
1-O-Hexyl-2,3,5-Trimethylhydroquinone Ameliorates l-DOPA-Induced Cytotoxicity in PC12 Cells

Abstract
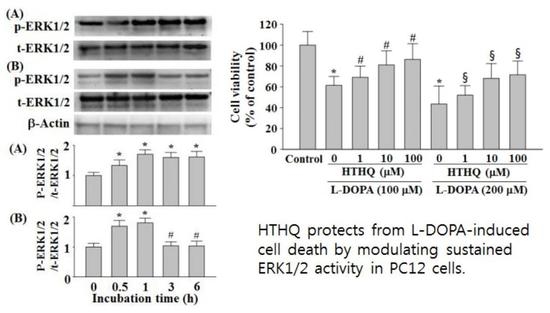
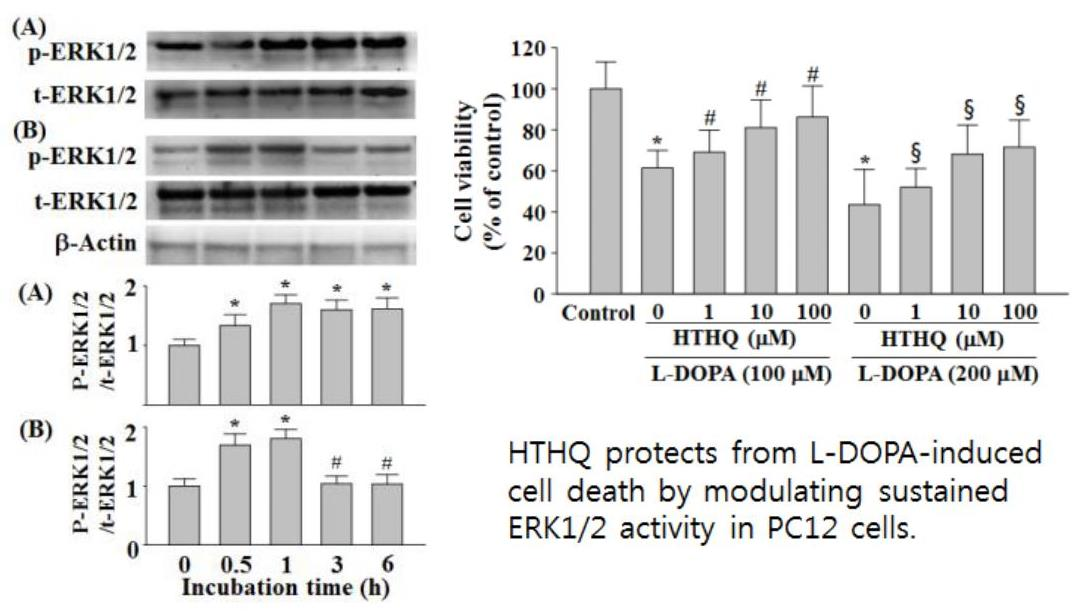{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
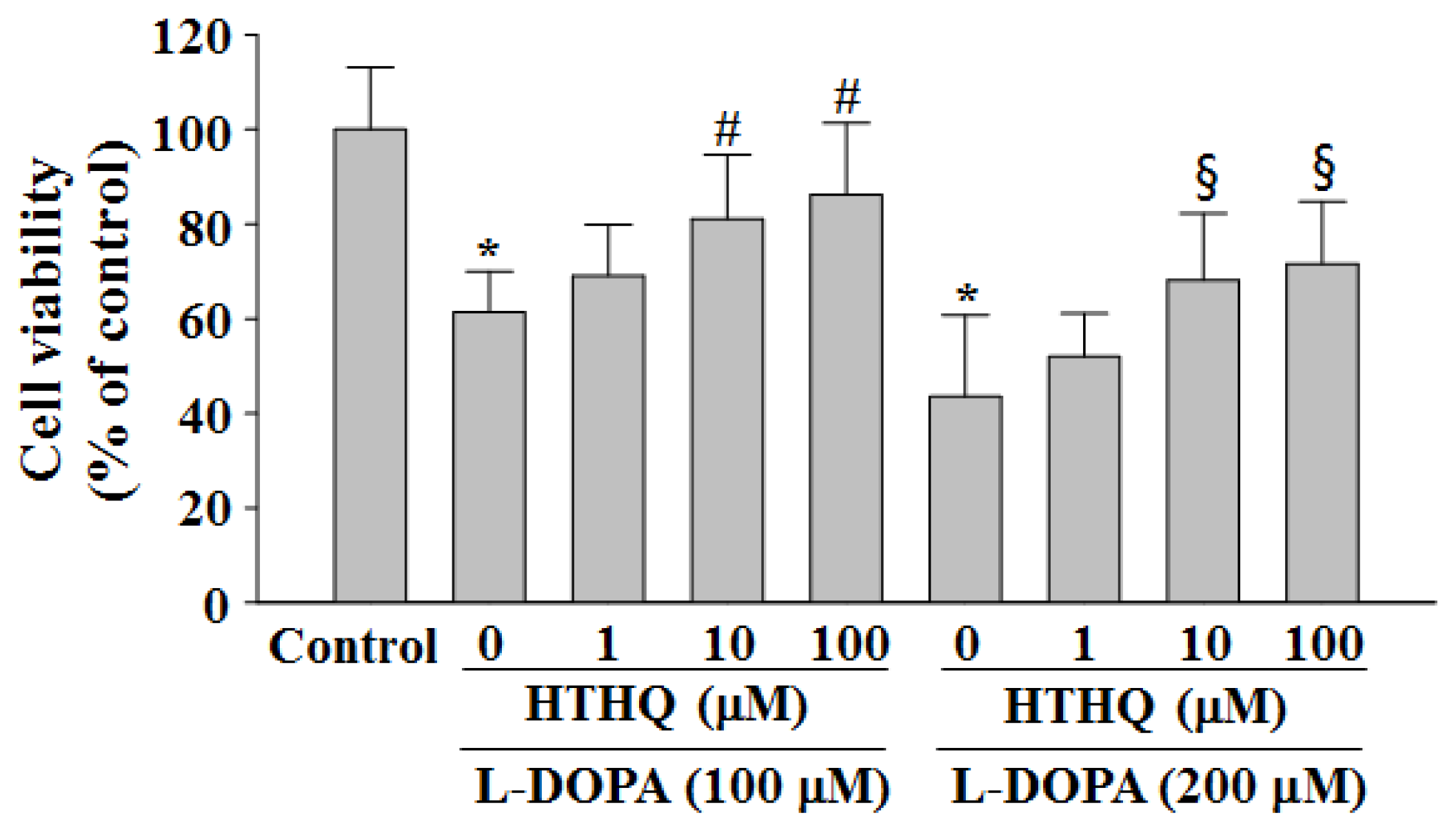
2.1. Cell Viability
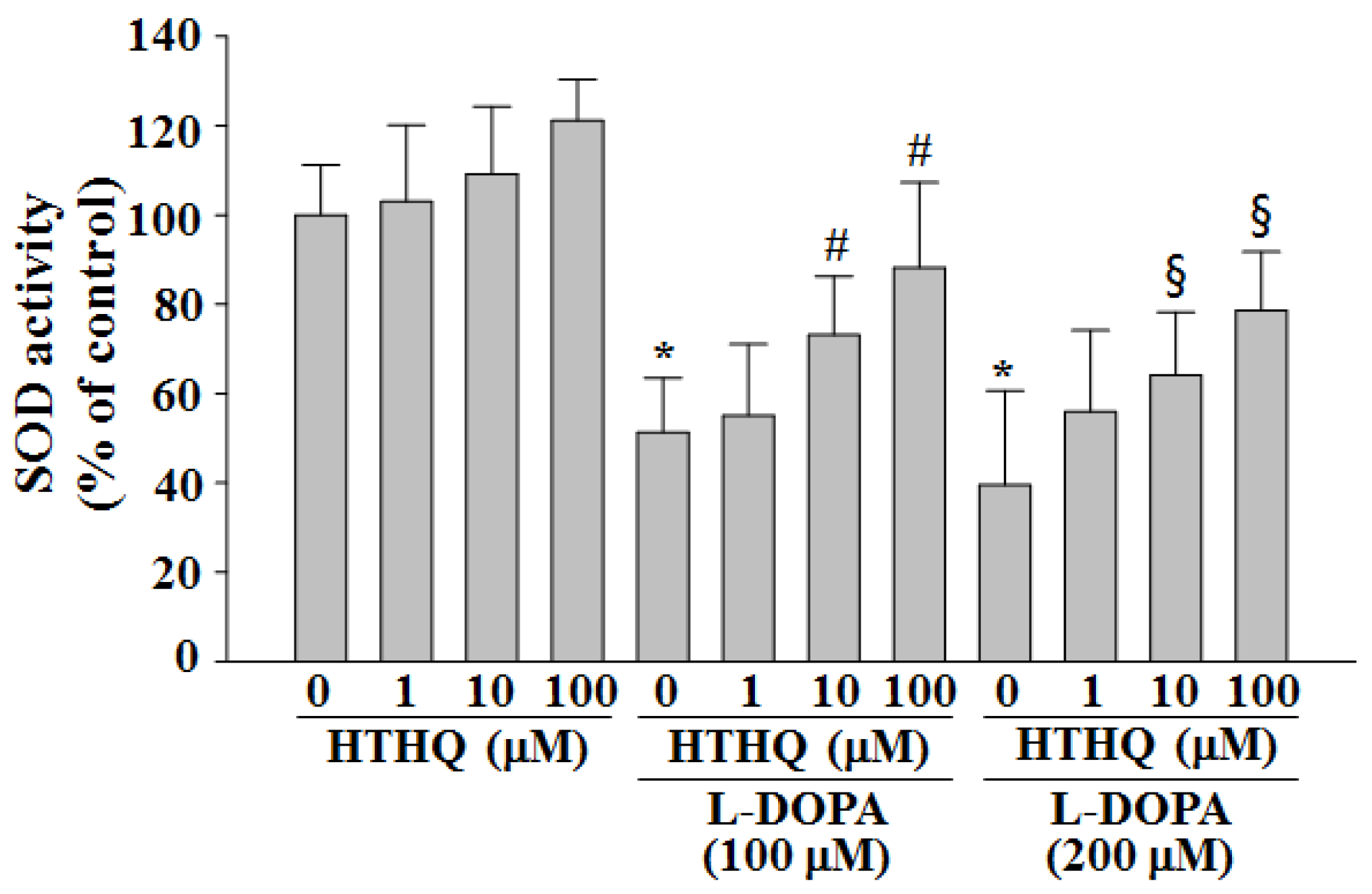
2.2. SOD Activity
2.3. Phosphorylation of ERK1/2
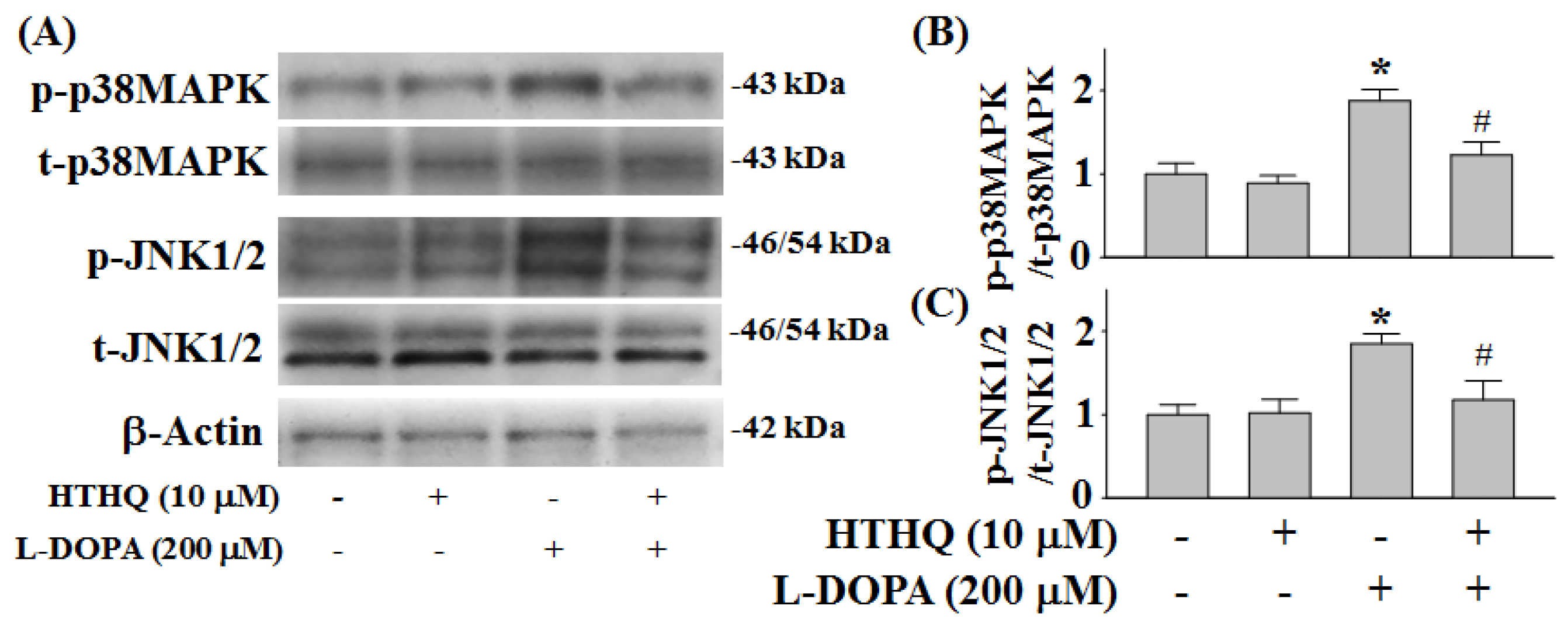
2.4. Phosphorylation of p38MAPK and JNK1/2
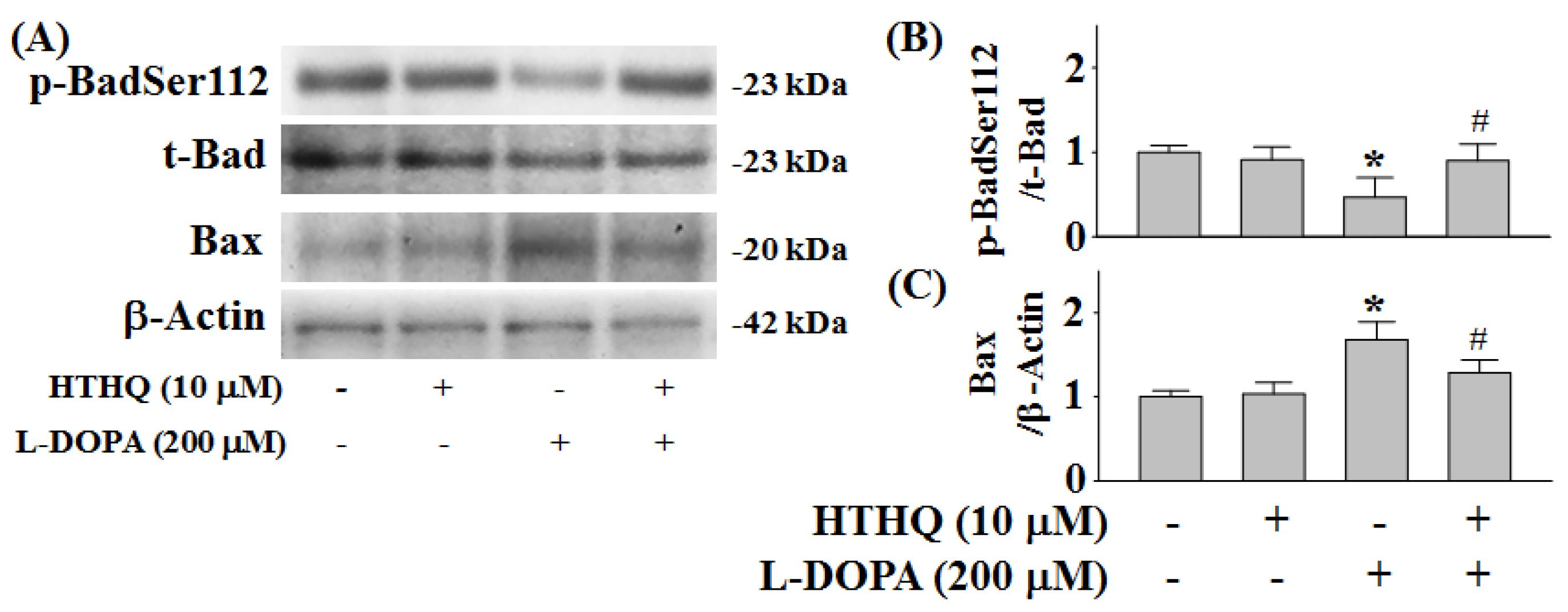
2.5. Phosphorylation of Bad and Expression of Bax
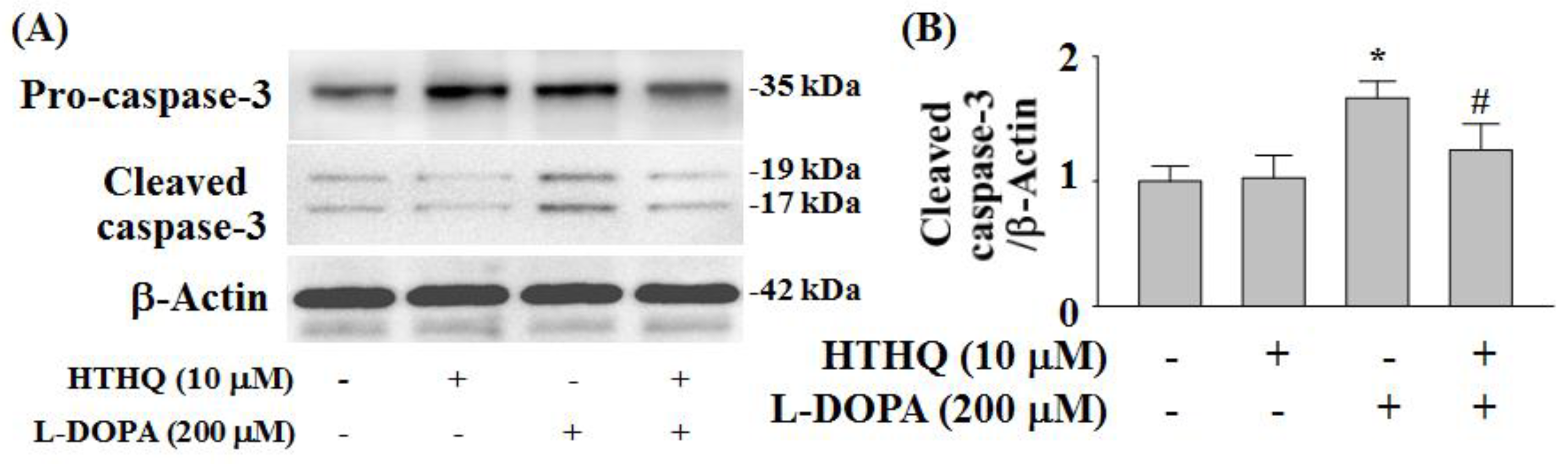
2.6. Expression of Cleaved Caspase-3
3. Discussion
4. Experimental
4.1. Materials
4.2. Cell Culture
4.3. Measurement of Cell Viability
4.4. Assay for Superoxide Dismutase (SOD) Activity
4.5. Western Blot Analysis
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hino, T.; Kawanishi, S.; Yasui, H.; Oka, S.; Sakurai, H. HTHQ (1-O-hexyl-2,3,5-trimethylhydroquinone), an anti-lipid-peroxydative compound: Its chemical and biological characterizations. Biochim. Biophys. Acta 1998, 1425, 47–60. [Google Scholar] [CrossRef]
- Hirose, M.; Akagi, T.; Hasegawa, R.; Yaono, M.; Satoh, T.; Hara, Y.; Wakabayashi, K.; Ito, N. Chemoprevention of 2-amino-1-methyl-6-phenylimidazole [4,5-b]-pyridine (PhIP)-induced mammary gland carcinogenesis by antioxidants in F344 female rats. Carcinogenesis 1995, 19, 217–221. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Walkinshaw, G.; Waters, C.M. Induction of apoptosis in catecholaminergic PC12 cells by L-DOPA. Implications for the treatment of Parkinson’s disease. J. Clin. Invest. 1995, 95, 2458–2464. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.N.; Maeda, T.; Kume, T.; Kaneko, S.; Kochiyama, H.; Akaike, A.; Goshima, Y.; Misu, Y. Differential neurotoxicity induced by L-DOPA and dopamine in cultured striatal neurons. Brain Res. 1996, 743, 278–283. [Google Scholar] [CrossRef]
- Dérijard, B.; Hibi, M.; Wu, I.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R. JNK1: A protein kinase stimulated by UV-light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; Ten Dkjke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, R.; Soares-da-Silva, P. Oxidative and non-oxidative mechanisms of neuronal cell death and apoptosis by L-3,4-dihydroxyphenylalanine (L-DOPA) and dopamine. Br. J. Pharmacol. 2002, 137, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.M.; Yang, Y.J.; Huang, H.S.; Kai, M.; Lee, M.K. Mechanisms of L-DOPA-induced cytotoxicity in rat adrenal pheochromocytoma cells: Implication of oxidative stress-related kinases and cyclic AMP. Neuroscience 2010, 170, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Zha, H.; Aime-Sempe, C.; Sato, T.; Reed, J.C. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J. Biol. Chem. 1996, 271, 7440–7444. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Demeter, M.R.; Ruan, H.; Comb, M.J. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J. Biol. Chem. 2000, 275, 25865–25869. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Park, H.J.; Shin, K.S.; Choi, H.S.; Kai, M.; Lee, M.K. Modulation of PC12 cell viability by forskolin-induced cyclic AMP levels through ERK and JNK pathways: An implication for L-DOPA-induced cytotoxicity in nigrostriatal dopamine neurons. Toxicol. Sci. 2012, 128, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Park, H.J.; Shin, K.S.; Lee, M.K. Multiple treatments with L-3,4-dihydroxyphenylalanine modulate dopamine biosynthesis and neurotoxicity through the protein kinase A-transient extracellular signal-regulated kinase and exchange protein activation by cyclic AMP-sustained extracellular signal-regulated kinase signaling pathways. J. Neurosci. Res. 2014, 92, 1746–1756. [Google Scholar] [PubMed]
- Tischler, A.S.; Perlman, R.L.; Morse, G.M.; Sheard, B.E. Glucocorticoids increase catecholamine synthesis and storage in PC12 pheochromocytoma cell cultures. J. Neurochem. 1983, 40, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Lambeng, N.; Michel, P.P.; Agid, Y.; Ruberg, M. The relationship between differentiation and survival in PC12 cells treated with cyclic adenosine monophosphate in the presence of epidermal growth factor or nerve growth factor. Neurosci. Lett. 2001, 297, 133–136. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.S.; York, R.D.; Stork, P.J. Extracellular-signal-regulated kinase signaling in neurons. Curr. Opin. Neurobiol. 1999, 9, 544–553. [Google Scholar] [CrossRef]
- Chu, C.T.; Levinthal, D.J.; Kulich, S.M.; Chalovich, E.M.; De Franco, D.B. Oxidative neuronal injury. The dark side of ERK1/2. Eur. J. Biochem. 2004, 271, 2060–2066. [Google Scholar] [CrossRef] [PubMed]
- Kiermayer, S.; Biondi, R.M.; Imig, J.; Plotz, G.; Haupenthal, J.; Zeuzem, S.; Piiper, A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol. Biol. Cell 2005, 16, 5639–5648. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Alcacer, C.; Cacciatore, S.; Heiman, M.; Herve, D.; Greengard, P.; Girault, J.A.; Valjent, E.; Fisone, G. L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 2009, 108, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Lipski, J.; Nistico, R.; Berretta, N.; Guayyeo, E.; Bernardi, G.; Mercuri, N.B. L-DOPA: A scapegoat for accelerated neurodegeneration in Parkinson’s disease? Prog. Neurobiol. 2011, 94, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Shin, K.S.; Zhao, T.T.; Park, H.J.; Lee, K.E.; Lee, M.K. L-DOPA modulates cell viability through the ERK-c-Jun system in PC12 and dopaminergic neuronal cells. Neuropharmacology 2016, 101, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, R.A.; Franklin, J.L. Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxid. Redox. Signal 2003, 5, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.; Watson, D.; Best, S.; Midgley, J.; Wenlong, H.; Petty, R. The determination of hydroxydopamines and other trace amines in the urine of parkinsonian patients and normal controls. Neurochem. Res. 1993, 18, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, K.S.; Zhao, T.T.; Lee, K.E.; Lee, M.K. Effects of asarinin on dopamine biosynthesis and 6-hydroxydopamine-induced cytotoxicity in PC12 cells. Arch. Pharm. Res. 2017, 40, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Standaert, D.G. Targets for neuroprotection in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.S.; Parker, W.D., Jr.; Bennett, J.P., Jr. L-DOPA increases nigral production of hydroxyl radicals in vivo: Potential L-DOPA toxicity? Neuro. Rep. 1994, 5, 1009–1011. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Langfort, J. The effect of subchronic, intermittent L-DOPA treatment on neuronal nitric oxide synthase and soluble guanylyl cyclase expression and activity in the striatum and midbrain of normal and MPTP-treated mice. Neurochem. Int. 2007, 50, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Saggu, H.; Cooksey, J.; Dexter, D.; Wells, F.R.; Lees, A.; Jenner, P.; Marden, C.D. A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J. Neurochem. 1989, 53, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Chuda, M.; Kuroda, S.; Hayabara, T. Hydroxyl radical and superoxide dismutase in blood of patients with Parkinson’s disease: Relationship to clinical data. J. Neurol. Sci. 1999, 170, 90–95. [Google Scholar] [CrossRef]
- Lai, C.-T.; Yu, P.H. Dopamine and L-β-3,4-dihydroxyphenylalanine hydrochloride (L-DOPA)-induced cytotoxicity towards catecholaminergic neuroblastoma SH-SY5Y cells. Effects of oxidative stress and antioxidative factors. Biochem. Pharmacol. 1997, 53, 363–372. [Google Scholar] [CrossRef]
- Davaasambuu, U.; Park, H.J.; Park, K.H.; Lee, C.K.; Hwang, B.Y.; Lee, M.K. Ombuoside from Gynostemma pentaphyllum protects PC12 cells from L-DOPA-induced neurotoxicity. Planta Med. 2018, 84, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Ohnishi, H.; Matsuura, S.; Manabe, T. Simple quantification of Cu, Zn-superoxide dismutase activity after separation by nondenaturing isoelectric focusing. Biochim. Biophys. Acta 2002, 1571, 245–248. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.J.; Kang, J.K.; Lee, M.K. 1-O-Hexyl-2,3,5-Trimethylhydroquinone Ameliorates l-DOPA-Induced Cytotoxicity in PC12 Cells. Molecules 2019, 24, 867. https://doi.org/10.3390/molecules24050867
Park HJ, Kang JK, Lee MK. 1-O-Hexyl-2,3,5-Trimethylhydroquinone Ameliorates l-DOPA-Induced Cytotoxicity in PC12 Cells. Molecules. 2019; 24(5):867. https://doi.org/10.3390/molecules24050867
Chicago/Turabian StylePark, Hyun Jin, Jong Koo Kang, and Myung Koo Lee. 2019. "1-O-Hexyl-2,3,5-Trimethylhydroquinone Ameliorates l-DOPA-Induced Cytotoxicity in PC12 Cells" Molecules 24, no. 5: 867. https://doi.org/10.3390/molecules24050867
APA StylePark, H. J., Kang, J. K., & Lee, M. K. (2019). 1-O-Hexyl-2,3,5-Trimethylhydroquinone Ameliorates l-DOPA-Induced Cytotoxicity in PC12 Cells. Molecules, 24(5), 867. https://doi.org/10.3390/molecules24050867