
Molecular and Functional Interaction of the Myokine Irisin with Physical Exercise and Alzheimer’s Disease


 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
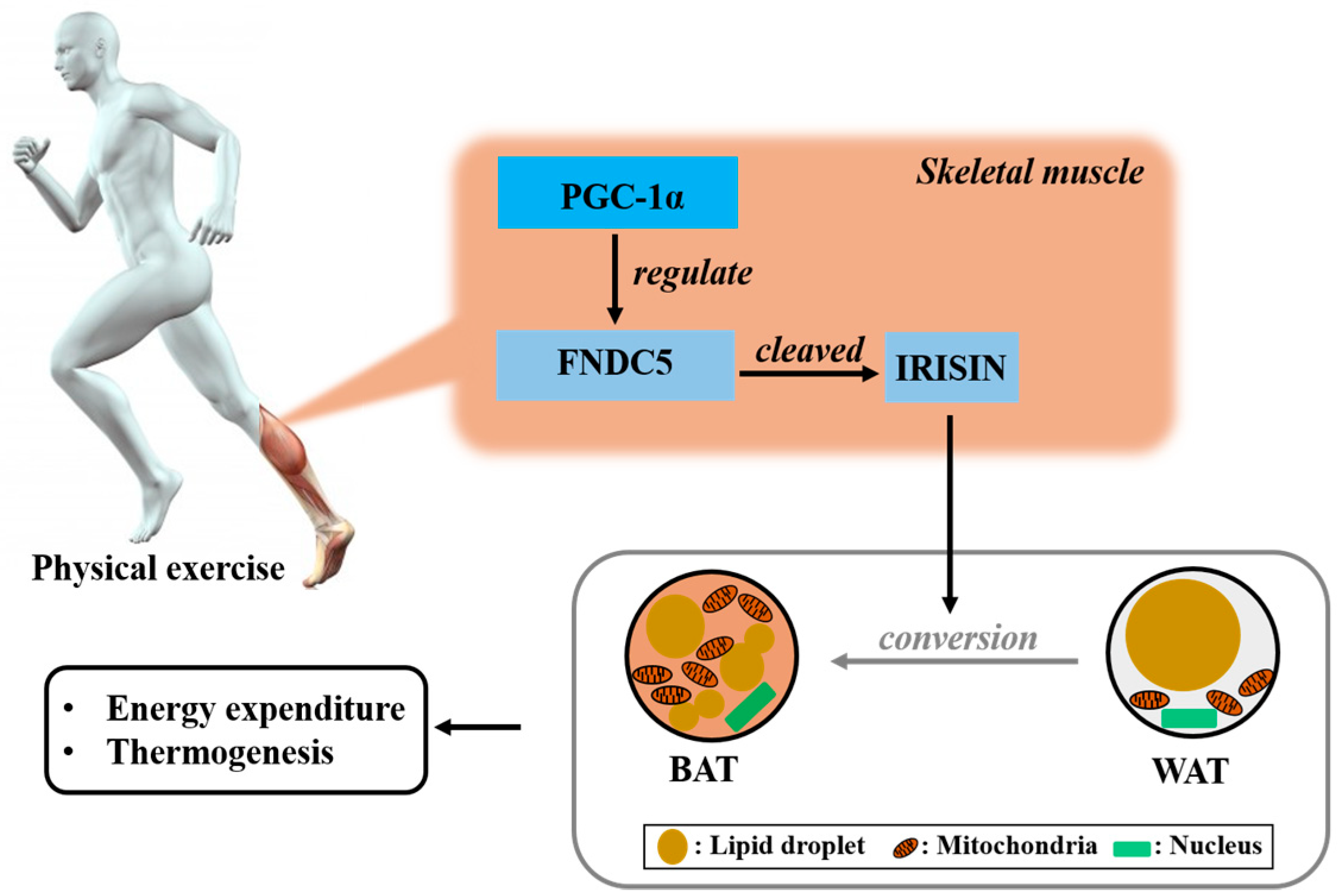
2. Irisin is the Hormone Induced by Physical Exercise
3. Neuroprotective Implications of Irisin via the Akt/ERK Signaling Pathway
4. The Potential Role of Irisin Protecting Hippocampus and Whole Nervous System
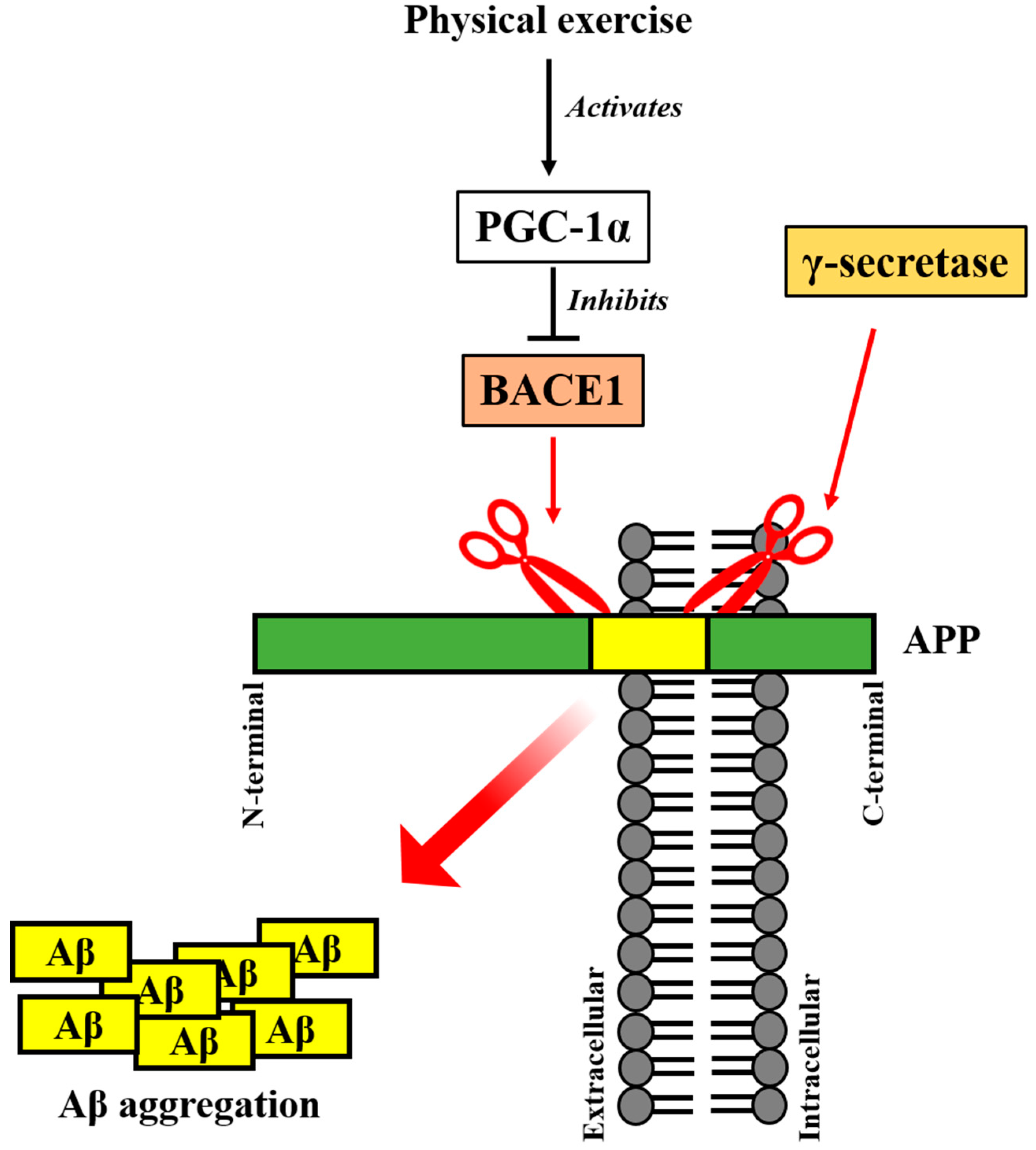
5. Irisin Precursor FNDC5 and Its Upstream PGC-1α Regulates AD Pathogenesis
6. Reduction of Aβ-Induced Endoplasmic Reticulum (ER) Stress by Exercise, and Potential Association with Irisin
7. Implications of Irisin for Age-Related Telomere Length (TL) Shortening and AD Pathogenesis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simonian, N.A.; Coyle, J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Behl, C.; Holsboer, F. Oxidative stress in the pathogenesis of Alzheimer’s disease and antioxidant neuroprotection. Fortschr. Neurol. Psychiatr. 1998, 66, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Koliatsos, V.E.; Kecojevic, A.; Troncoso, J.C.; Gastard, M.C.; Bennett, D.A.; Schneider, J.A. Early involvement of small inhibitory cortical interneurons in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Akama, K.T.; Van Eldik, L.J. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNF alpha)-dependent, and involves a TNF alpha receptor-associated factor- and NF kappa B-inducing kinase-dependent signaling mechanism. J. Biol. Chem. 2000, 275, 7918–7924. [Google Scholar] [PubMed]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Griffin, W.S.; Mrak, R.E. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J. Leukocyte Biol. 2002, 72, 233–238. [Google Scholar] [PubMed]
- Kim, S.E.; Ko, I.G.; Park, C.Y.; Shin, M.S.; Kim, C.J.; Jee, Y.S. Treadmill and wheel exercise alleviate lipopolysaccharide-induced short-term memory impairment by enhancing neuronal maturation in rats. Mol. Med. Rep. 2013, 7, 31–36. [Google Scholar] [CrossRef]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef]
- Tanaka, K.; Quadros, A.C., Jr.; Santos, R.F.; Stella, F.; Gobbi, L.T.; Gobbi, S. Benefits of physical exercise on executive functions in older people with Parkinson’s disease. Brain Cogn. 2009, 69, 435–441. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Bortoluzzi, S.; Scannapieco, P.; Cestaro, A.; Danieli, G.A.; Schiaffino, S. Computational reconstruction of the human skeletal muscle secretome. Proteins 2006, 62, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Deng, X.; Huang, W.; Yu, J.H.; Wang, J.X.; Wang, J.P.; Yang, S.B.; Liu, X.; Wang, L.; Zhang, Y.; et al. Irisin protects against neuronal injury induced by oxygen-glucose deprivation in part depends on the inhibition of ROS-NLRP3 inflammatory signaling pathway. Mol. Immunol. 2017, 91, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Novelle, M.G.; Contreras, C.; Romero, P.A.; Lopez, M.; Dieguez, C. Irisin, two years later. Int. J. Endocrinol. 2013, 2013, 746281. [Google Scholar] [CrossRef] [PubMed]
- Enerbäck, S. The origins of brown adipose tissue. N. Engl. J. Med. 2009, 360, 2021–2023. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Mougios, V.; Kabasakalis, A.; Fatouros, I.; Siopi, A.; Douroudos, I.I.; Filippaios, A.; Panagiotou, G.; Park, K.H.; Mantzoros, C.S. Exercise-induced irisin secretion is independent of age or fitness level and increased irisin may directly modulate muscle metabolism through AMPK activation. J. Clin. Endocrinol. Metab. 2014, 99, E2154–E2161. [Google Scholar] [CrossRef]
- Norheim, F.; Langleite, T.M.; Hjorth, M.; Holen, T.; Kielland, A.; Stadheim, H.K. The effects of acute and chronic exercise on PGC-1alpha, irisin and browning of subcutaneous adipose tissue in humans. FEBS J. 2014, 281, 739–749. [Google Scholar] [CrossRef]
- Park, M.J.; Kim, D.I.; Choi, J.H.; Heo, Y.R.; Park, S.H. New role of irisin in hepatocytes: The protective effect of hepatic steatosis in vitro. Cell Signal. 2015, 27, 1831–1839. [Google Scholar] [CrossRef]
- Lu, J.; Xiang, G.; Liu, M.; Mei, W.; Xiang, L.; Dong, J. Irisin protects against endothelial injury and ameliorates atherosclerosis in apolipoprotein E-Null diabetic mice. Atherosclerosis 2015, 243, 438–448. [Google Scholar] [CrossRef]
- Han, F.; Zhang, S.; Hou, N.; Wang, D.; Sun, X. Irisin improves endothelial function in obese mice through the AMPK-eNOS pathway. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1501–H1508. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, E.; Norheim, F.; Thiede, B.; Holen, T.; Ohashi, T.; Schering, L.; Lee, S.; Brenmoehl, J.; Thomas, S.; Drevon, C.A.; et al. Irisin—A myth rather than an exercise-inducible myokine. Sci. Rep. 2015, 5, 8889. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Linderman, J.D.; Smith, S.; Brychta, R.J.; Wang, J.; Idelson, C.; Perron, R.M.; Werner, C.D.; Phan, G.Q.; Kammula, U.S.; et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014, 19, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Dun, S.L.; Lyu, R.M.; Chen, Y.H.; Chang, J.K.; Luo, J.J.; Dun, N.J. Irisin-immunoreactivity in neural and non-neural cells of the rodent. Neuroscience 2013, 240, 155–162. [Google Scholar] [CrossRef]
- Li, D.J.; Li, Y.H.; Yuan, H.B.; Qu, L.F.; Wang, P. The novel exercise-induced hormone irisin protects against neuronal injury via activation of the Akt and ERK1/2 signaling pathways and contributes to the neuroprotection of physical exercise in cerebral ischemia. Metabolism 2017, 68, 31–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef]
- Song, H.; Wu, F.; Zhang, Y.; Zhang, Y.; Wang, F.; Jiang, M.; Wang, Z.; Zhang, M.; Li, S.; Yang, L.; et al. Irisin promotes human umbilical vein endothelial cell proliferation through the ERK signaling pathway and partly suppresses high glucose-induced apoptosis. PLoS ONE 2014, 9, e110273. [Google Scholar] [CrossRef]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Di Comite, M.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 11, 12157–12162. [Google Scholar] [CrossRef]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. 2015, 129, 839–850. [Google Scholar] [CrossRef]
- Shamim, D.; Laskowski, M. Inhibition of inflammation mediated through the tumor necrosis factor α biochemical pathway can lead to favorable outcomes in Alzheimer disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517722512. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Berchtold, N.C.; Christie, L.A. Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends Neurosci. Sci. 2007, 30, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 2012, 16, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.W.; Vivar, C.; Kramer, A.F.; Van, P.H. Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn. Sci. 2013, 17, 525–544. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology 2011, 77, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Arida, R.M.; Cavalheiro, E.A.; Silva, A.C.; Scorza, F.A. Physical activity and epilepsy: Proven and predicted benefits. Sports Med. 2008, 38, 607–615. [Google Scholar] [CrossRef]
- Buchman, A.S.; Boyle, P.A.; Yu, L.; Shah, R.C.; Wilson, R.S.; Bennett, D.A. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 2012, 78, 1323–1329. [Google Scholar] [CrossRef]
- Russo, N.A.; Beard, R.C.; Cotman, C.W. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology 1999, 21, 679–682. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, Y.; Zhang, P.; Sha, H.; Jia, J.; Hu, Y. Exercise induces mitochondrial biogenesis after brain ischemia in rats. Neuroscience 2012, 205, 10–17. [Google Scholar] [CrossRef]
- Futoshi, A.; Takayuki, M.; Tsuyoshi, S.; Minoru, F.; Yoshikazu, U.; Nobuoki, E.; Tetsunori, S.; Hironobu, Y. High-sensitivity C-reactive protein is associated with hippocampus volume in nondementia patients with type 2 diabetes mellitus. Metabolism 2011, 60, 460–466. [Google Scholar]
- Vanitallie, T.B. Preclinical sporadic Alzheimer’s disease: Target for personalized diagnosis and preventive intervention. Metabolism 2013, 62 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Zsuga, J.; Tajti, G.; Papp, C.; Juhasz, B.; Gesztelyi, R. FNDC5/irisin, a molecular target for boosting reward-related learning and motivation. Med. Hypotheses 2016, 90, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Martinez Munoz, I.Y.; Camarillo Romero, E.D.S.; Garduno Garcia, J.J. Irisin a Novel Metabolic Biomarker: Present Knowledge and Future Directions. Int. J. Endocrinol. 2018, 7816806. [Google Scholar] [CrossRef] [PubMed]
- Piya, M.K.; Harte, A.L.; Sivakumar, K.; Tripathi, G.; Voyias, P.D.; James, S.; Sabico, S.; Al-Daghri, N.M.; Saravanan, P.; Barber, T.M.; et al. The identification of irisin in human cerebrospinal fluid: Influence of adiposity, metabolic markers, and gestational diabetes. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E512–E518. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.S.; Dincer, F.; Mantzoros, C.S. Pharmacological concentrations of irisin increase cell proliferation without influencing markers of neurite outgrowth and synaptogenesis in mouse H19-7 hippocampal cell lines. Metabolism 2013, 62, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik, B.D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Zhang, L.; Ruan, J.; Zhang, X.; Chen, J.; Ma, C.; Yu, Z. Detection and quantitation of irisin in human cerebrospinal fluid by tandem mass spectrometry. Peptides 2018, 103, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Z.; Nusslock, R. Exercise-Mediated Neurogenesis in the Hippocampus via BDNF. Front. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, D.Y. Aquarobic exercises improve the serum blood irisin and brain-derived neurotrophic factor levels in elderly women. Exp. Gerontol. 2018, 104, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, H.; Wang, H.; Wang, J.H.; Song, F.; Sun, Y. Irisin Exerts Neuroprotective Effects on Cultured Neurons by Regulating Astrocytes. Mediat. Inflamm. 2018, 2018, 9070341. [Google Scholar] [CrossRef]
- Papp, C.; Pak, K.; Erdei, T.; Juhasz, B.; Seres, I.; Szentpeteri, A.; Kardos, L.; Szilasi, M.; Gesztelyi, R.; Zsuga, J. Alteration of the irisin–brain-derived neurotrophic factor axis contributes to disturbance of mood in COPD patients. Int. J. Chron. Obstruct. Pulm. Dis. 2017, 12, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Bistué Millón, M.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A.; et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Kumar, A. Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of β-amyloid animal model of Alzheimer’s disease. Neurotox. Res. 2014, 25, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Yarrow, J.F.; White, L.J.; McCoy, S.C.; Borst, S.E. Training augments resistance exercise induced elevation of circulating brain derived neurotrophic factor (BDNF). Neurosci. Lett. 2010, 479, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Song, J. The role of irisin in Alzheimer’s disease. J. Clin. Med. 2018, 7, 407. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, S.J.; Sinn, D.I.; Kim, S.U.; Kim, M.; Roh, J.K. Granulocyte colony-stimulating factor stimulates neurogenesis via vascular endothelial growth factor with STAT activation. Brain Res. 2006, 1073, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Jodeiri Farshbaf, M.; Ghaedi, K.; Megraw, T.L.; Curtiss, J.; Shirani Faradonbeh, M.; Vaziri, P.; Nasr-Esfahani, M.H. Does PGC1α/FNDC5/BDNF elicit the beneficial effects of exercise on neurodegenerative disorders? Neuromol. Med. 2016, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, B. BDNF (I)rising from exercise. Cell Metab. 2013, 18, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jager, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef]
- Lammich, S.; Kojro, E.; Postini, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [PubMed]
- Gertsik, N.; Chiu, D.; Li, Y.M. Complex regulation of γ-secretase: From obligatory to modulatory subunits. Front. Aging Neurosci. 2015, 6, 342. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C. Intracellular and extracellular Aβ, a tale of two neuropathologies. Brain Pathol. 2005, 15, 66–71. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar] [CrossRef]
- Um, H.S.; Kang, E.B.; Leem, Y.H.; Cho, I.H.; Yang, C.H.; Chae, K.R.; Hwang, D.Y.; Cho, J.Y. Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer’s disease in an NSE/APPsw-transgenic model. Int. J. Mol. Med. 2008, 22, 529–539. [Google Scholar]
- Komulainen, P.; Pedersen, M.; Hanninen, T.; Bruunsgaard, H.; Lakka, T.A.; Kivipelto, M.; Hassinen, M.; Rauramaa, T.H.; Pedersen, B.K.; Rauramaa, R. BDNF is a novel marker of cognitive function in ageing women: The DR’s EXTRA Study. Neurobiol. Learn. Mem. 2008, 90, 596–603. [Google Scholar] [CrossRef]
- Bruno, M.A.; Leon, W.C.; Fragoso, G.; Mushynski, W.E.; Almazan, G.; Cuello, A.C. Amyloid β-induced nerve growth factor dysmetabolism in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 857–869. [Google Scholar] [CrossRef]
- Radak, Z.; Hart, N.; Sarga, L.; Koltai, E.; Atalay, M.; Ohno, H.; Boldogh, I. Exercise plays a preventive role against Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Chae, C.H.; Jung, S.L.; An, S.H.; Park, B.Y.; Wang, S.W.; Cho, I.H.; Cho, J.Y.; Kim, H.T. Treadmill exercise improves cognitive function and facilitates nerve growth factor signaling by activating mitogen-activated protein kinase/extracellular signal regulated kinase1/2 in the streptozotocin-induced diabetic rat hippocampus. Neuroscience 2009, 164, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, J.J.; Diao, S.; Kwak, Y.D.; Liu, L.; Zhi, L.; Bueler, H.; Bhat, N.R.; Williams, R.W.; Park, E.A.; et al. Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013, 17, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Kovacs, D.M.; Wong, P.C. The β-secretase enzyme BACE in health and Alzheimer’s disease: Regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009, 29, 12787–12794. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Kuzuya, A.; Tanigawa, K.; Araki, M.; Kawai, R.; Ma, B.; Sasakura, Y.; Maesako, M.; Tashiro, Y.; Miyamoto, M.; et al. Fibronectin type III domain-containing protein 5 interacts with APP and decreases amyloid β production in Alzheimer’s disease. Mol. Brain 2018, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Chafekar, S.M.; Hoozemans, J.J.; Zwart, R.; Baas, F.; Scheper, W. A β 1-42 Induces Mild Endoplasmic Reticulum Stress in an Aggregation State–Dependent Manner. Antioxid. Redox Signal. 2007, 9, 2245–2254. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 85–392. [Google Scholar] [CrossRef]
- Paschen, W.; Mengesdorf, T. Cellular abnormalities linked to endoplasmic reticulum dysfunction in cerebrovascular disease therapeutic potential. Pharmacol. Ther. 2005, 108, 362–375. [Google Scholar] [CrossRef]
- Scheper, W.; Hoozemans, J. Endoplasmic reticulum protein quality control in neurodegenerative disease: The good, the bad and the therapy. Curr. Med. Chem. 2009, 16, 615–626. [Google Scholar] [CrossRef]
- Ohri, S.S.; Maddie, M.A.; Zhao, Y.; Qiu, M.S.; Hetman, M.; Whittemore, S.R. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia 2011, 59, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, V.; Collyer, E.; Armentano, D.; Parsons, G.B.; Court, F.A.; Hetz, C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 2012, 3, 272. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Sakurai, M.; Abe, K.; Matsumiya, G.; Sawa, Y. Impact of the endoplasmic reticulum stress response in spinal cord after transient ischemia. Brain Res. 2007, 1169, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Park, K.G. Endoplasmic reticulum stress and diabetes. J. Korean Endocr. Soc. 2008, 23, 1–8. [Google Scholar] [CrossRef][Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Kang, E.B.; Kwon, I.S.; Koo, J.H.; Kim, E.J.; Kim, C.H.; Lee, J.; Yang, C.H.; Lee, Y.I.; Cho, I.H.; Cho, J.Y. Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis 2013, 18, 1332–1347. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Epel, E.S. Telomeres and adversity: Too toxic to ignore. Nature 2012, 490, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.L.; Mezuk, B.; Bareis, N.; Lin, J.; Blackburn, E.H.; Epel, E.S. Depression, anxiety and telomere length in young adults: Evidence from the National Health and Nutrition Examination Survey. Mol. Psychiatry 2015, 20, 520–528. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J.; Ratka, A. Telomere shortening and Alzheimer’s disease. Neuromol. Med. 2013, 15, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Schupf, N.; Lee, J.H.; Tang, M.X.; Mayeux, R. Shorter telomeres are associated with mortality in those with APOE epsilon4 and dementia. Ann. Neurol. 2006, 60, 181–187. [Google Scholar] [CrossRef]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence that aging and amyloid promote microglial cell senescence. Rejuv. Res. 2007, 10, 61–74. [Google Scholar] [CrossRef]
- Floris, S.; Ruuls, S.R.; Wierinckx, A.; van der Pol, S.M.; Döpp, E.; van der Meide, P.H.; Dijkstra, C.D.; De Vries, H.E. Interferon-beta directly influences monocyte infiltration into the central nervous system. J. Neuroimmunol. 2002, 127, 69–79. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Feliciani, C.; Gambi, D. Peripheral chemokine receptors, their ligands, cytokines and Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; von Figura, G.; Liu, Y.; Kraus, J.M.; Torrice, C.; Dillon, P.; Rudolph-Watabe, M.; Ju, Z.; Kestler, H.A.; Sanoff, H.; et al. Lifestyle impacts on the aging-associated expression of biomarkers of DNA damage and telomere dysfunction in human blood. Aging Cell 2010, 9, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Rana, K.S.; Arif, M.; Hill, E.J.; Aldred, S.; Nagel, D.A.; Nevill, A.; Randeva, H.S.; Bailey, C.J.; Bellary, S.; Brown, J.E. Plasma irisin levels predict telomere length in healthy adults. Age 2014, 36, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Parks, C.G.; DeRoo, L.A.; Chen, H.; Taylor, J.A.; Cawthon, R.M.; Sandler, D.P. Obesity and weight gain in adulthood and telomere length. Cancer Epidemiol. Biomark. Prev. 2009, 18, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, A.T.; Zimmerman, J.B.; Witkowski, S.; Hearn, J.W.; Hatfield, B.D.; Roth, S.M. Relationship between physical activity level, telomere length, and telomerase activity. Med. Sci. Sports Exerc. 2008, 40, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Cherkas, L.F.; Hunkin, J.L.; Kato, B.S.; Richards, J.B.; Gardner, J.P.; Surdulescu, G.L.; Kimura, M.; Lu, X.; Spector, T.D.; Aviv, A. The association between physical activity in leisure time and leukocyte telomere length. Arch. Intern. Med. 2008, 168, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Shimose, S.; Kubo, T.; Fujimori, J.; Yasunaga, Y.; Sugita, T.; Ochi, M. Correlation between p38 mitogen-activated protein kinase and human telomerase reverse transcriptase in sarcomas. J. Exp. Clin. Cancer Res. 2012, 31, 5. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Sumsuzzman, D.M.; Choi, J.; Kang, H.; Lee, S.-R.; Hong, Y. Molecular and Functional Interaction of the Myokine Irisin with Physical Exercise and Alzheimer’s Disease. Molecules 2018, 23, 3229. https://doi.org/10.3390/molecules23123229
Jin Y, Sumsuzzman DM, Choi J, Kang H, Lee S-R, Hong Y. Molecular and Functional Interaction of the Myokine Irisin with Physical Exercise and Alzheimer’s Disease. Molecules. 2018; 23(12):3229. https://doi.org/10.3390/molecules23123229
Chicago/Turabian StyleJin, Yunho, Dewan Md. Sumsuzzman, Jeonghyun Choi, Hyunbon Kang, Sang-Rae Lee, and Yonggeun Hong. 2018. "Molecular and Functional Interaction of the Myokine Irisin with Physical Exercise and Alzheimer’s Disease" Molecules 23, no. 12: 3229. https://doi.org/10.3390/molecules23123229
APA StyleJin, Y., Sumsuzzman, D. M., Choi, J., Kang, H., Lee, S.-R., & Hong, Y. (2018). Molecular and Functional Interaction of the Myokine Irisin with Physical Exercise and Alzheimer’s Disease. Molecules, 23(12), 3229. https://doi.org/10.3390/molecules23123229