1. Introduction
The medical industry has begun to utilize active pharmaceutical ingredients (APIs) in a co-crystalline form to enhance the functionalities of their products [
1,
2,
3]. These solids are unique, as they contain two non-ionic components, an API and a coformer. The API and coformer can bind through van der Waals forces and non-covalent interactions such as hydrogen bonding, similar to biologics such as DNA and proteins [
4,
5]. Binding through non-ionic forces can enable more flexibility in crystalline engineering, removing the need to form an ionic bond. Understanding the intermolecular forces holding these co-crystals together can help to better understand, predict and control their physical properties. The creation of co-crystal APIs is a strategy that has shown to influence solubility and bioavailability [
6], thermal stability [
7], and has also found use in the control of product concentrations in the development of new types of therapeutics [
8].
To better understand the structures of co-crystals with APIs, researchers have utilized terahertz (THz) spectroscopy and low frequency Raman spectroscopy (LFRS). Both LFRS and THz spectroscopy probe frequencies between 3–300 cm
, where the prominent vibrational modes are related to the packing of the constituent organics in a crystal and the intermolecular forces that hold them in place [
9,
10]. These spectroscopic techniques have already proven useful for identifying different polymorphs that form when mixing multiple organic compounds [
10,
11,
12], for the in situ monitoring of crystallization and structural transformations [
13,
14], as well as uncovering solid state transition mechanisms [
15,
16]. THz spectroscopy is potentially a powerful tool in probing biological systems, because it is compatible with online measurements, remote sampling, and three-dimensional imaging [
17], although little work has been published on large complex systems beyond binary and tertiary co-crystals. Most THz research has focused on smaller organic-based systems (≈50–150 atoms/unit cell) where several groups have worked towards using theory and modeling to assign the vibrational modes found in both THz absorption and low frequency Raman spectroscopies [
10,
18,
19,
20].
An open question in the theory and modeling of materials is: “How closely can calculated properties match experimentally determined information?” This is especially difficult to answer when computing the measurable properties of a complex material with multiple components and potentially nonlinear behavior, or if changes in the chemical environment, such as the formation of hydrogen bonds or solvation, must be taken into account. The atomistic detail needed to address these issues can be gleaned from using methods based on quantum mechanics, specifically density functional theory (DFT). DFT methods have been around since the mid-1960s [
21,
22] and allowed for calculation of properties in the solid state for simple systems, such as the ordering and electronic structure of Si [
23] and ZnS semiconductors [
24]. Since the 1990s improvements in pseudopotential design and construction [
25,
26,
27,
28,
29,
30] and exchange-correlation functionals [
31,
32,
33], as well as the distribution of open source software packages [
34,
35] and the advent of highly parallelized computing have placed DFT methods at the forefront of accurate and reliable methods based in quantum mechanics.
How organic molecules pack into crystal structures is ultimately governed by intermolecular interaction patterns. While chemists can qualitatively identify systems in which, for example,
–
stacking, hydrogen bonding, and long-range dispersion forces occur, it is extremely difficult to predict from such an analysis what crystal structure will be preferred for a given organic molecule, either individually or with a co-former. Here, first-principles modeling based on DFT calculations becomes attractive as a method known to be reliable for modeling the structure and properties of inorganic crystal structure materials [
36,
37,
38,
39,
40,
41].
DFT calculations are upheld by materials chemistry as the gold-standard in quantum mechanical modeling owing to desirable balance between computational speed and accuracy. However, directly applying these methods to organic crystalline materials is complicated by the differing intermolecular forces active in these materials, noted above. While DFT is in principle an exact method, in practice the local density approximation, LDA (local density approximation) (and extensions to it, such as the generalized-gradient approximation, GGA) to the true, universal exchange-correlation functional are employed. In effect, this means that, while all non-relativistic electronic effects are included in the formal exact DFT functional, including van der Waals interactions, the long-range, non-local correlations that contribute to van der Waals forces are not accounted for in calculations that use LDA or GGA functionals. Solutions to this problem are complicated, as local effects are, by construction, included. Therefore, extending DFT approximations to include long-range forces is not as simple as just adding additional terms. Indeed, while such efforts to extend DFT came to the forefront in the early-2000s, the methods themselves [
42,
43,
44,
45,
46,
47], as well as best-practices for their applications [
48,
49], are still active areas of research.
Of the various approaches to extend DFT to include long-range electron correlation interactions, D2 (and more recently D3) dispersion coefficient methods, [
43,
46,
50], referred to as DFT-D, are popular based on the relatively simple mathematical form, improved predictions, and use of adjustable parameters. While it is beyond the scope of this manuscript to review the methods (those this has been done elsewhere, for example, Ref. [
51]), it can be stated briefly that DFT-D empirical parameters are determined by fits done to conformational and interaction energies computed using high-level quantum mechanical calculations. While in some implementations the fit parameters are adjustable, this is not universally the case.
Here, we present calculations that use open source DFT packages (that employ pseudopotentials and a planewave basis set and include dispersion corrections) to obtain the lattice parameters and vibrational modes of organic co-crystals and their constituent components. It is impossible to know which empirically tuned variant of DFT-D will offer the best performance for the organic co-crystals under study here. Given the lack of benchmarking information in the literature, we elect to test the performance of Grimme-D2 DFT-D calculations, compared to standard DFT-GGA. Our choice of Grimme-D2 DFT-D for this study is motivated by the fact that, in some implementations, the empirical parameter controlling the dispersion corrections is tunable by the user, enabling us to explore how its variation affects predictions of the bulk lattice constants. Future studies could go on to test other DFT-D functionals, and this would contribute to the developing understanding of which correction schemes offer optimal performance for organic crystals. We compare our DFT calculated results, both with and without dispersion corrections, to experimentally determined lattice constants and absorptions over THz frequencies. We use the information obtained with DFT to analyze the dielectric response of the systems studied here in order to elucidate trends in the types of functional groups present, crystal packing, and ultimately how one could use dielectric response as a tailorable property when designing and understanding complex organic co-crystal API formulations. We focus on identifying infrared (IR) active modes, their contributions to the overall dielectric response, and how closely they match the IR active modes measured in THz absorption spectroscopy. We use the work presented here to establish a baseline for future studies where recently developed methods to model organic solids could be used to improve upon the disagreements between theory and experimental endeavors.
2. Results and Discussion
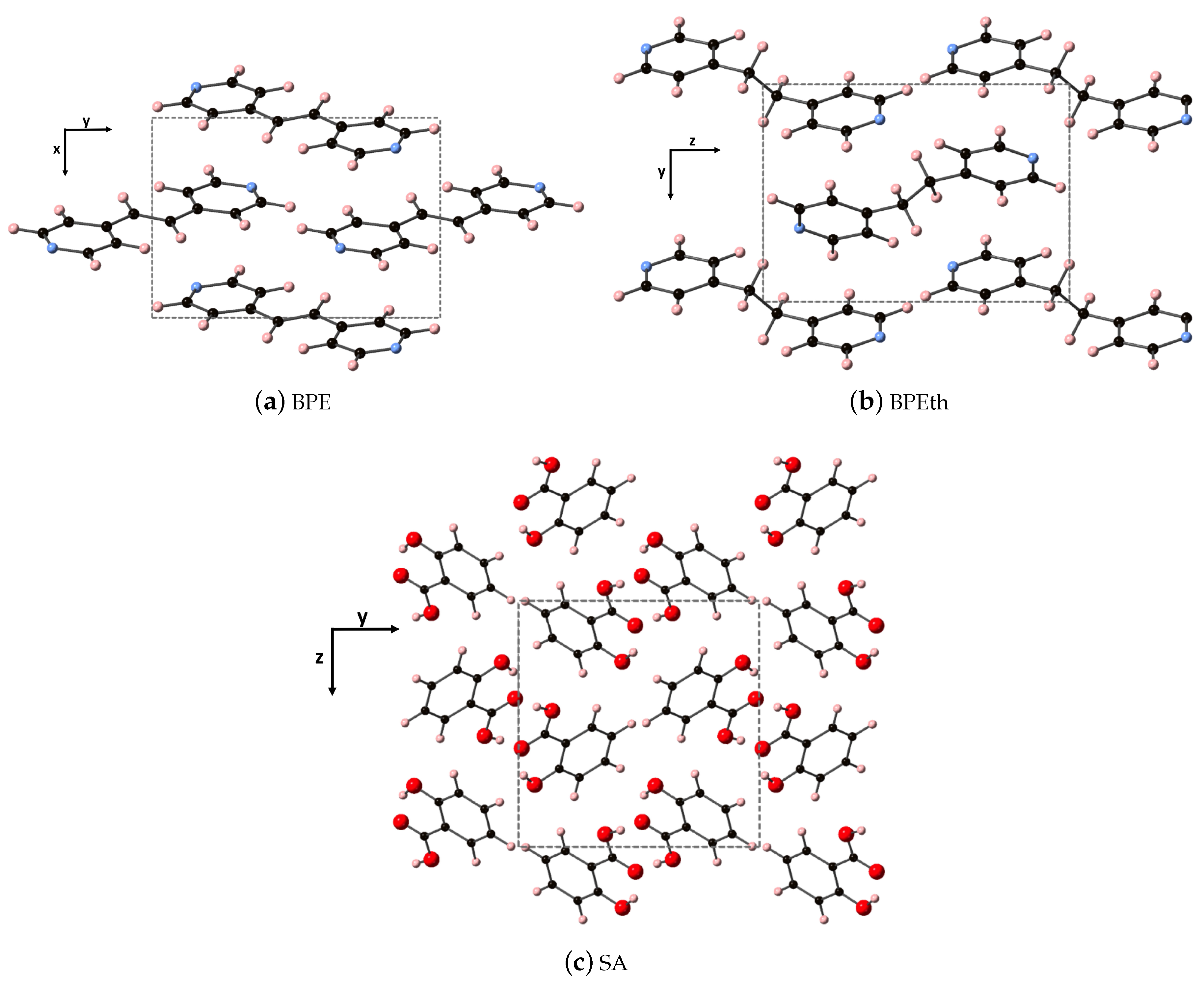
The fully relaxed monoclinic crystal structures of the components used to create the co-crystals (discussed in the next subsection) are depicted in
Figure 1. All three are indexed in space group 14, where all three have monoclinic angle
> 90
. Shown are the repeat units of 1,2-bis(4-pyridyl)ethylene (BPE), 1,2-bis(4-pyridyl)ethane (BPEth), and salicylic acid (SA) where the direction containing the greatest dispersion is marked by the vertical axis. The coformers BPE and BPEth differ in the functional group connecting the planar C
N ring units; inspection of
Figure 1a,b shows that BPE contains an alkene C
H
unit and BPEth contains an alkane C
H
unit, and that this influences the relative orientation of packing of these structures. In neither case is there a significant number of intermolecular (primary) hydrogen bonds, and a consequence of this is packing directions in which dispersion is significant.
Table 1 contains the computed crystallographic parameters of the fully relaxed systems.
a,
b, and
c are the lattice constants of the primitive unit cell, and
is the angle between
a and
c.
Inspection of
Table 1 shows that DFT without dispersion corrections overestimated BPE lattice constants
a and
c by ≈ 40 and 20 %, and BPEth lattice constant
b by ≈ 20%, relative to experiments. Inclusion of dispersion (DFT-D) reduces these errors to ≈−7 and −3 %, and ≈−4 %, for BPE and BPEth, respectively. Another error that is not typically reported in DFT studies is that of
, the angle between
a and
c. For the monoclinic BPE unit cell, DFT overestimates this angle by over 25%, and this is because the errors in
a and
c are so large. DFT-D decreases the error in
to ≈−2.5%. The experimentally determined lattice parameters for BPE [
52], BPEth [
53], and SA [
54] are given in the parenthesis of
Table 1, followed by the % deviation of each lattice parameter from experiments. The experimental structures in Refs. [
52,
53,
54] were determined using X-ray diffraction at room temperature.
The SA units in
Figure 1c display a packing that is influenced by hydrogen bonding between the carboxyls of each unit (mainly along
y, but also present in
x) and this results in a network of packed dimeric units where dispersion is greatest along
z. DFT overestimates the lattice constants
a and
c by ≈ 6 and 32%, and this is improved using DFT-D; the difference between DFT-D computed lattice constants and experiment is reduced to −0.8 and −0.2% for
a and
c, respectively. Much like BPE, DFT-D also improves the agreement between computed and experimentally determined angle
; inclusion of dispersion changes
from 75.39
to 93.54
, more in line with experiment.
Beyond agreement of lattice parameters, inclusion of dispersion also affects the vibrational modes of a material. We now focus on comparing DFT and DFT-D computed vibrational modes, as well as their relative contributions to the dielectric constant,
, for each of the three co-crystal components, BPE, BPEth, and SA.
Table 2 reports the isotropically averaged values of
for DFT and DFT-D, compared to the experimentally determined values. We find that the DFT-D values are closer to experiment than DFT values, where for BPE and BPEth DFT underestimates
by −22 and −14%, respectively, while for DFT-D the values are overestimated by +14 and 4%. The opposite trend is observed for SA, where DFT overestimates
by −20%, and DFT-D underestimates it by 10%. Both experiment and DFT-D show that
increases in the order BPE > BPEth > SA.
We now turn to an analysis of the frequencies and oscillator strength of the DFT and DFT-D computed vibrational modes that govern . Details of how to compute from first-principles using this information are given in the Materials and Methods subsection called Computational Details.
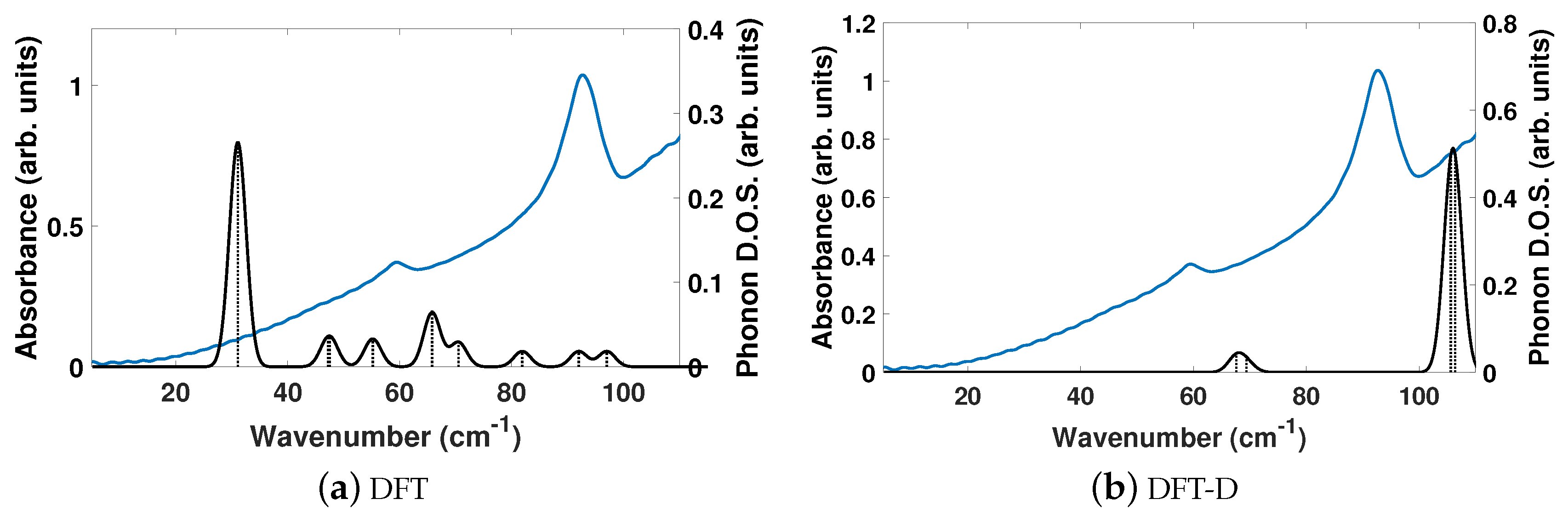
The experimentally determined THz spectra of BPE contains two peaks centered at 59.4 and 92.73 cm
, as depicted by the blue curves in
Figure 2a,b.
Figure 2 is a comparison of the vibrational modes determined by an analysis of the dielectric response, as discussed in the Methodology section, between DFT (a) and DFT-D (b). We find that, in the range of 20–110 cm
, DFT predicts nine total IR active modes with a contribution to
of more than 0.01, and that the peak heights (scaled by contribution to
in this region) are very much not in alignment.
Table 3 shows that the largest contribution to
in DFT is a mode at 31.1 cm
, which accounts for more than 50% of the total response along
y. This is not the case using DFT-D, which predicts four total IR active modes with a contribution larger than 0.01 to
in the range of 20–110 cm
. These four peaks occur as two sets of peaks at 67.6/69.5 cm
and 105.6/106.4 cm
, where they are within 1–2 cm
of each other and most likely not resolvable in experiments.
Figure 2b shows that DFT-D with an
= 0.75 obtains two sets of modes that are shifted by +10 and +13 cm
from the experimentally determined spectra.
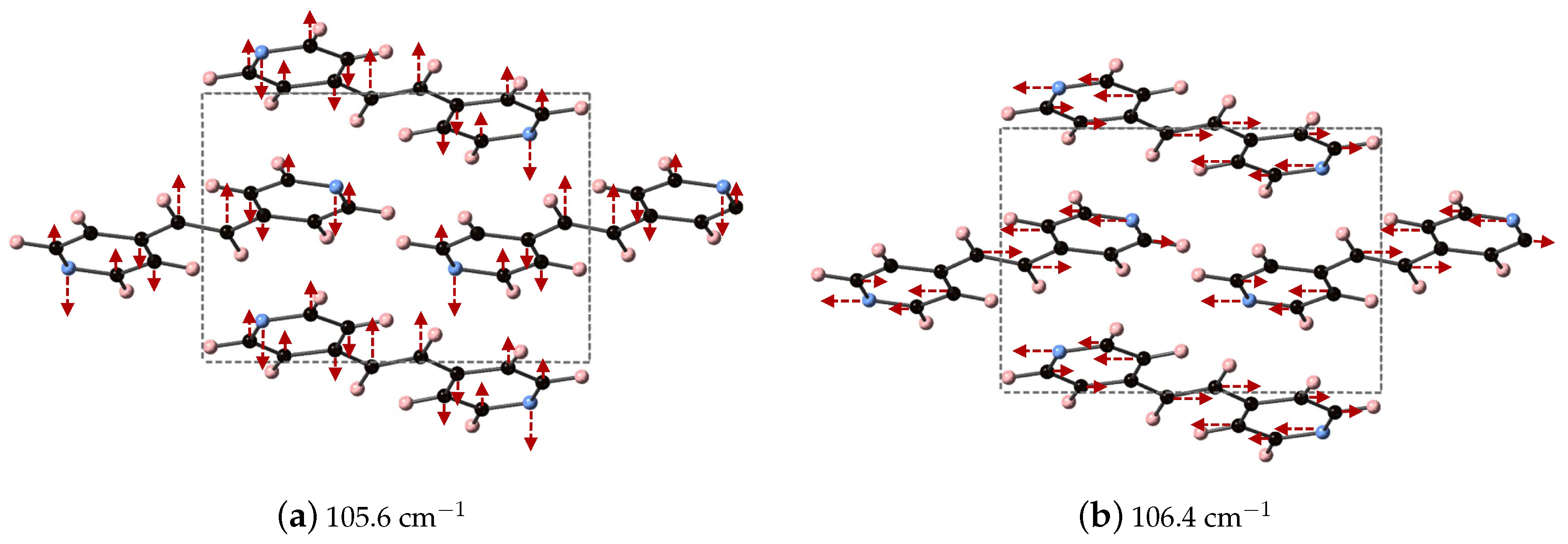
We are able to differentiate the peaks computed by DFT-D into contributions to the
x (
a),
y (
b), and
z (
c) directions of
, as shown in
Table 3 and focus on the modes at 105.6 and 106.4 cm
, which contribute ≈ 33 and 50% of the overall response in
x- and
y-directions, respectively. These vibrational modes are depicted in
Figure 3 and can be described as asymmetric stretches where the largest displacements are localized on nitrogen and the carbon in the C
H
unit, where these atoms displace in opposite directions.
While it is helpful to separate the components of by direction for an analysis of the vibrational modes, most experiments will report an isotropically averaged value, as discussed in the Methodology section. The DFT and DFT-D computed isotropically averaged (ionic response) of BPE are 0.27 and 0.29, while the directionally averaged (electronic response) are 2.41 and 3.63. Inclusion of dispersion in DFT-D has increased by 7.4% and by 50.6%. Summed together, the averaged components of for BPE using DFT and DFT-D are 2.68 and 3.92, respectively.
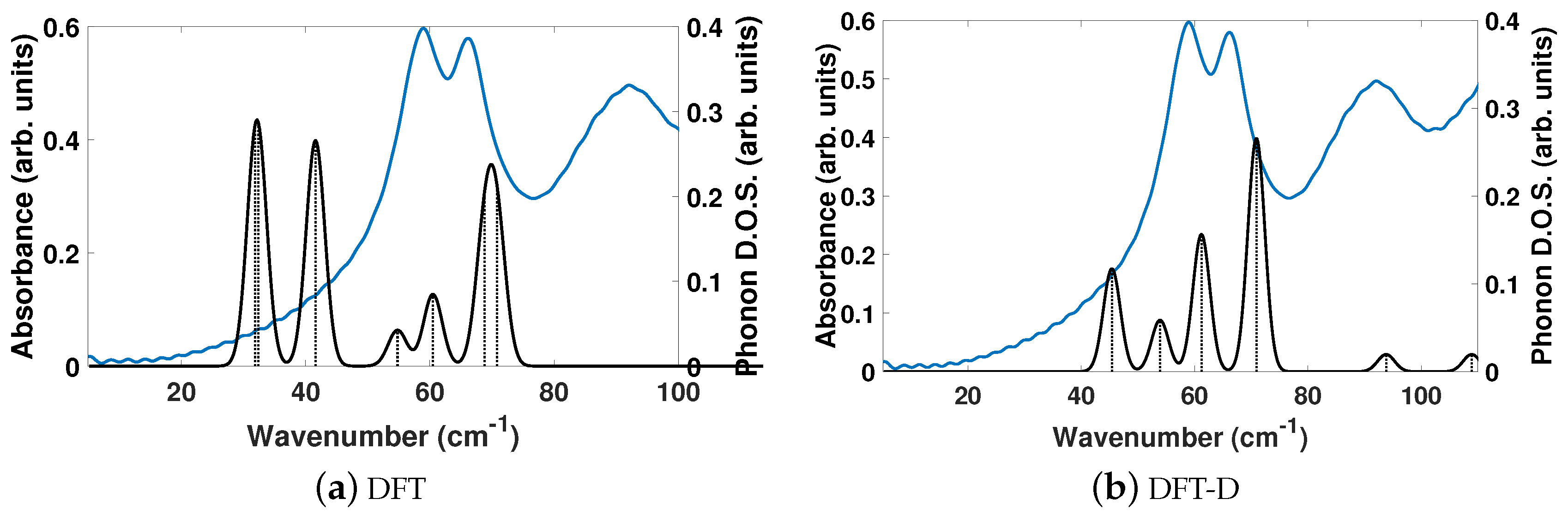
The experimentally determined THz spectra of BPEth contains three peaks centered at 58.99, 66.26, and 91.92 cm
, as depicted by the blue curves in
Figure 4a,b.
Figure 4 is a comparison of the vibrational modes determined by an analysis of the dielectric response, as discussed in the Methodology section, between DFT (a) and DFT-D (b). We find that in the range of 20–110 cm
, DFT predicts seven total IR active modes with a contribution to
of more than 0.01, and that the peak heights (scaled by contribution to
in this region) are not in alignment, much like for BPE.
Table 4 shows that the largest contributions to
in DFT are three modes below 42 cm
(at 31.8, 32.5, and 41.6 cm
) and two modes at 68.8 and 70.8 cm
, where all five vibrational modes have a comparable contribution to
. Inclusion of dispersion in DFT-D decreases the number of IR active vibrational modes to six and changes the distribution to higher frequencies, as well as their respective contribution to
. This case is depicted in
Figure 4b. For example, the lowest frequency peak is at 45.4 cm
, and the modes with the largest contribution to
occur at 61.2 and 70.9 cm
, in line with the experimentally determined spectra. Also of note in
Figure 4b is the appearance of two peaks at 93.8 and 108.9 cm
in the DFT-D computed spectra, which were absent in the DFT calculation without dispersion, and coincidental with peaks in the experimentally determined THz spectra. While the DFT-D spectra of BPEth has more inconsistencies when compared to the experimental THz spectra than BPE, the frequency distribution and contribution is better than when using only DFT.
The vibrational modes of BPEth at 61.2 and 70.9 cm
, which are the two largest contributors to
in the
y- and
x-directions, respectively, are shown in
Figure 5. The vibrational mode at 61.2 cm
can be described as an asymmetric stretch in which the largest displacements are localized on the nitrogen in the aromatic ring and the carbon in the C
H
unit connected the two aromatic rings. Unlike the mode identified for BPE, the motion of these atoms are in the same direction, and not opposed. The vibrational mode at 70.9 cm
has displacements in the
-direction and can be described as an asymmetric stretching mode mixed with some aromatic ring rotation. In this mode, some of the aromatic carbon displace just as far as the nitrogen and the carbon in the C
H
unit.
The DFT and DFT-D computed isotropically averaged (ionic response) of BPEth are 0.32 and 0.27, while the directionally averaged (electronic response) are 2.36 and 2.98. Inclusion of dispersion in DFT-D has decreased by 15.6% and increased by 26.3%. Summed together, the averaged components of for BPEth using DFT and DFT-D are 2.68 and 3.25, respectively, comparable to the values of 2.68 and 3.92 for BPE.
Turning from the nitrogen-containing heterocycles BPE and BPEth to the carboxylic acid component of the co-crystals, we investigate the dielectric response of salicylic acid. As depicted in
Figure 1c, the unit cell for SA has hydrogen bonding that takes place primarily along the
y-axis, and this manifests as an increased contribution to
along
y relative to the other two axes for both DFT and DFT-D.
Table 5 shows that for DFT the
y component of
is 1.01, while for
x and
z the sums are 0.79, and 0.44, respectively. Adding dispersion changes, the magnitudes of these three values, but not the trend; the trend in
is still
y >
x >
z.
Figure 6 shows that neither DFT or DFT-D computed spectra are a good match to the experimentally determined THz spectra of SA. The seven experimentally determined THz peaks of SA are centered at 36.8, 46.1, 53.5, 60.0, 68.9, 77.0, and 89.3 cm
in the range of 20–110 cm
. DFT computes too many vibrational modes at lower frequencies (30–50 cm
), resulting in too many total modes, and no modes above 80 cm
. DFT-D has relative peaks whose contributions to
are not aligned to the experimental structure, but computed the correct number of absorption features. If we compare them to the seven DFT-D computed peaks at 37.9, 49.7, 66.5, 76.1, 78.4, 92.3, and 100.2 cm
, then the differences in frequency are 1.1, 3.6, 13.0, 16.1, 9.5, 15.3, 10.9 cm
going from 20 to 110 cm
.
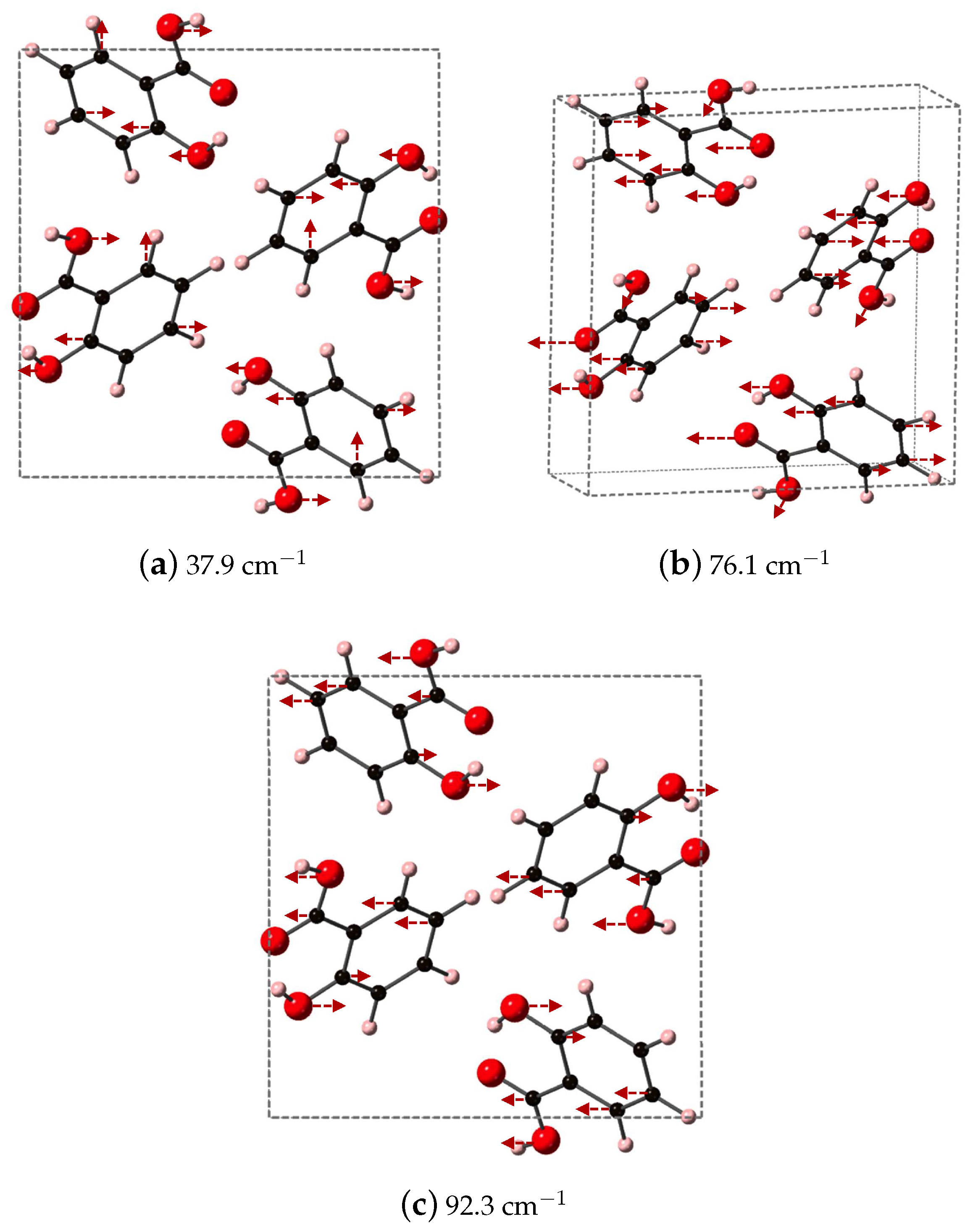
We identify the three vibrational modes of SA that contribute a majority of the dielectric response, which occur at 37.9, 76.1, and 92.3 cm
and are shown in
Figure 7. All three modes have significant contributions from the oxygen atoms of both the phenolic OH and COOH acid functional groups. At 37.9 cm
, the OH of the COOH acid group displaces along
y, opposite to OH on the aromatic ring, while the carbon in the aromatic ring breathes in and out. Higher in frequency, at 76.1 cm
, the oxygen of the COOH carbonyl and the OH on the aromatic ring displace in the same direction along
while carbon in the aromatic ring undergo asymmetric stretches. The highest frequency in
Figure 7, 92.3
, has the OH of the COOH acid group moving along
y, opposed to the OH on the aromatic ring. In this vibrational mode, the carbon of CO functional group also moves, and overall the mode represents a partial rotation of SA.
For SA, we find that the difference between DFT and DFT-D computed contributions to are opposite that of BPE and BPEth; both and decrease significantly with the addition of dispersion. decreases from 0.75 to 0.51, and decreases from 2.95 to 2.28. This means that the overall averaged goes from 3.70 to 2.79, a decrease of 24.6% when including dispersion corrections. The directionally averaged value of (0.51) is ≈20% of the DFT-D calculated dielectric response.
In general, all three comparisons between DFT and DFT-D in
Figure 2,
Figure 4, and
Figure 6 show that DFT will yield high-intensity, low frequency modes in the range of ≈25–40 cm
whose intensity and frequency change with the application of dispersion corrections in DFT-D. While the agreement between DFT-D and experimentally determined THz spectra is better for BPE than for BPEth and SA, DFT-D does yield results in which the three components are differentiable; BPE has IR active modes at 106 cm
, BPEth has modes in the range of 60–70 cm
, and SA has modes that span the range of 40–90 cm
, where the modes at 38, 76, and 92 cm
all involve displacements of the oxygen in both the OH and COOH functional groups. The numerical values of dielectric response of BPE and BPEth are more similar to each other than to SA, and this may be because the only difference between them is the alkene (C
H
) vs alkane (C
H
) connection between heterocyclic rings. The contributions to
are greatest for SA, which may be explained by the increased hydrogen bonding of SA relative to BPE and BPEth.
2.1. Co-Crystals
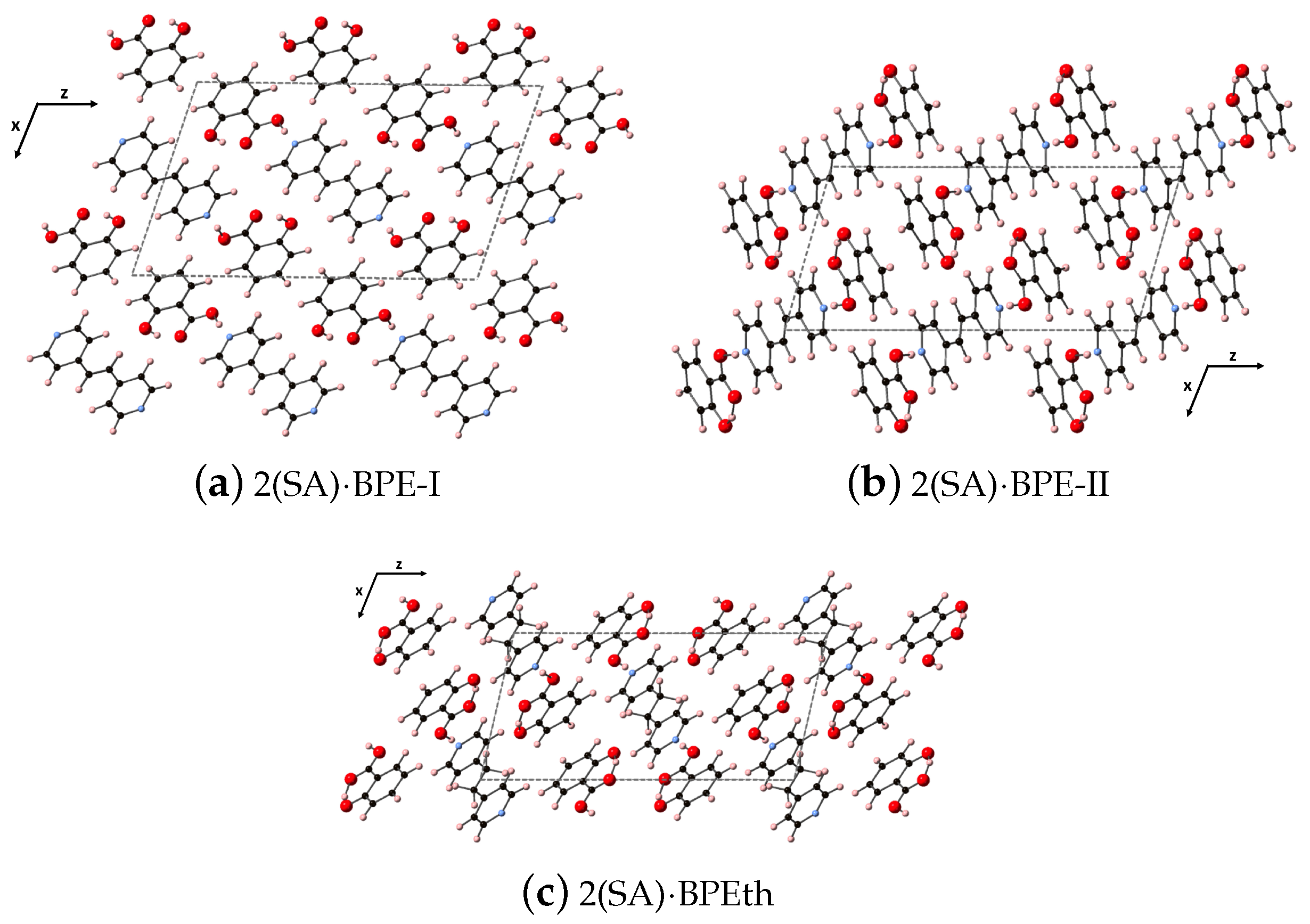
Here, we perform the same types of analyses as in the previous section but for the larger co-crystal systems 2(SA)·BPE Forms I and II, and 2(SA)·BPEth. As will be discussed elsewhere, all three form monoclinic crystal structures (space group 14), but 2(SA)·BPE-II and 2(SA)·BPEth display packing similar to each other, while 2(SA)·BPE-I is different. The synthesis and characterization of these three co-crystals systems will be described elsewhere. While all three co-crystal structures display hydrogen bonding between the carboxyl unit of SA and the nitrogen of BPE and BPEth, 2(SA)·BPE-II and 2(SA)·BPEth also have the H of the aromatic OH of SA in close proximity to the carbonyl O of the COO unit creating a stable six member ring via resonance. This additional hybridization is absent in 2(SA)·BPE-I. The DFT-D relaxed structures are shown in
Figure 8 for (a) 2(SA)·BPE-I, (b) 2(SA)·BPE-II and (c) 2(SA)·BPEth, looking down the
y-axis at the
plane.
We relax the co-crystal compositions using both DFT and DFT-D, and the lattice parameters of these relaxations are tabulated in
Table 6. Much like the components BPE, BPEth, and SA discussed in the previous section of the Results, relaxations using DFT yield deviations from experimentally determined structures on the order of 5–25% overestimation, and the largest errors coincide with the directions where dispersion will be the greatest. This is observed for lattice constant
a of 2(SA)·BPE-I and lattice constant
c of 2(SA)·BPE-II and 2(SA)·BPEth. Inclusion of dispersion corrections in DFT-D decreases the error of the lattice parameters, in most cases to within the 1–2% acceptable for DFT-GGA calculations in the solid state, but DFT-D underestimates lattice constant
c of 2(SA)·BPE-II and 2(SA)·BPEth by ≈ 8%.
As the disagreement between experimentally determined and DFT-computed lattice parameters with no dispersion correction is significantly large, and we showed earlier that DFT-D computed vibrational modes of the components were needed for a better description of the THz spectral features, we compute the vibrational modes of the co-crystals using DFT-D.
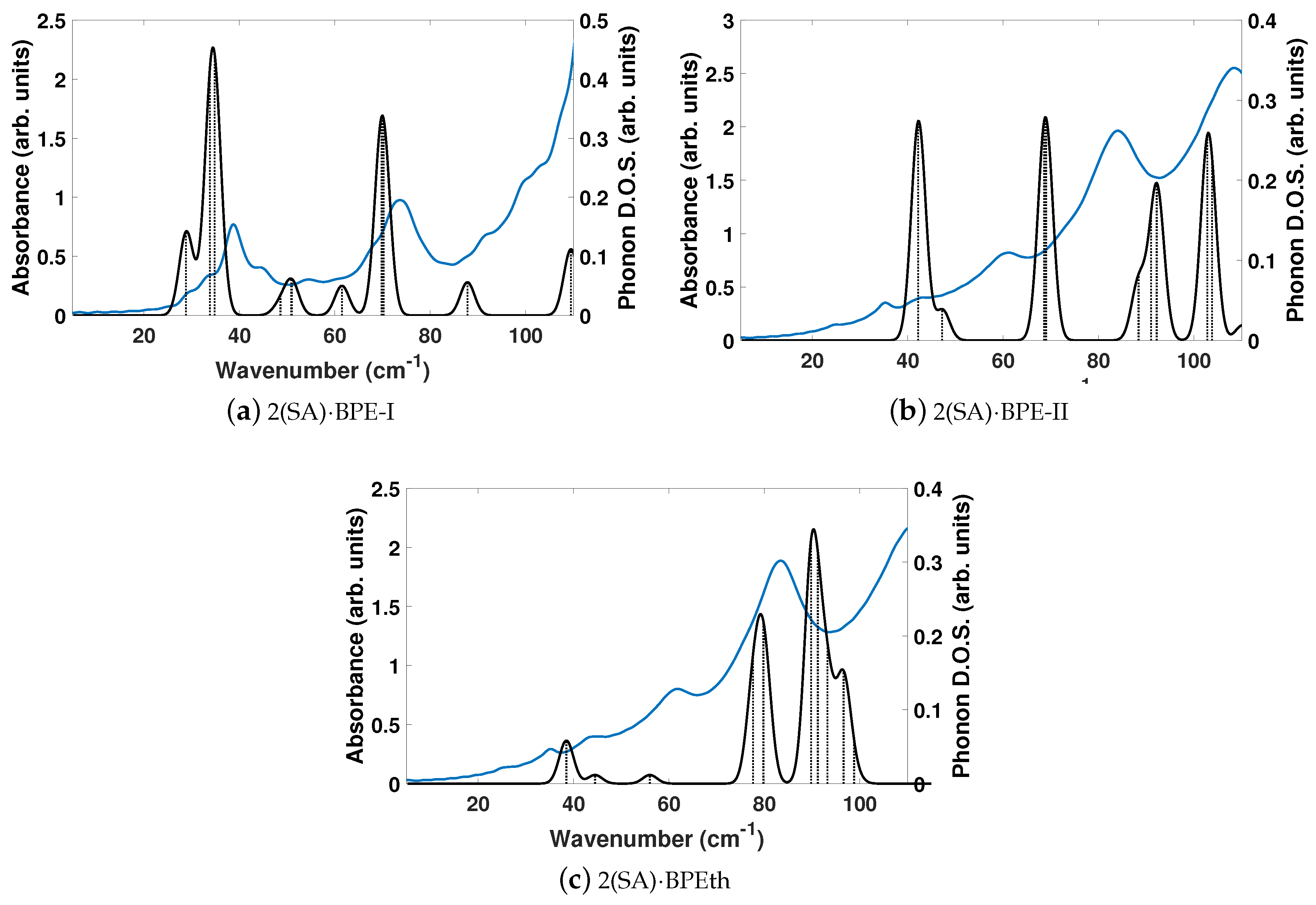
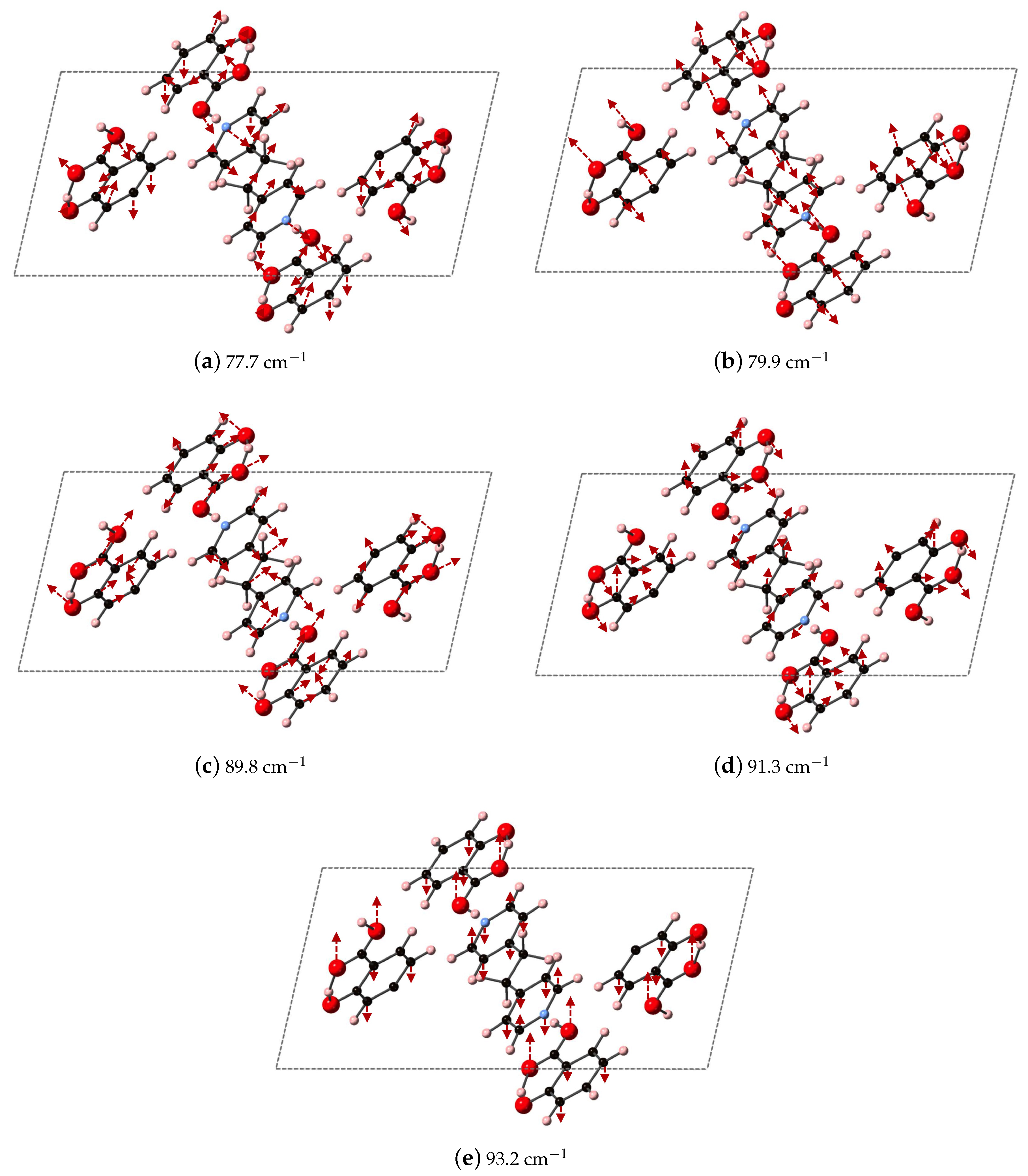
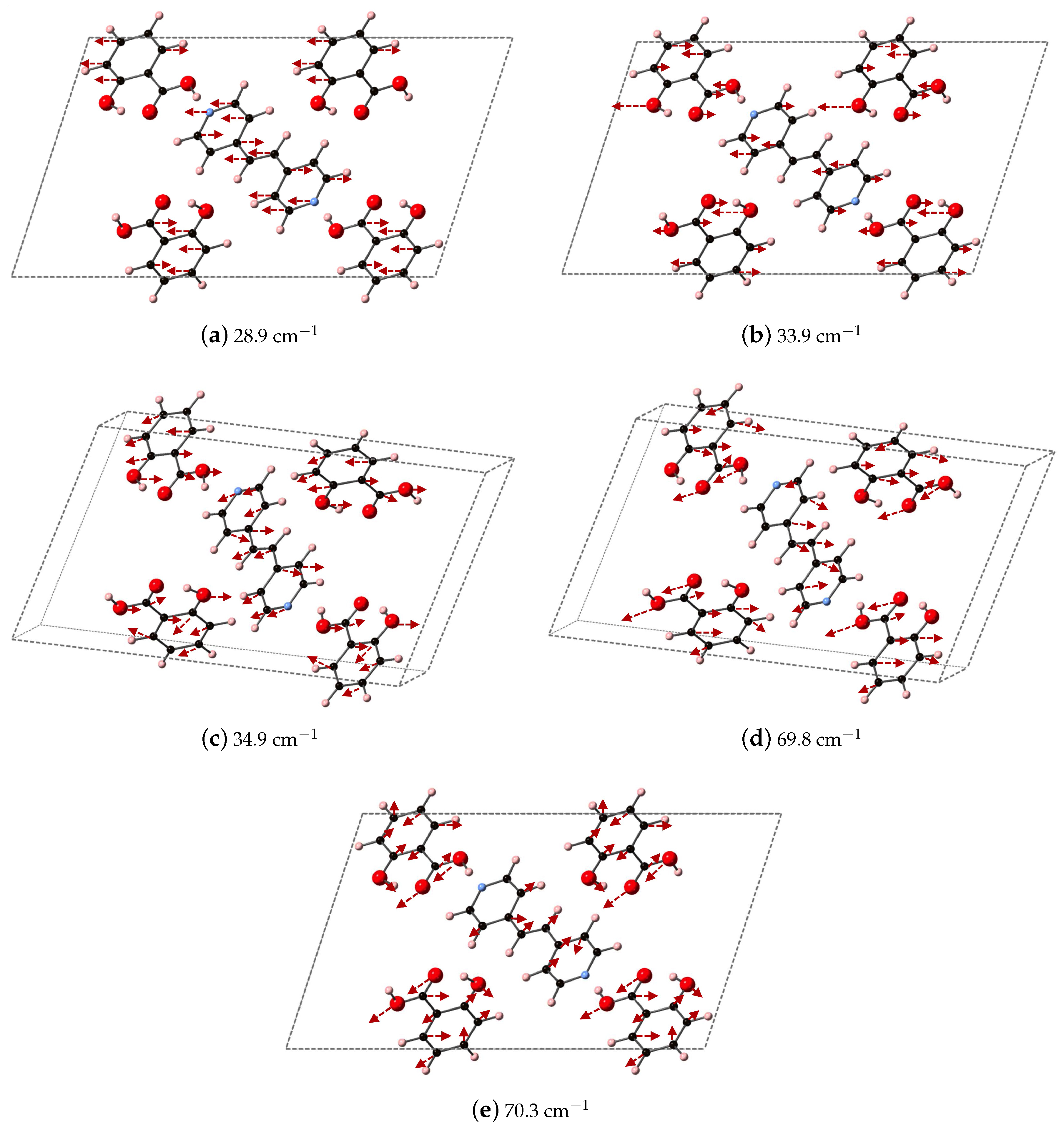
Figure 9 depicts the DFT-D computed vibrational modes of the co-crystals, compared to experimentally determined THz spectra. The comparison of vibrational modes in
Figure 9a shows that DFT-D predicts low frequency modes for 2(SA)·BPE-I (below 40 cm
) close to experiment. Experiment shows ten THz peaks of 2(SA)·BPE-I centered at 29.5, 33.5, 38.8, 44.9, 54.6, 68.1, 73.7, 92.7, 99.6, and 102.4 cm
, compared to DFT-D which predicts IR active modes at 28.9, 33.9, 34.9, 48.5, 50.9/51.1, 61.5, 69.8/70.3, 87.8, 109.5 cm
. The smallest difference in frequencies between THz spectroscopy and DFT-D is on the order of 1–2 cm
and the largest difference in frequencies is on the order of 10 cm
. The minor deviations in vibrational mode frequencies, from the experimentally determined values, may be related to the close agreement between experimentally determined and DFT-D computed lattice parameters, which are on the order of ± 1.5%. The experimentally determined THz spectra of 2(SA)·BPE-II (
Figure 9b) and 2(SA)·BPEth (
Figure 9c) are similar with frequencies centered at 35.2, 43.6, 61.6, 83.4, and 110.7 cm
for 2(SA)·BPEth and at 24.9, 35.4, 42.8, 60.8, 84.0, and 108.7 cm
for 2(SA)·BPE-II. The experimentally determined frequencies are listed in
Table 7.
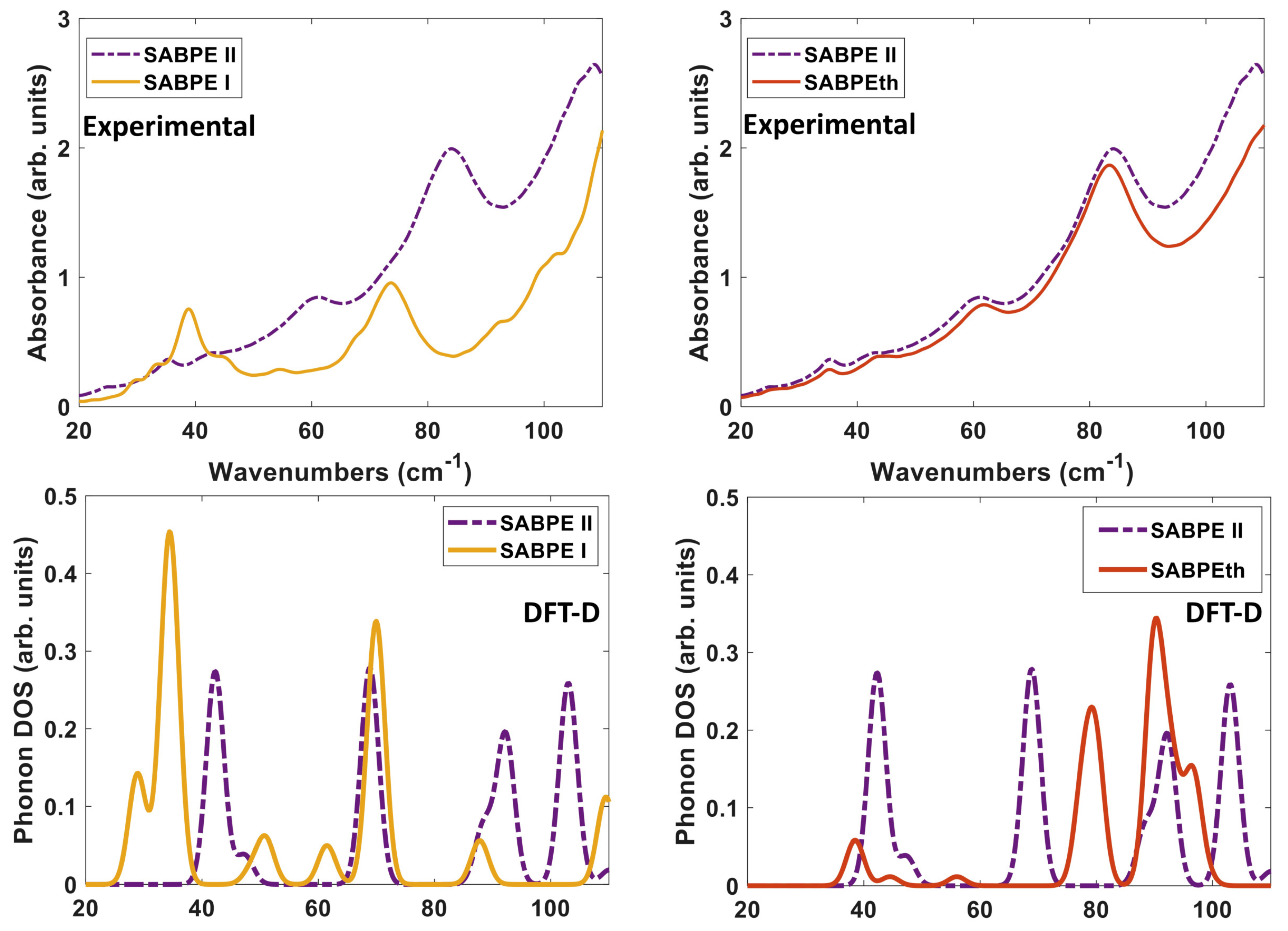
We compare both the experimentally determined and DFT-D computed spectra of the three co-crystals to each other in
Figure 10. When comparing the experimental spectra (top of
Figure 10), it seems that orientation and packing effects dominate the THz moreso than the difference in ligand identity as BPE and BPEth. The comparison of 2(SA)·BPE-I and 2(SA)·BPE-II in
Figure 10 (left) highlights the difference between co-crystals with the same components, but different crystal packing and, thus, different lattice parameters. The experimental spectra show coincidental peaks only at ≈35 and 43 cm
, while the DFT-D do not coincide at low frequency (30–60 cm
), coincide in the range of 70–90 cm
, and begin to differ again above 90 cm
.
For 2(SA)·BPE-II and 2(SA)·BPEth, the DFT-D calculated IR active modes are consistently shifted higher, at least 5–8 cm
than experiments, if they match up at all. This mismatch between the THz and DFT-D for 2(SA)·BPEth and 2(SA)·BPE-II co-crystals may be caused by the ≈ 8% underestimation of lattice constant
c.
Figure 10 (right) shows that the experimental THz spectra of 2(SA)·BPE-II and 2(SA)·BPEth coincide at all frequencies above 30 cm
and that for the DFT-D computed spectra they (roughly) coincide at low frequencies (30–60 cm
), do not coincide in the range of 70–90 cm
, and begin to coincide again above 90 cm
.
We compare the three ranges of frequencies computed via DFT-D to determine if our methods could yield spectral resolution that goes beyond the resolution capabilities of experimental THz data collection at room temperature, and our analysis points towards characteristic frequency ranges for these co-crystal systems that may indicate where differences in packing vs. differences in bipyridyl coformer (as ligand identity) will dictate vibrational modes. We find that packing arrangement may dominate between 30–60 cm
, ligand substitutions may dominate in the range of 70–90 cm
, and then packing may dominate again above 90 cm
. Example vibrational modes computed using DFT-D in these three regions are shown in
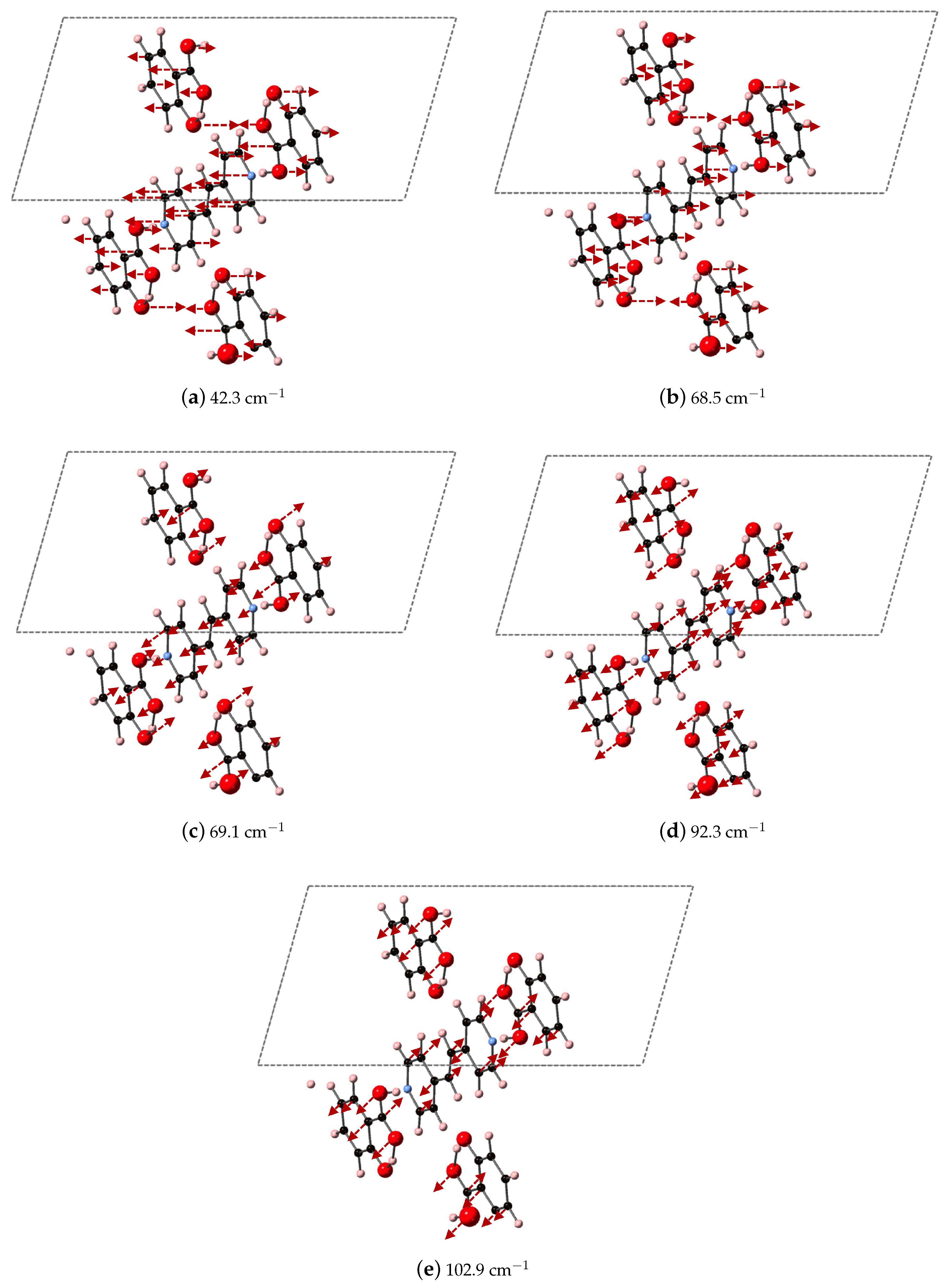
Figure 11,
Figure 12 and
Figure 13 for 2(SA)·BPEth, 2(SA)·BPE-I, and 2(SA)·BPE-II, respectively. The differences between the the experimentally determined and DFT-D computed vibrational modes motivate the need for further investigations that could include low temperature THz spectra collection and methodology that goes beyond the Grimme D2 formalism used here, which will be discussed in the next section.
The DFT-D calculated contributions to
for 2(SA)·BPEth are tabulated in
Table 8 and
Table 9 for 2(SA)·BPE polymorphs I and II. The directionally averaged values of
and
for 2(SA)·BPEth are 1.14 and 2.97, 1.48 and 3.05 for 2(SA)·BPE-I, and 0.81 and 3.33 for 2(SA)·BPE-II, and when added together yield
that are 4.11, 4.53, and 4.14, respectively.
for the co-crystals is 20–30% of the total dielectric response, an increase when compared to the behaviors of their components, such as SA (18%) and BPEth (8%). The isotropically averaged values of
for the three co-crystals are tabulated in
Table 10, compared to the experimentally determined values. We find that all three DFT-D computed
are underestimated compared to experiments, and that, unlike the
of the components, the trends in magnitude of
do not agree between experiments and DFT-D. Moreover, the DFT-D calculated values are closer to each other than those measured experimentally.
The overall dielectric responses of the co-crystals and their components also demonstrate that intermolecular interactions, highly dependent on packing orientation, result in a dielectric response that is not merely an average of the components. For example, the directionally averaged values of for SA and BPE are 0.51 and 0.29, respectively, and an average of those two numbers is 0.40. The values for 2(SA)·BPE-I and 2(SA)·BPE-II are 1.48 and 0.81, which is more than even when the are added together (0.79). This implies a tunable, nonlinear dielectric response can be achieved, and potentially optimized, in co-crystal formulations.
Analysis of the vibrational modes of the co-crystals show that all modes in the range of 20–110 cm
have significant contributions from both the nitrogen containing heterocycles BPE and BPEth, and SA. A majority of the largest displacements in each mode above 40 cm
is localized on both of the O atoms of the COOH acid, and the phenolic OH of SA, and in many cases the nitrogen of the heterocyclic ring as well. Below 40 cm
, such as the modes of 2(SA)·BPE-I in
Figure 12, O motion contributes less than the motions present in the heterocyclic ring. Vibrational modes with frequencies higher than 90 cm
, such as the modes of 2(SA)·BPE-II in
Figure 13 also tend to have larger carbon displacements than modes at lower frequencies.
2.2. Improving Agreement with Experiment
In the two previous sections of the Results and Discussion, we showed that using DFT-D to compute the lattice parameters and vibrational spectra of the 2(SA)·BPE and 2(SA)·BPEth co-crystals, as well as their components, results in a marked improvement over the same parameters computed using DFT. There still remains some inconsistencies between DFT-D computed spectra and experimentally determined lattice parameters and THz spectra, such as 8% lattice constant underestimation for 2(SA)·BPE-II and 2(SA)·BPEth and the upshift in frequency for the IR active modes of BPE.
One method to improve agreement between experimental THz spectra and DFT-D modeling is to adjust the global dispersion scaling factor,
, from the default value of 0.75. Systematically changing
results in changes in lattice parameters and frequencies of IR-active modes, where better agreement can be reached between modeling efforts and THz spectra [
4,
18,
19,
55]. Here, we turn to another open source planewave DFT code in which varying
is allowed in the input file. We use Quantum Espresso [
35] and the GBRV (Garrity-Bennett-Rabe-Vanderbilt) [
29] set of ultrasoft pseudopotentials [
27] to map out how changing the global dispersion correction will affect lattice parameters for BPE, BPEth, SA, and 2(SA)·BPEth. We keep the
k-grid sampling and energy convergence criteria the same as for the calculations employing ABINIT [
34] and the ONCV-type (optimized norm-conserving Vanderbilt) of pseudopotential [
28].
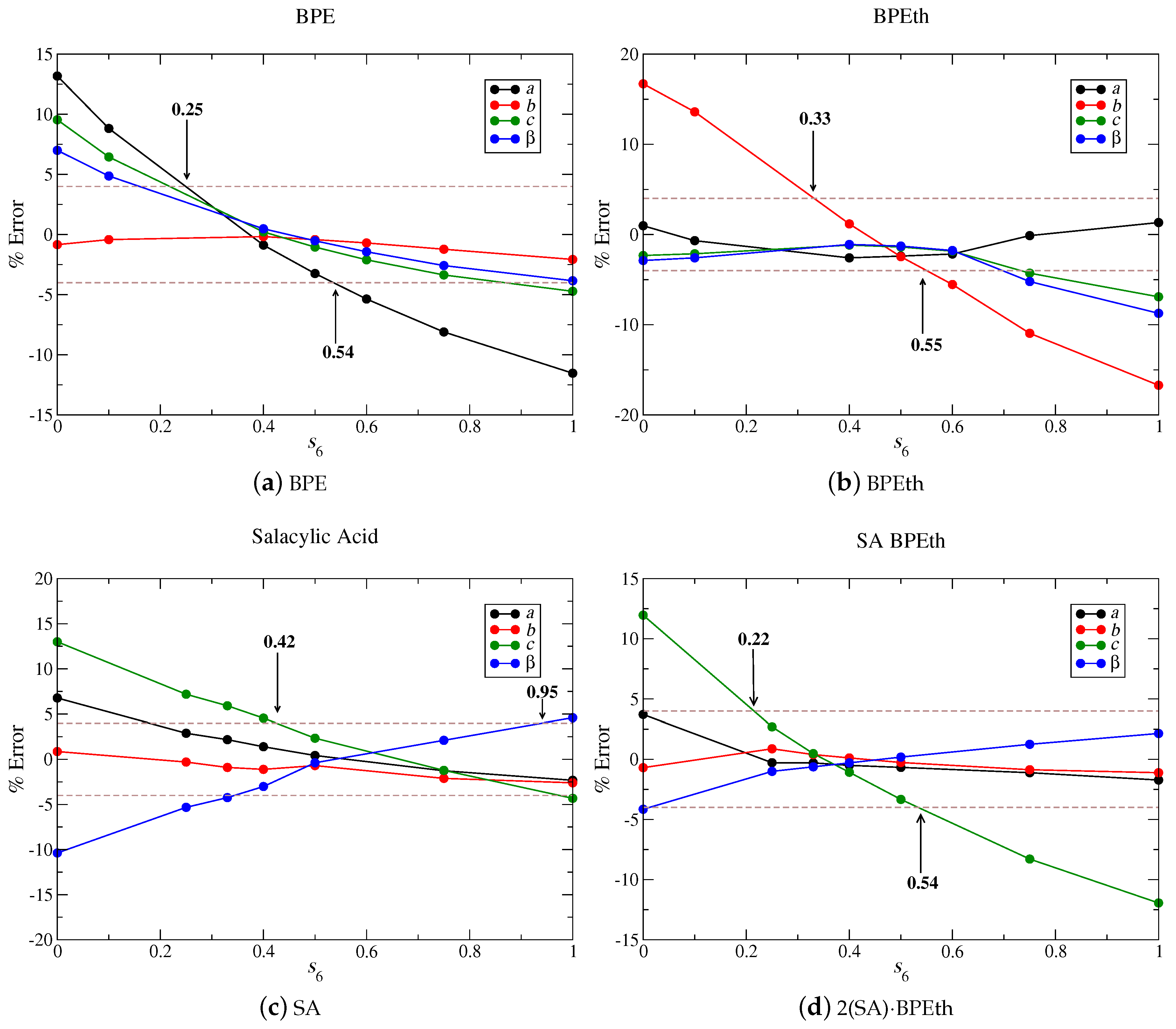
Figure 14 shows that
values of 0.4, 0.5, 0.6, and 0.35 for BPE (a), BPEth (b), SA (c), and 2(SA)·BPEth (d) independently minimize the percent error when lattice parameters are compared to experiment. If we increase the maximum % error to ± 4%, then we obtain ranges of
values of 0.25–0.54 for BPE, 0.33–0.55 for BPEth, 0.42–0.95 for SA, and 0.22–0.54 for 2(SA)·BPEth. In all cases, but SA, an
= 0.75 is not present in any of these ranges. Using these four test cases as an example, we obtain a range of 0.42 <
< 0.54, which is in line with the values used in previous studies of organic co-crystals [
4,
18,
19,
55].
4. Conclusions
The comparison of DFT to DFT-D for the organic compounds BPE, BPEth, and SA shows that the inclusion of dispersion using the semiempirical Grimme-D2 dispersion correction will yield lattice parameters and vibrational modes whose values are closer to those measured in experiment. We find that the agreement between DFT-D computed vibrational modes and THz spectra was best for BPE and worst for SA. As pointed out in the final subsection of the Results and Discussion, this may be caused by using an
global scaling factor that was too large, 0.75 vs. ≈0.50, or could also be because of the increased amount of hydrogen bonding in the SA crystal structure. With regards to the co-crystals 2(SA)·BPEth and 2(SA)·BPE forms I and II, DFT-D did pick out differentiable trends between the systems, but the lattice constant disagreement (for 2(SA)·BPEth and 2(SA)·BPE-II) between theory and experiment was still too large to be able to assign specific vibrational features to the THz spectra. Moreover, the experimental spectra showed that atomic packing and overall crystal lattice structure dominated the THz region moreso than changes in ligand identity as BPE vs. BPEth, while DFT-D showed that both atomic packing and ligand identity led to differentiable regions where each tended to dominate. One way to obtain more information about the DFT-D computed vibrational modes, and to match more closely to experiments, would be to use what is presented here as input for molecular dynamics calculations that take into account effects such as temperature or by quasi-harmonic DFT approximations [
69] that map out the effects of temperature and volume changes on solid-state properties related to low frequency vibrations [
70,
71,
72].
Our study determines the optimal ranges of for the set of materials under investigation and we are able to discern differences in that may be correlated to crystal structure/property relations and be used as a guide for further investigations. Organic heterocycles like BPE and BPEth with – stacking interactions can be optimized with ≈ 0.3–0.5, and organics that demonstrate significant H-bonding, like SA, can be optimized with ≈ 0.4–0.9. Comparing the of the combination, these two different types of bonding schemes (to create co-crystals) also yield insights; the lattice parameters of 2(SA)·BPEth co-crystal are closest to experiments when approaches the values used for organic materials with – stacking and not those with significant hydrogen-bonding. This suggests that, in complex materials, containing multiple functional groups or packing behavior, at least two or three different should be used for preliminary structural optimizations. Determining which range of may be best will allow for a better understanding of the relative forces that stabilize a complex structure.
Our analysis of the dielectric response of BPE, BPEth, and SA shows that functional group identity, degree of hydrogen bonding, and crystal packing/arrangement will influence the dielectric constant, . This work could be extended to analyze and compare the effects of fluoride, cyano, aldehyde, and amide substituents on aromatic heterocycles similar to what is presented here, or on different types of heterocycles (pyrazine, imidazole, furan, thiophene) and 3D bonding architectures (fullerenes, substituted adamantanes) on dielectric behavior, to be used as a design parameter for co-crystal APIs.
Here, we establish a baseline of DFT computed dielectric response and THz spectroscopy for the organic co-crystals 2(SA)·BPEth and 2(SA)·BPE forms I and II, as well as their constituent components, using dispersion corrected planewave DFT methods that employ the GBRV set of ultrasoft pseudopotentials. Further development of the work presented here could include using our Grimme-D2 results as a starting point for calculations that take into account molecular environments, such as the parameter free approach developed by Tkatchenko and Scheffler [
73] or the Grimme-D3 methodology, which employs a three-body dispersion term [
74] and fractional coordination numbers. Recently developed dispersion corrected hybrid functionals [
75,
76] may also improve the agreement between DFT modeling and THz measurements beyond what is presented here using a combination of DFT-GGA and Grimme-D2 dispersion correction.

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}