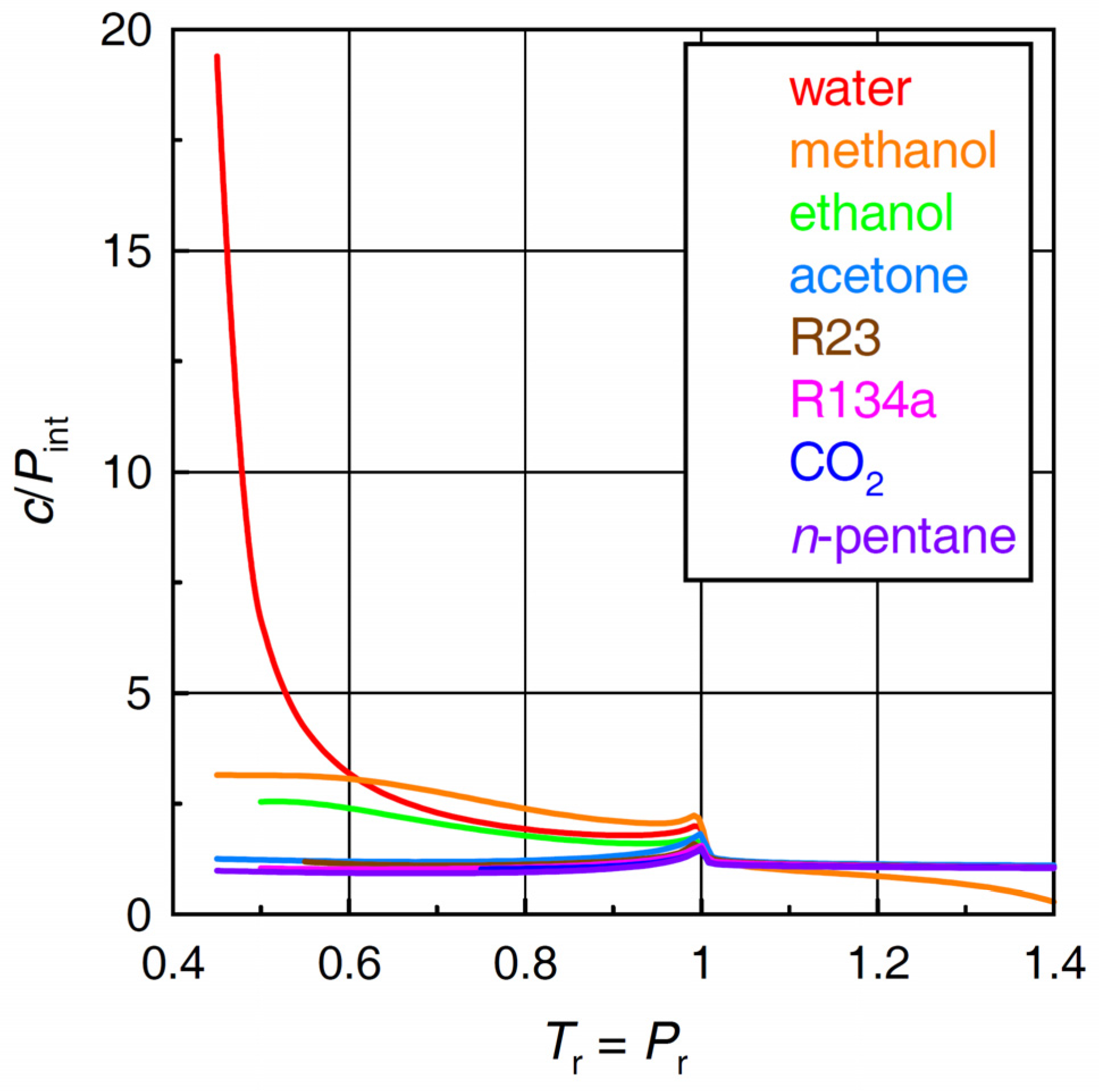
2.4. Ratio of Cohesive Energy Density to Internal Pressure
3. Materials and Methods
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Derivatives of c and Pint with Respect to T, P and v
List of Symbols
| c | cohesive energy density |
| cP | molar isobaric heat capacity |
| cV | molar isochoric heat capacity |
| cV0 | ideal-gas molar isochoric heat capacity |
| P | pressure |
| Pint | internal pressure [= (∂u/∂v)T] |
| T | temperature |
| u | molar internal energy |
| u0 | ideal-gas molar internal energy |
| V | volume |
| v | molar volume |
Greek Symbols
| αP | isobaric expansivity [= (1/v)(∂v/∂T)P] |
| βT | isothermal compressibility [= −(1/v)(∂v/∂P)T] |
| γV | thermal pressure coefficient [= (∂P/∂T)V] |
| δ | solubility parameter [= c1/2] |
References
- Harvey, A.H.; Friend, D.G. Physical properties of water. In Aqueous Systems at Elevated Temperatures and Pressures. Physical Chemistry in Water, Steam and Hydrothermal Solutions; Palmer, D.A., Fernández-Prini, R., Harvey, A.H., Eds.; Elsevier–Academic Press: London, UK, 2004; Chapter 1; pp. 1–27. [Google Scholar]
- Weingärtner, H.; Franck, E.U. Supercritical water as a solvent. Angew. Chem. Int. Ed. 2005, 44, 2672–2692. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, M.; Franck, E.U. Static dielectric constant of water and steam. J. Phys. Chem. Ref. Data 1980, 9, 1291–1304. [Google Scholar] [CrossRef]
- Fernández, D.P.; Goodwin, A.R.H.; Lemmon, E.W.; Levelt Sengers, J.M.H.; Williams, R.C. A formulation for the static permittivity of water and steam at temperatures from 238 K to 873 K at pressures up to 1200 MPa, including derivatives and Debye–Hückel coefficients. J. Phys. Chem. Ref. Data 1997, 26, 1125–1166. [Google Scholar] [CrossRef]
- Marshall, W.L.; Franck, E.U. Ion product of water substance, 0–1000 °C, 1–10,000 bars. New international formulation and its background. J. Phys. Chem. Ref. Data 1981, 10, 295–304. [Google Scholar] [CrossRef]
- Bandura, A.V.; Lvov, S.N. The ionization constant of water over wide ranges of temperature and density. J. Phys. Chem. Ref. Data 2006, 35, 15–30. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003; Chapter 6; pp. 329–388. [Google Scholar]
- Wiehe, I.A.; Bagley, E.B. Estimation of dispersion and hydrogen bonding energies in liquids. AIChE J. 1967, 13, 836–838. [Google Scholar] [CrossRef]
- Prausnitz, J.M.; Lichtenthaler, R.N.; Gomes de Azevedo, E. Molecular Thermodynamics of Fluid-Phase Equilibria, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 1999; Section 7.2; pp. 313–326. [Google Scholar]
- Wagner, W.; Overhoff, U. ThermoFluids. Interactive Software for the Calculation of Thermodynamic Properties for More Than 60 Pure Substances; Springer-Verlag: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- NIST Reference Fluid Thermodynamic and Transport Properties Database (REFPROP). Available online: https://www.nist.gov/srd/refprop (accessed on 7 February 2019).
- Lemmon, E.W.; Span, R. Short fundamental equations of state for 20 industrial fluids. J. Chem. Eng. Data 2006, 51, 785–850. [Google Scholar] [CrossRef]
- Span, R.; Wagner, W. A new equation of state for carbon dioxide covering the fluid region from the triple-point temperature to 1100 K at pressures up to 800 MPa. J. Phys. Chem. Ref. Data 1996, 25, 1509–1596. [Google Scholar] [CrossRef]
- Dillon, H.E.; Penoncello, S.G. A fundamental equation for calculation of the thermodynamic properties of ethanol. Int. J. Thermophys. 2004, 25, 321–335. [Google Scholar] [CrossRef]
- de Reuck, K.M.; Craven, R.J.B. International Thermodynamic Tables of the Fluid State–Vol. 12: Methanol; Blackwell Scientific Publications: Oxford, UK, 1993. [Google Scholar]
- Span, R.; Wagner, W. Equations of state for technical applications. II. Results for nonpolar fluids. Int. J. Thermophys. 2003, 24, 41–109. [Google Scholar] [CrossRef]
- Penoncello, S.G.; Shan, Z.; Jacobsen, R.T. A fundamental equation for the calculation of the thermodynamic properties of trifluoromethane (R-23). ASHRAE Trans. 2000, 106, 739–756. [Google Scholar]
- Tillner-Roth, R.; Baehr, H.D. An international standard equation of state for the thermodynamic properties of 1,1,1,2-tetrafluoroethane (HFC-134a) for temperatures from 170 K to 455 K at pressures up to 70 MPa. J. Phys. Chem. Ref. Data 1994, 23, 657–729. [Google Scholar] [CrossRef]
- Wagner, W.; Pruss, A. The IAPWS formulation 1995 for the thermodynamic properties of ordinary water substance for general and scientific use. J. Phys. Chem. Ref. Data 2002, 31, 387–535. [Google Scholar] [CrossRef]
- Lewis, W.C.M. Internal, molecular, or intrinsic pressure—A survey of the various expressions proposed for its determination. Trans. Faraday Soc. 1911, 7, 94–115. [Google Scholar] [CrossRef]
- Hildebrand, J.H. Solubility III. Relative values of internal pressures and their practical application. J. Am. Chem. Soc. 1919, 41, 1067–1080. [Google Scholar] [CrossRef]
- Dack, M.R.J. The importance of solvent internal pressure and cohesion to solution phenomena. Chem. Soc. Rev. 1975, 4, 211–229. [Google Scholar] [CrossRef]
- Marcus, Y. Internal pressure of liquids and solutions. Chem. Rev. 2013, 113, 6536–6551. [Google Scholar] [CrossRef] [PubMed]
- Chorążewski, M.; Postnikov, E.B.; Oster, K.; Polishuk, I. Thermodynamic properties of 1,2-dichloroethane and 1,2-dibromoethane under elevated pressures: Experimental results and predictions of a novel DIPPR-based version of FT-EoS, PC-SAFT, and CP-PC-SAFT. Ind. Eng. Chem. Res. 2015, 54, 9645–9656. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, M. Cohesive Energy Densities Versus Internal Pressures of Near and Supercritical Fluids. Molecules 2019, 24, 961. https://doi.org/10.3390/molecules24050961
Roth M. Cohesive Energy Densities Versus Internal Pressures of Near and Supercritical Fluids. Molecules. 2019; 24(5):961. https://doi.org/10.3390/molecules24050961
Chicago/Turabian StyleRoth, Michal. 2019. "Cohesive Energy Densities Versus Internal Pressures of Near and Supercritical Fluids" Molecules 24, no. 5: 961. https://doi.org/10.3390/molecules24050961
APA StyleRoth, M. (2019). Cohesive Energy Densities Versus Internal Pressures of Near and Supercritical Fluids. Molecules, 24(5), 961. https://doi.org/10.3390/molecules24050961