Natural Products Containing ‘Rare’ Organophosphorus Functional Groups




Abstract
1. Introduction
2. Natural Products Containing a P–N Bond (Phosphoramidates)
2.1. Small-Molecule Natural Products Containing a P–N Bond








2.1.1. Phosphagens
- N-phosphoagmatine (24) is a phosphagen identified in protozoa Euglena oracilis and Ochromonas danica [90]. As in the case of other phosphagens, a specific agmatine kinase (EC 2.7.3.10) is responsible for the synthesis of N-phosphoagmatine (24), with l-arginine also being phosphorylated but much less efficiently [90].
- N-phosphoopheline (26) is a phosphagen identified in the marine annelid Ophelia neglecta. The main function of the opheline kinase (EC 2.7.3.7) is the synthesis of the phosphagen N-phosphoopheline, however the substrate specificity of (EC 2.7.3.7) is much broader than other phosphagen kinases and (EC 2.7.3.7) can also phosphorylate taurocyamine, lombricine, and taurocyamine albeit with lower efficiency [94].
- N-phospholombricine (25) is a phosphagen identified in several invertebrate species, mostly annelids, e.g., earthworms [95,96,97,98]. The compound 25 is synthetized by the lombricine kinase (EC 2.7.3.5) which specificity varies with respect to the source species the enzyme was isolated from [63,99,100,101]. It is worth noting that different, evolutionarily distant, organisms can produce the same phosphagens that only differ in the stereoisomer of one component residue. For example, N-phospholombricine in majority of annelids contains a d-serine residue while the N-phospholombricine of echiuroids, a group of marine worms contains an l-serine moiety [51,79].
- N-phosphoguanidinoacetate (N-phosphoglycocyamine) (23) is a phosphagen identified in many invertebrate species, mainly annelids. The compound 23 is synthetized by the guanidinoacetate kinase (also named glycocyamine kinase; EC 2.7.3.1). Guanidinoacetate kinase participates in arginine and proline metabolism in the cell and is widely distributed across the invertebrate branch of the tree of life. The glycocyamine kinases (EC 2.7.3.1) from the annelid Hediste diversicolor was also shown to be responsible for the synthesis of the N-phosphoguanidine (28), however it is unclear if N-phosphoguanidine (28) is a true endogenous phosphagen of Hediste diversicolor or any other species [102]. While it is theoretically possible for N-phosphoguanidine to be formed in vivo and for compound 28 to be an important metabolite in the cell its importance in the cellular metabolism remains to be proven.
- N-phosphothalassemine (27) is a phosphagen structurally similar to lombricine. N-phosphothalassemine (27) was isolated from a common earthworm Lumbricus terrestris and an unsegmented marine worm Thalassema thalassema [103]. The phosphagen kinase EC 2.7.3.5, responsible for phosphorylation of lombricine, is also responsible for phosphorylation of methylated lombricines such as thalassemine [103].
- N-phosphohypotaurocyamine (22) is a rare sulfinic acid phosphagen so far identified only in peanut worms (Golfingia sp.) [78,104]. Hypotaurocyamine kinase (EC 2.7.3.6) responsible for the synthesis of N-phosphohypotaurocyamine has high preference towards hypotaurocyamine, although it can also phosphorylate taurocyamine, albeit with diminished efficiency [77]. It is suggested that this unusual phosphagen system evolved from molluscan N-phosphoarginine kinase [78].
- N-phosphotaurocyamine (21) is a sulfonic acid phosphagen synthesized by a widespread taurocyamine kinase (EC 2.7.3.4) identified in a large number of annelid species [59,81,82,83,84,85]. However, recent identification of taurocyamine kinases in a large number of non-annelid species, including trematodes Paragonimus westermani, Schistosoma japonicum, Clonorchis sinensis [82,83,86,87,88,89] and, in two isolated cases in unicellular oomycetes [82,83,86,87,88,89], suggests that the evolutionary and phylogenetic scope of alternative substrate specificities of phosphagen kinases may be more widespread than previously thought [105].
- N-phosphoarginine (19) phosphagen is as widespread among invertebrates as N-phosphocreatine (20) is widespread among vertebrate species. Arginine kinase (EC 2.7.3.3), responsible for phosphorylation of arginine, also occurs in unicellular organisms like protists and even bacteria which could suggest evolutionary ancient origins of N-phosphoarginine phosphagen system. Indeed, N-phosphoarginine phosphagen system appears to be the earliest one developed by life on Earth and at least couple of other phosphagen systems derive their evolutionary history from an earlier version of the N-phosphoarginine phosphagen system [51,54,58,75,105,106,107,108]. Interestingly d-arginine is a substrate for Sabellastarte indicad-arginine kinase [109,110].
- N-phosphocreatine (20) is a phosphagen synthesized by creatine kinase (EC 2.7.3.2). N-phosphocreatine (20) is produced both by vertebrates and invertebrates (Table 1). Compound 20 functions as a rapid reserve of high-energy phosphates to recycle ATP in high-energy demand tissues such as the brain or skeletal muscle and was mostly studied in mammals [111,112,113]. The chemical properties, functions, and clinical relevance of N-phosphocreatine (20) and its corresponding kinase were reviewed extensively elsewhere and will not be expanded here [106,114,115,116,117]. In brief, N-phosphocreatine is crucial for normal vertebrate physiology, not only on the whole organ level, e.g., in normal muscle activity, but also on the individual cellular level, e.g., in the formation of the ‘creatine kinase circuit’ that is essential for high-sensitivity hearing, as demonstrated by an unexpected hearing loss in creatine kinase knockout mice experiments [118].


2.1.2. Natural Phosphoramidate Nucleotides









2.2. N-phosphorylation of Proteins and Peptides
2.2.1. N-phosphorylation of l-arginine

2.2.2. N-phosphorylation of l-lysine

2.2.3. N-phosphorylation of l-histidine

2.2.4. N-phosphorylation of Other Amino Acids?
3. Natural Products Containing a P–S Bond (Phosphorothioates)
3.1. Small-Molecule Natural Products Containing a P–S Bond

3.2. S-phosphorylation of Proteins and Peptides

3.3. Phosphorothioate DNA Modifications

4. Natural Products Containing a P–C Bond
4.1. Phosphonates






4.2. Phosphinates


4.3. Phosphines

5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Westheimer, F. Why nature chose phosphates. Science 1987, 6, 1173–1178. [Google Scholar] [CrossRef]
- Hunter, T. Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2513–2516. [Google Scholar] [CrossRef] [PubMed]
- Kamerlin, S.C.L.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why nature really chose phosphate. Q. Rev. Biophys. 2013, 46, 1–132. [Google Scholar] [CrossRef] [PubMed]
- Rossomando, E.F.; Hadjimichael, J. Characterization and cAMP inhibition of a lysyl-(N-epsilon-5′-phospho) adenosyl phosphoamidase in Dictyostelium discoideum. Int. J. Biochem. 1986, 18, 481–484. [Google Scholar] [CrossRef]
- Masako, K.; Tsuyoshi, O.; Hitoshi, O.; Akira, K. Nucleoside monophosphoramidate hydrolase from rat liver: Purification and characterization. Int. J. Biochem. 1994, 26, 235–245. [Google Scholar] [CrossRef]
- Roush, R.F. Biosynthesis of Nitrogen-Phosphorus Bond Containing Peptide Natural Products; Harvard University: Cambridge, MA, USA, 2011; ISBN 1124729585. [Google Scholar]
- Kuroda, Y.; Goto, T.; Okamoto, M.; Yamashita, M.; Iguchi, E.; Kohsaka, M.; Aoki, H.; Imanaka, H. FR-900137, a new antibiotic. I. Taxonomy and fermentation of the organism, and isolation and characterization of the antibiotic. J. Antibiot. 1980, 33, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Tanaka, H.; Okamoto, M.; Goto, T.; Kohsaka, M.; Aoki, H.; Imanaka, H. FR-900137, a new antibiotic. II. Structure determination of FR-900137. J. Antibiot. 1980, 33, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Ogita, T.; Gunji, S.; Fukazawa, Y.; Terahara, A.; Kinoshita, T.; Nagaki, H.; Beppu, T. The structures of fosfazinomycins A and B. Tetrahedron Lett. 1983, 24, 2283–2286. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, K.-K.A.; Lee, J.; van der Donk, W.A. Biosynthesis of fosfazinomycin is a convergent process. Chem. Sci. 2015, 6, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, K.-K.A.; van der Donk, W.A. New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 2016, 7, 5219–5223. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-K.A.; Ng, T.L.; Wang, P.; Huang, Z.; Balskus, E.P.; van der Donk, W.A. Glutamic acid is a carrier for hydrazine during the biosyntheses of fosfazinomycin and kinamycin. Nat. Commun. 2018, 9, 3687. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.M.; Sperry, J. Natural Products Containing a Nitrogen–Nitrogen Bond. J. Nat. Prod. 2013, 76, 794–812. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.J.; Ng, T.L.; Wang, P.; Balskus, E.P. Heteroatom–Heteroatom Bond Formation in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5784–5863. [Google Scholar] [CrossRef]
- Schiessl, K.; Roller, A.; Hammerschmidt, F. Determination of absolute configuration of the phosphonic acid moiety of fosfazinomycins. Org. Biomol. Chem. 2013, 11, 7420–7426. [Google Scholar] [CrossRef] [PubMed]
- Pruess, D.L.; Scannell, J.P.; Ax, H.A.; Kellett, M.; Weiss, F.; Demny, T.C.; Stempel, A. Antimetabolites produced by microorganisms. VII. J. Antibiot. (Tokyo) 1973, 26, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Diddens, H.; Dorgerloh, M.; Zahner, H. Metabolic products of microorganisms 176. On the transport of small peptide antibiotics in bacteria. J. Antibiot. (Tokyo) 1979, 32, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Petkowski, J.J.; Bains, W.; Seager, S. Natural Products Containing a Nitrogen-Sulfur Bond. J. Nat. Prod. 2018, 81, 423–446. [Google Scholar] [CrossRef] [PubMed]
- Petkowski, J.J.; Bains, W.; Seager, S. An Apparent Binary Choice in Biochemistry: Mutual Reactivity Implies Life Chooses Thiols or Nitrogen-Sulfur Bonds, but not Both. Astrobiology 2018. [Google Scholar] [CrossRef] [PubMed]
- DiNovi, M.; Trainor, D.A.; Nakanishi, K.; Sanduja, R.; Alam, M. The structure of PB-1, an unusual toxin isolated from the red tide dinoflagellate Ptychodiscus brevis. Tetrahedron Lett. 1983, 24, 855–858. [Google Scholar] [CrossRef]
- Murao, S.; Katsura, M.; Fukuhara, K.; Oda, K. New metallo proteinase inhibitor (MK-I) produced by Streptomyces mozunensis MK-23. Agric. Biol. Chem. 1980, 44, 701–703. [Google Scholar] [CrossRef]
- Kitagishi, K.; Hiromi, K. Binding between thermolysin and its specific inhibitor, phosphoramidon. J. Biochem. 1984, 95, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Kitagishi, K.; Hiromi, K.; Oda, K.; Murao, S. Equilibrium study on the binding between thermolysin and Streptomyces metalloprotease inhibitor, talopeptin (MKI). J. Biochem. 1983, 93, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, K.; Murao, S.; Nozawa, T.; Hatano, M. Structural elucidation of talopeptin (MK-I), a novel metallo proteinase inhibitor produced by streptomyces mozunensis MK-23. Tetrahedron Lett. 1982, 23, 2319–2322. [Google Scholar] [CrossRef]
- Kitagishi, K.; Hiromi, K. Inhibitory Spectrum of Talopeptin (MKI), a Specific Inhibitor of Thermolysin. Agric. Biol. Chem. 1984, 48, 1287–1291. [Google Scholar]
- Weaver, L.H.; Kester, W.R.; Matthews, B.W. A crystallographic study of the complex of phosphoramidon with thermolysin. A model for the presumed catalytic transition state and for the binding of extended substrates. J. Mol. Biol. 1977, 114, 119–132. [Google Scholar] [CrossRef]
- Giraldi, T.; Sava, G.; Perissin, L.; Zorzet, S. Primary tumor growth and formation of spontaneous lung metastases in mice bearing Lewis carcinoma treated with proteinase inhibitors. Anticancer Res. 1984, 4, 221–224. [Google Scholar] [PubMed]
- Wu-Wong, J.R.; Chiou, W.J.; Opgenorth, T.J. Phosphoramidon modulates the number of endothelin receptors in cultured Swiss 3T3 fibroblasts. Mol. Pharmacol. 1993, 44, 422–429. [Google Scholar] [PubMed]
- Vemulapalli, S.; Chiu, P.J.S.; Chintala, M.; Bernardino, V. Attenuation of Ischemic Acute Renal Failure by Phosphoramidon in Rats. Pharmacology 1993, 47, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Datta, I.; Banerjee, M.; Mukherjee, S.K.; Majumdar, S.K. JU-2, a novel phosphorous-containing antifungal antibiotic from Streptomyces kanamyceticus M 8. Indian J. Exp. Biol. 2001, 39, 604–606. [Google Scholar] [PubMed]
- Quitschau, M.; Schuhmann, T.; Piel, J.; von Zezschwitz, P.; Grond, S. The New Metabolite (S)-Cinnamoylphosphoramide from Streptomyces sp. and Its Total Synthesis. European, J. Org. Chem. 2008, 2008, 5117–5124. [Google Scholar] [CrossRef]
- Kasai, N.; Fukuhara, K.; Murao, S. Purification and some properties of FMPI, a novel metallo-proteinase inhibitor produced by Streptomyces rishiriensis NK-122. Agric. Biol. Chem. 1982, 46, 2979–2985. [Google Scholar] [CrossRef]
- Kasai, N.; Fukuhara, K.; Oda, K.; Murao, S. Inhibition of Angiotensin I Converting Enzyme and Carboxypeptidase A by FMPI, Talopeptin, and Their Derivatives. Agric. Biol. Chem. 1983, 47, 2915–2916. [Google Scholar] [CrossRef]
- Kasai, N.; Fukuhara, K.; Murao, S. Purification of an Aspergillus oryzae Metallo-proteinase by Talopeptin-aminohexyl-Sepharose and Its Properties. Agric. Biol. Chem. 1984, 48, 1533–1538. [Google Scholar] [CrossRef]
- Murao, S.; Kasai, N.; Kimura, Y.; Oda, K.; Fukuhara, K. A New Metallo-proteinase Inhibitor (FMPI) Produced by Streptomyces rishiriensis NK-122. Agric. Biol. Chem. 1982, 46, 855–857. [Google Scholar] [CrossRef]
- Murao, S.; Kasai, N.; Kimura, Y.; Oda, K. Isolation of metallo-proteinase inhibitor (FMPI) producing microorganism. Agric. Biol. Chem. 1982, 46, 2697–2703. [Google Scholar]
- Guerry, P.; Poly, F.; Riddle, M.; Maue, A.; Chen, Y.-H.; Monteiro, M. Campylobacter Polysaccharide Capsules: Virulence and Vaccines. Front. Cell. Infect. Microbiol. 2012, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.C.H.; van Alphen, L.B.; Harboe, A.; Li, J.; Christensen, B.B.; Szymanski, C.M.; Brondsted, L. Bacteriophage F336 recognizes the capsular phosphoramidate modification of Campylobacter jejuni NCTC11168. J. Bacteriol. 2011, 193, 6742–6749. [Google Scholar] [CrossRef] [PubMed]
- van Alphen, L.B.; Wenzel, C.Q.; Richards, M.R.; Fodor, C.; Ashmus, R.A.; Stahl, M.; Karlyshev, A.V.; Wren, B.W.; Stintzi, A.; Miller, W.G.; et al. Biological roles of the O-methyl phosphoramidate capsule modification in Campylobacter jejuni. PLoS ONE 2014, 9, e87051. [Google Scholar] [CrossRef] [PubMed]
- Pequegnat, B.; Laird, R.M.; Ewing, C.P.; Hill, C.L.; Omari, E.; Poly, F.; Monteiro, M.A.; Guerry, P. Phase-Variable Changes in the Position of O-Methyl Phosphoramidate Modifications on the Polysaccharide Capsule of Campylobacter jejuni Modulate Serum Resistance. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.; van Alphen, L.; Frodor, C.; Crowley, S.; Christensen, B.; Szymanski, C.; Brøndsted, L. Phase Variable Expression of Capsular Polysaccharide Modifications Allows Campylobacter jejuni to Avoid Bacteriophage Infection in Chickens. Front. Cell. Infect. Microbiol. 2012, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Karlyshev, A.V.; Linton, D.; Gregson, N.A.; Lastovica, A.J.; Wren, B.W. Genetic and biochemical evidence of a Campylobacter jejuni capsular polysaccharide that accounts for Penner serotype specificity. Mol. Microbiol. 2000, 35, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Karlyshev, A.V.; Champion, O.L.; Churcher, C.; Brisson, J.-R.; Jarrell, H.C.; Gilbert, M.; Brochu, D.; St Michael, F.; Li, J.; Wakarchuk, W.W.; et al. Analysis of Campylobacter jejuni capsular loci reveals multiple mechanisms for the generation of structural diversity and the ability to form complex heptoses. Mol. Microbiol. 2005, 55, 90–103. [Google Scholar] [CrossRef] [PubMed]
- St Michael, F.; Szymanski, C.M.; Li, J.; Chan, K.H.; Khieu, N.H.; Larocque, S.; Wakarchuk, W.W.; Brisson, J.-R.; Monteiro, M.A. The structures of the lipooligosaccharide and capsule polysaccharide of Campylobacter jejuni genome sequenced strain NCTC 11168. Eur. J. Biochem. 2002, 269, 5119–5136. [Google Scholar] [CrossRef] [PubMed]
- Thota, V.N.; Ferguson, M.J.; Sweeney, R.P.; Lowary, T.L. Synthesis of the Campylobacter jejuni 81-176 Strain Capsular Polysaccharide Repeating Unit Reveals the Absolute Configuration of its O-Methyl Phosphoramidate Motif. Angew. Chem. Int. Ed. Engl. 2018, 57, 15592–15596. [Google Scholar] [CrossRef] [PubMed]
- Taylor, Z.W.; Brown, H.A.; Holden, H.M.; Raushel, F.M. Biosynthesis of Nucleoside Diphosphoramidates in Campylobacter jejuni. Biochemistry 2017, 56, 6079–6082. [Google Scholar] [CrossRef] [PubMed]
- Taylor, Z.W.; Brown, H.A.; Narindoshvili, T.; Wenzel, C.Q.; Szymanski, C.M.; Holden, H.M.; Raushel, F.M. Discovery of a Glutamine Kinase Required for the Biosynthesis of the O-Methyl Phosphoramidate Modifications Found in the Capsular Polysaccharides of Campylobacter jejuni. J. Am. Chem. Soc. 2017, 139, 9463–9466. [Google Scholar] [CrossRef] [PubMed]
- Taylor, Z.W.; Raushel, F.M. Cytidine Diphosphoramidate Kinase: An Enzyme Required for the Biosynthesis of the O-Methyl Phosphoramidate Modification in the Capsular Polysaccharides of Campylobacter jejuni. Biochemistry 2018, 57, 2238–2244. [Google Scholar] [CrossRef]
- Taylor, Z.W.; Chamberlain, A.R.; Raushel, F.M. Substrate Specificity and Chemical Mechanism for the Reaction Catalyzed by Glutamine Kinase. Biochemistry 2018, 57, 5447–5455. [Google Scholar] [CrossRef]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The “phosphocreatine circuit” for cellular energy homeostasis. Biochem. J. 1992, 281 (Pt 1), 21–40. [Google Scholar] [CrossRef]
- Ellington, W.R. Evolution and physiological roles of phosphagen systems. Annu. Rev. Physiol. 2001, 63, 289–325. [Google Scholar] [CrossRef] [PubMed]
- Jarilla, B.R.; Agatsuma, T. Phosphagen kinases of parasites: Unexplored chemotherapeutic targets. Korean J. Parasitol. 2010, 48, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.D.; Graham, J.; Snider, M.J.; Fraga, D. Characterization of a novel bacterial arginine kinase from Desulfotalea psychrophila. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2008, 150, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Soga, S.; Inoue, M.; Uda, K. Characterization and origin of bacterial arginine kinases. Int. J. Biol. Macromol. 2013, 57, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Fannin, L.; Aryal, M.; Stock, K.; Snider, M.; Fraga, D. Characterization of Bacterial Arginine Kinases in Species from the Order Myxococcales. FASEB J. 2018, 32, 655.10. [Google Scholar] [CrossRef]
- Michibata, J.; Okazaki, N.; Motomura, S.; Uda, K.; Fujiwara, S.; Suzuki, T. Two arginine kinases of Tetrahymena pyriformis: Characterization and localization. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2014, 171, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.R. Evolution of Cilia. Cold Spring Harb Perspect Biol 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Conejo, M.; Bertin, M.; Pomponi, S.A.; Ellington, W.R. The early evolution of the phosphagen kinases—insights from choanoflagellate and poriferan arginine kinases. J. Mol. Evol. 2008, 66, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Merceron, R.; Awama, A.M.; Montserret, R.; Marcillat, O.; Gouet, P. The Substrate-free and -bound Crystal Structures of the Duplicated Taurocyamine Kinase from the Human Parasite Schistosoma mansoni. J. Biol. Chem. 2015, 290, 12951–12963. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Pullalarevu, S.; Surabian, K.T.; Howard, A.; Suzuki, T.; Moult, J.; Herzberg, O. Structural basis for the mechanism and substrate specificity of glycocyamine kinase, a phosphagen kinase family member. Biochemistry 2010, 49, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Bruschweiler-Li, L.; Davulcu, O.; Skalicky, J.J.; Brüschweiler, R.; Chapman, M.S. Arginine Kinase: Joint Crystallographic and NMR RDC Analyses Link Substrate-Associated Motions to Intrinsic Flexibility. J. Mol. Biol. 2011, 405, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Jourden, M.J.; Clarke, C.N.; Palmer, A.K.; Barth, E.J.; Prada, R.C.; Hale, R.N.; Fraga, D.; Snider, M.J.; Edmiston, P.L. Changing the substrate specificity of creatine kinase from creatine to glycocyamine: Evidence for a highly evolved active site. Biochim. Biophys. Acta - Proteins Proteomics 2007, 1774, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Bush, D.J.; Kirillova, O.; Clark, S.A.; Davulcu, O.; Fabiola, F.; Xie, Q.; Somasundaram, T.; Ellington, W.R.; Chapman, M.S. The structure of lombricine kinase: Implications for phosphagen kinase conformational changes. J. Biol. Chem. 2011, 286, 9338–9350. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ye, S.; Guo, S.; Yan, W.; Bartlam, M.; Rao, Z. Structural basis for a reciprocating mechanism of negative cooperativity in dimeric phosphagen kinase activity. FASEB J. 2009, 24, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Clark, S.A.; Ellington, W.R.; Chapman, M.S. The role of phosphagen specificity loops in arginine kinase. Protein Sci. 2003, 13, 575–585. [Google Scholar] [CrossRef]
- Clark, S.A.; Davulcu, O.; Chapman, M.S. Crystal structures of arginine kinase in complex with ADP, nitrate, and various phosphagen analogs. Biochem. Biophys. Res. Commun. 2012, 427, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qiao, Z.; Ye, S.; Zhang, R. Structure of a double-domain phosphagen kinase reveals an asymmetric arrangement of the tandem domains. Acta Crystallogr. Sect. D 2015, 71, 779–789. [Google Scholar] [CrossRef] [PubMed]
- López-Zavala, A.A.; García-Orozco, K.D.; Carrasco-Miranda, J.S.; Sugich-Miranda, R.; Velázquez-Contreras, E.F.; Criscitiello, M.F.; Brieba, L.G.; Rudiño-Piñera, E.; Sotelo-Mundo, R.R. Crystal structure of shrimp arginine kinase in binary complex with arginine—a molecular view of the phosphagen precursor binding to the enzyme. J. Bioenerg. Biomembr. 2013, 45, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Haouz, A.; Pereira, C.A.; Aguilar, C.; Alzari, P.M. The crystal structure of Trypanosoma cruzi arginine kinase. Proteins Struct. Funct. Bioinforma. 2007, 69, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Laino, A.; Lopez-Zavala, A.A.; Garcia-Orozco, K.D.; Carrasco-Miranda, J.S.; Santana, M.; Stojanoff, V.; Sotelo-Mundo, R.R.; Garcia, C.F. Biochemical and structural characterization of a novel arginine kinase from the spider Polybetes pythagoricus. PeerJ 2017, 5, e3787. [Google Scholar] [CrossRef] [PubMed]
- Kuby, S.A.; Noda, L.; Lardy, H.A. Adenosinetriphosphate-creatine transphosphorylase I. Isolation of the crystalline enzyme from rabbit muscle. J. Biol. Chem. 1954, 209, 191–201. [Google Scholar] [PubMed]
- Keutel, H.J.; Jacobs, H.K.; Okabe, K.; Yue, R.H.; Kuby, S.A. Studies on adenosine triphosphate transphosphorylases. VII. Isolation of the crystalline adenosine triphosphate--creatine transphosphorylase from calf brain. Biochemistry 1968, 7, 4283–4290. [Google Scholar] [CrossRef] [PubMed]
- Ennor, A.H.; Rosenberg, H.; Armstrong, M.D. Specificity of creatine phosphokinase. Nature 1955, 175, 120. [Google Scholar] [CrossRef] [PubMed]
- Uda, K.; Kuwasaki, A.; Shima, K.; Matsumoto, T.; Suzuki, T. The role of Arg-96 in Danio rerio creatine kinase in substrate recognition and active center configuration. Int. J. Biol. Macromol. 2009, 44, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Uda, K.; Fujimoto, N.; Akiyama, Y.; Mizuta, K.; Tanaka, K.; Ellington, W.R.; Suzuki, T. Evolution of the arginine kinase gene family. Comp. Biochem. Physiol. Part D Genomics Proteomics 2006, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Yano, D.; Suzuki, T.; Hirokawa, S.; Fuke, K.; Suzuki, T. Characterization of four arginine kinases in the ciliate Paramecium tetraurelia: Investigation on the substrate inhibition mechanism. Int. J. Biol. Macromol. 2017, 101, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Van Thoai, N.; Robin, Y.; Pradel, L.-A. Hypotaurocyamine phosphokinase comparaison avec la taurocyamine phosphokinase. Biochim. Biophys. Acta (BBA)-Specialized Sect. Enzymol. Subj. 1963, 73, 437–444. [Google Scholar] [CrossRef]
- Uda, K.; Iwai, A.; Suzuki, T. Hypotaurocyamine kinase evolved from a gene for arginine kinase. FEBS Lett. 2005, 579, 6756–6762. [Google Scholar] [CrossRef] [PubMed]
- Robin, Y. Phosphagens and molecular evolution in worms. Biosystems 1974, 6, 49–56. [Google Scholar] [CrossRef]
- Morrison, J.F. 13 Arginine Kinase and Other Invertebrate Guanidino Kinases. In The Enzymes; Elsevier: Amsterdam, The Netherlands, 1973; Volume 8, pp. 457–486. ISBN 1874-6047. [Google Scholar]
- Uda, K.; Tanaka, K.; Bailly, X.; Zal, F.; Suzuki, T. Phosphagen kinase of the giant tubeworm Riftia pachyptila: Cloning and expression of cytoplasmic and mitochondrial isoforms of taurocyamine kinase. Int. J. Biol. Macromol. 2005, 37, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.-Y.; Lee, J.-Y.; Tokuhiro, S.; Nagataki, M.; Jarilla, B.R.; Nomura, H.; Kim, T.I.; Hong, S.-J.; Agatsuma, T. Molecular cloning and characterization of taurocyamine kinase from Clonorchis sinensis: A candidate chemotherapeutic target. PLoS Negl. Trop. Dis. 2013, 7, e2548. [Google Scholar] [CrossRef] [PubMed]
- Jarilla, B.R.; Tokuhiro, S.; Nagataki, M.; Uda, K.; Suzuki, T.; Acosta, L.P.; Agatsuma, T. The role of Y84 on domain 1 and Y87 on domain 2 of Paragonimus westermani taurocyamine kinase: Insights on the substrate binding mechanism of a trematode phosphagen kinase. Exp. Parasitol. 2013, 135, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Surholt, B. Taurocyamine kinase from body-wall musculature of the lugworm Arenicola marina. Eur. J. Biochem. 1979, 93, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsumoto, T.; Suzuki, T. Identification of amino acid residues responsible for taurocyamine binding in mitochondrial taurocyamine kinase from Arenicola brasiliensis. Biochim. Biophys. Acta (BBA) Proteins Proteomics 2011, 1814, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Tokuhiro, S.; Nagataki, M.; Jarilla, B.R.; Uda, K.; Suzuki, T.; Sugiura, T.; Agatsuma, T. Phosphagen kinase in Schistosoma japonicum: II. Determination of amino acid residues essential for substrate catalysis using site-directed mutagenesis. Mol. Biochem. Parasitol. 2014, 194, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Jarilla, B.R.; Tokuhiro, S.; Nagataki, M.; Uda, K.; Suzuki, T.; Acosta, L.P.; Agatsuma, T. Gene structure of the two-domain taurocyamine kinase from Paragonimus westermani: Evidence for a distinct lineage of trematode phosphagen kinases. FEBS Lett. 2013, 587, 2278–2283. [Google Scholar] [CrossRef] [PubMed]
- Tokuhiro, S.; Uda, K.; Yano, H.; Nagataki, M.; Jarilla, B.R.; Suzuki, T.; Agatsuma, T. Phosphagen kinase in Schistosoma japonicum: Characterization of its enzymatic properties and determination of its gene structure. Mol. Biochem. Parasitol. 2013, 188, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Jarilla, B.R.; Tokuhiro, S.; Nagataki, M.; Hong, S.-J.; Uda, K.; Suzuki, T.; Agatsuma, T. Molecular characterization and kinetic properties of a novel two-domain taurocyamine kinase from the lung fluke Paragonimus westermani. FEBS Lett. 2009, 583, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Piccinni, E.; Coppellotti, O. Phosphagens in protozoa—II. Presence of phosphagen kinase in Ochromonas danica. Comp. Biochem. Physiol. Part B Comp. Biochem. 1979, 62, 287–289. [Google Scholar] [CrossRef]
- Sauer, U.; Schlattner, U. Inverse metabolic engineering with phosphagen kinase systems improves the cellular energy state. Metab. Eng. 2004, 6, 220–228. [Google Scholar] [CrossRef]
- Canonaco, F.; Schlattner, U.; Pruett, P.S.; Wallimann, T.; Sauer, U. Functional expression of phosphagen kinase systems confers resistance to transient stresses in Saccharomyces cerevisiae by buffering the ATP pool. J. Biol. Chem. 2002, 277, 31303–31309. [Google Scholar] [CrossRef] [PubMed]
- Canonaco, F.; Schlattner, U.; Wallimann, T.; Sauer, U. Functional expression of arginine kinase improves recovery from pH stress of Escherichia coli. Biotechnol. Lett. 2003, 25, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Van Thoai, N.; Di Jeso, F.; Robin, Y.; Der Terrossian, E. On new adenosine-5′-triphosphoric acid: Guanidine phosphotransferase, opheline kinase. Biochim. Biophys. Acta 1966, 113, 542–550. [Google Scholar] [CrossRef]
- Ennor, A.H.; Rosenberg, H. The isolation of N-phosphoryl-lombricine from earthworms. Biochem. J. 1962, 83, 14 LP–17 LP. [Google Scholar] [CrossRef] [PubMed]
- Euerby, M.R.; Partridge, L.Z.; Gibbons, W.A. High-performance liquid chromatographic determination of lombricine and N-phosphoryl lombricine in the earthworm by pre-column fluorescence derivatisation with o-phthaldialdehyde-ethanethiol. J. Chromatogr. A 1988, 445, 433–440. [Google Scholar] [CrossRef]
- Thoai, N.; Robin, Y. Métabolisme des dérivés guanidylés: IV. Sur une nouvelle guanidine monosubstituée biologique: L’ester guanidoéthylsérylphosphorique (lombricine) et le phosphagène correspondant. Biochim. Biophys. Acta 1954, 14, 76–79. [Google Scholar] [CrossRef]
- Pant, R. Isolation of lombricine and its enzymic phosphorylation. Biochem. J. 1959, 73, 30 LP–33 LP. [Google Scholar] [CrossRef] [PubMed]
- Doumen, C. cDNA identification, comparison and phylogenetic aspects of lombricine kinase from two oligochaete species. Comp. Biochem. Physiol. Part B 2010, 156, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Doumen, C. Variable intron/exon structure in the oligochaete lombricine kinase gene. Gene 2012, 505, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kawasaki, Y.; Furukohri, T.; Ellington, W.R. Evolution of phosphagen kinase. VI. Isolation, characterization and cDNA-derived amino acid sequence of lombricine kinase from the earthworm Eisenia foetida, and identification of a possible candidate for the guanidine substrate recognition site. Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol. 1997, 1343, 152–159. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishimura, Y.; Umekawa, M.; Yamamoto, Y.; Kawamichi, H.; Furukohri, T. Evolution of phosphagen kinase VII. Isolation of glycocyamine kinase from the polychaete Neanthes diversicolor and the cDNA-derived amino acid sequences of alpha and beta chains. J. Protein Chem. 1999, 18, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Van Thoai, N.; Robin, Y.; Guillou, Y. A new phosphagen, N′-phosphorylguanidinoethylphospho-O-( -N,N-dimethyl)serine (phosphothalassemine). Biochemistry 1972, 11, 3890–3895. [Google Scholar] [CrossRef] [PubMed]
- Robin, Y.; van Thoai, N. Sur une nouvelle guanidine monosubstituée biologique, l’hypotaurocyamine (acide 2-guanidoéthanesulfinique) et le phosphagène correspondant. Biochim. Biophys. Acta 1962, 63, 481–488. [Google Scholar] [CrossRef]
- Uda, K.; Saishoji, N.; Ichinari, S.; Ellington, W.R.; Suzuki, T. Origin and properties of cytoplasmic and mitochondrial isoforms of taurocyamine kinase. FEBS J. 2005, 272, 3521–3530. [Google Scholar] [CrossRef]
- Bertin, M.; Pomponi, S.M.; Kokuhuta, C.; Iwasaki, N.; Suzuki, T.; Ellington, W.R. Origin of the genes for the isoforms of creatine kinase. Gene 2007, 392, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Ellington, W.R.; Suzuki, T. Evolution and divergence of creatine kinase genes. Mol. Anat. Physiol. Proteins Creat. Kinase. Nov. Sci. N. Y. 2006, 1–27. [Google Scholar]
- Suzuki, T.; Uda, K.; Adachi, M.; Sanada, H.; Tanaka, K.; Mizuta, C.; Ishida, K.; Ellington, W.R. Evolution of the diverse array of phosphagen systems present in annelids. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2009, 152, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Uda, K.; Matumoto, A.; Suzuki, T. Identification of the key amino acid residues in Sabellastarte arginine kinase for distinguishing chiral guanidino substrates (d- and l-arginine). J. Mol. Catal. B Enzym. 2010, 64, 75–80. [Google Scholar] [CrossRef]
- Uda, K.; Suzuki, T. A novel arginine kinase with substrate specificity towards d-arginine. Protein J. 2007, 26, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef] [PubMed]
- East, D.B. Biochemical Pathways of Creatine and Creatine Phosphate. Ph.D. Thesis, University of Tennessee, Knoxville, TN, USA, 2002. [Google Scholar]
- Rockstein, M. The distribution of phosphoarginine and phosphocreatine in marine invertebrates. Biol. Bull. 1971, 141, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Barclay, C.J. Energy demand and supply in human skeletal muscle. J. Muscle Res. Cell Motil. 2017, 38, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kitzenberg, D.; Colgan, S.P.; Glover, L.E. Creatine kinase in ischemic and inflammatory disorders. Clin. Transl. Med. 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Ferreira, L. Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein (Sao Paulo) 2014, 12, 126–131. [Google Scholar] [CrossRef] [PubMed]
- McLeish, M.J.; Kenyon, G.L. Relating structure to mechanism in creatine kinase. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 1–20. [Google Scholar] [CrossRef]
- Shin, J.-B.; Streijger, F.; Beynon, A.; Peters, T.; Gadzala, L.; McMillen, D.; Bystrom, C.; Van der Zee, C.E.E.M.; Wallimann, T.; Gillespie, P.G. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron 2007, 53, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Shelby, K.S.; Coudron, T.A. Bacterial Elicitation of Transcriptional Response of Female Squash Bug, Anasa tristis (De Geer). Southwest. Entomol. 2017, 42, 37–47. [Google Scholar] [CrossRef]
- Brown, A.E.; Grossman, S.H. The mechanism and modes of inhibition of arginine kinase from the cockroach (Periplaneta americana). Arch. Insect Biochem. Physiol. 2004, 57, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Zhang, L.; Lin, Q.; Liu, N. Arginine kinase: Differentiation of gene expression and protein activity in the red imported fire ant, Solenopsis invicta. Gene 2009, 430, 38–43. [Google Scholar] [CrossRef]
- Tanaka, K.; Ichinari, S.; Iwanami, K.; Yoshimatsu, S.; Suzuki, T. Arginine kinase from the beetle Cissites cephalotes (Olivier). Molecular cloning, phylogenetic analysis and enzymatic properties. Insect Biochem. Mol. Biol. 2007, 37, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.; Mahler, V.; Hayek, B.; Sperr, W.R.; Schöller, M.; Prozell, S.; Wiedermann, G.; Valent, P.; Valenta, R.; Duchêne, M. Molecular and immunological characterization of arginine kinase from the Indianmeal moth, Plodia interpunctella, a novel cross-reactive invertebrate pan-allergen. J. Immunol. 2001, 167, 5470–5477. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, M. Mitochondrial arginine kinase in the midgut of the tobacco hornworm (Manduca sexta). J. Exp. Biol. 1997, 200, 2789–2796. [Google Scholar] [PubMed]
- Rosenthal, G.A.; Dahlman, D.L.; Robinson, G.W. l-Arginine kinase from tobacco hornworm, Manduca sexta (L.). Purification, properties, and interaction with l-canavanine. J. Biol. Chem. 1977, 252, 3679–3683. [Google Scholar] [PubMed]
- Wu, Q.-Y.; Li, F.; Zhu, W.-J.; Wang, X.-Y. Cloning, expression, purification, and characterization of arginine kinase from Locusta migratoria manilensis. Comp. Biochem. Physiol. Part B 2007, 148, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ilg, T.; Werr, M. Arginine kinase of the sheep blowfly Lucilia cuprina: Gene identification and characterization of the native and recombinant enzyme. Pestic. Biochem. Physiol. 2012, 102, 115–123. [Google Scholar] [CrossRef]
- Cheung, A.C. Kinetic properties of arginine phosphokinase from honeybees, Apis mellifera L. (Hymenoptera, Apidae). Arch. Biochem. Biophys. 1973, 154, 28–39. [Google Scholar] [CrossRef]
- Kucharski, R.; Maleszka, R. Arginine kinase is highly expressed in the compound eye of the honey bee, Apis mellifera. Gene 1998, 211, 343–349. [Google Scholar] [CrossRef]
- Kang, L.; Shi, H.; Liu, X.; Zhang, C.; Yao, Q.; Wang, Y.; Chang, C.; Shi, J.; Cao, J.; Kong, J.; et al. Arginine kinase is highly expressed in a resistant strain of silkworm (Bombyx mori, Lepidoptera): Implication of its role in resistance to Bombyx mori nucleopolyhedrovirus. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2011, 158, 230–234. [Google Scholar] [CrossRef]
- Werr, M.; Cramer, J.; Ilg, T. Identification and characterization of two arginine kinases from the parasitic insect Ctenocephalides felis. Insect Biochem. Mol. Biol. 2009, 39, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Eppenberger, H.M. Properties of arginine kinase from Drosophila melanogaster. Eur. J. Biochem. 1973, 38, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Rockstein, M.; Kumar, S.S. Arginine kinase from the housefly, Musca domestica: Purification and properties. Insect Biochem. 1972, 2, 344–352. [Google Scholar] [CrossRef]
- Baker III, G.T. Purification and some properties of arginine phosphokinase from the blow fly, Phormia regina. Insect Biochem. 1976, 6, 449–456. [Google Scholar] [CrossRef]
- Dong, F.; Zhang, N.; Xie, Z.; Meng, X.; Qian, K.; Ji, C.; Lu, M.; Du, Y.; Wang, J. Characterization and in vitro expression of arginine kinase gene in the invasive western flower thrips, Frankliniella occidentalis. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2019, 229, 51–57. [Google Scholar] [CrossRef]
- Bobolea, I.; Barranco, P.; Pastor-Vargas, C.; Iraola, V.; Vivanco, F.; Quirce, S. Arginine kinase from the cellar spider (Holocnemus pluchei): A new asthma-causing allergen. Int. Arch. Allergy Immunol. 2011, 155, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Arjunwadkar, A.V.; Reddy, S.R.R. Characterization and distribution of arginine kinase in the tissues of the scorpion, Palamneus phipsoni. Can. J. Zool. 1985, 63, 2262–2266. [Google Scholar] [CrossRef]
- Lyu, K.; Zhang, L.; Zhu, X.; Cui, G.; Wilson, A.E.; Yang, Z. Arginine kinase in the cladoceran Daphnia magna: cDNA sequencing and expression is associated with resistance to toxic Microcystis. Aquat. Toxicol. 2015, 160, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Somasundaram, T.; Blanc, E.; Parthasarathy, G.; Ellington, W.R.; Chapman, M.S. Transition state structure of arginine kinase: Implications for catalysis of bimolecular reactions. Proc. Natl. Acad. Sci. USA 1998, 95, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Strong, S.J.; Ellington, W.R. Isolation and sequence analysis of the gene for arginine kinase from the chelicerate arthropod, Limulus polyphemus: Insights into catalytically important residues. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 1995, 1246, 197–200. [Google Scholar] [CrossRef]
- Zhang, G.; Yan, G.; Yang, X.; Wong, Y.; Sun, J.; Zhang, Y.; He, L.; Xu, Y.; Qian, P. Characterization of arginine kinase in the barnacle Amphibalanus amphitrite and its role in the larval settlement. J. Exp. Zool. Part B Mol. Dev. Evol. 2016, 326, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Zavala, A.A.; Sotelo-Mundo, R.R.; Garcia-Orozco, K.D.; Isac-Martinez, F.; Brieba, L.G.; Rudiño-Piñera, E. Crystallization and X-ray diffraction studies of arginine kinase from the white Pacific shrimp Litopenaeus vannamei. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Gattis, J.L.; Ruben, E.; Fenley, M.O.; Ellington, W.R.; Chapman, M.S. The active site cysteine of arginine kinase: Structural and functional analysis of partially active mutants. Biochemistry 2004, 43, 8680–8689. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-L.; Mao, H.-Y.; Cao, M.-J.; Cai, Q.-F.; Su, W.-J.; Zhang, Y.-X.; Liu, G.-M. Purification, physicochemical and immunological characterization of arginine kinase, an allergen of crayfish (Procambarus clarkii). Food Chem. Toxicol. 2013, 62, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Cao, M.-J.; Cai, Q.-F.; Su, W.-J.; Yu, H.-L.; Ruan, W.-W.; Liu, G.-M. Purification, cloning, expression and immunological analysis of Scylla serrata arginine kinase, the crab allergen. J. Sci. Food Agric. 2011, 91, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-S.; Zheng, Z.-L.; Lei, J.; Pan, J.-C.; Zou, G.-L. Cloning, expression, characterization and phylogenetic analysis of arginine kinase from greasyback shrimp (Metapenaeus ensis). Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2009, 153, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Zavala, A.A.; Sotelo-Mundo, R.R.; Hernandez-Flores, J.M.; Lugo-Sanchez, M.E.; Sugich-Miranda, R.; Garcia-Orozco, K.D. Arginine kinase shows nucleoside diphosphate kinase-like activity toward deoxythymidine diphosphate. J. Bioenerg. Biomembr. 2016, 48, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-L.; Ji, P.-F.; Kong, P.; Wang, Z.-Y.; Xiang, J.-H. Arginine kinase from Litopenaeus vannamei: Cloning, expression and catalytic properties. Fish Shellfish Immunol. 2009, 26, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Iwanami, K.; Iseno, S.; Uda, K.; Suzuki, T. A novel arginine kinase from the shrimp Neocaridina denticulata: The fourth arginine kinase gene lineage. Gene 2009, 437, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.-Y.; Lee, J.; Yin, S.-J.; Wang, W.; Wang, Z.-J.; Yang, J.-M.; Qian, G.-Y.; Si, Y.-X.; Park, Y.-D. Effects of osmolytes on arginine kinase from Euphausia superba: A study on thermal denaturation and aggregation. Process Biochem. 2014, 49, 936–947. [Google Scholar] [CrossRef]
- Si, Y.-X.; Song, J.-J.; Fang, N.-Y.; Wang, W.; Wang, Z.-J.; Yang, J.-M.; Qian, G.-Y.; Yin, S.-J.; Park, Y.-D. Purification, characterization, and unfolding studies of arginine kinase from Antarctic krill. Int. J. Biol. Macromol. 2014, 67, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Arockiaraj, J.; Vanaraja, P.; Easwvaran, S.; Singh, A.; Alinejaid, T.; Othman, R.Y.; Bhassu, S. Gene profiling and characterization of arginine kinase-1 (MrAK-1) from freshwater giant prawn (Macrobrachium rosenbergii). Fish Shellfish Immunol. 2011, 31, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Livera, W.C.D.; Shimizu, C. Comparison and Characterization of Arginine Kinases Purified from the Prawn Penaeus japonicus (Kurumaebi) and the Swimming Crab Portunus trituberculatus (Gazami). Agric. Biol. Chem. 1989, 53, 2377–2386. [Google Scholar] [CrossRef]
- Jiang, S.; Jia, Z.; Chen, H.; Wang, L.; Song, L. The modulation of haemolymph arginine kinase on the extracellular ATP induced bactericidal immune responses in the Pacific oyster Crassostrea gigas. Fish Shellfish Immunol. 2016, 54, 282–293. [Google Scholar] [CrossRef]
- Fujimoto, N.; Tanaka, K.; Suzuki, T. Amino acid residues 62 and 193 play the key role in regulating the synergism of substrate binding in oyster arginine kinase. FEBS Lett. 2005, 579, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Mizuta, C.; Uda, K.; Fujimoto, N.; Okamoto, M.; Suzuki, T. Unique evolution of Bivalvia arginine kinases. Cell. Mol. Life Sci. C. 2004, 61, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, L.; Zhou, Z.; Yang, C.; Gao, Y.; Wang, L.; Song, L. The arginine kinase in Zhikong scallop Chlamys farreri is involved in immunomodulation. Dev. Comp. Immunol. 2012, 37, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Compaan, D.M.; Ellington, W.R. Functional consequences of a gene duplication and fusion event in an arginine kinase. J. Exp. Biol. 2003, 206, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Uda, K.; Yamamoto, K.; Iwasaki, N.; Iwai, M.; Fujikura, K.; Ellington, W.R.; Suzuki, T. Two-domain arginine kinase from the deep-sea clam Calyptogena kaikoi--evidence of two active domains. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2008, 151, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, K.; Tada, H.; Uda, K. Cold-adapted features of arginine kinase from the deep-sea clam Calyptogena kaikoi. Mar. Biotechnol. 2012, 14, 294–303. [Google Scholar] [CrossRef]
- Suzuki, T.; Sugimura, N.; Taniguchi, T.; Unemi, Y.; Murata, T.; Hayashida, M.; Yokouchi, K.; Uda, K.; Furukohri, T. Two-domain arginine kinases from the clams Solen strictus and Corbicula japonica: Exceptional amino acid replacement of the functionally important D(62) by G. Int. J. Biochem. Cell Biol. 2002, 34, 1221–1229. [Google Scholar] [CrossRef]
- Suzuki, T.; Tomoyuki, T.; Uda, K. Kinetic properties and structural characteristics of an unusual two-domain arginine kinase of the clam Corbicula japonica. FEBS Lett. 2003, 533, 95–98. [Google Scholar] [CrossRef]
- Reddy, S.R.; Roustan, C.; Benyamin, Y. Purification and properties of two molecular forms of arginine kinase from the adductor muscle of the scallop, Pecten maximus. Comp. Biochem. Physiol. B. 1991, 99, 387–394. [Google Scholar] [CrossRef]
- Kong, X.; Liu, H.; Zhang, H. Positive selection adaptation of two-domain arginine kinase (AK) from cold seep Vesicomyidae clams. Mol. Biol. Rep. 2018, 45, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.-Y.; Zhang, L.-L.; Wu, F.; Fu, Y.-Y.; Yin, S.-J.; Si, Y.-X.; Park, Y.-D. Kinetics for Cu2+ induced Sepia pharaonis arginine kinase inactivation and aggregation. Int. J. Biol. Macromol. 2016, 91, 926–933. [Google Scholar] [CrossRef]
- Si, Y.-X.; Lee, J.; Cheng, J.-G.; Yin, S.-J.; Park, Y.-D.; Qian, G.-Y.; Jiang, X.-M. Kinetics for Zinc Ion Induced Sepia Pharaonis Arginine Kinase Inactivation and Aggregation. Protein Pept. Lett. 2016, 23, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-W.; Cao, M.-J.; Cai, Q.-F.; Ruan, M.-M.; Mao, H.-Y.; Su, W.-J.; Liu, G.-M. Purification, cloning, and immunological characterization of arginine kinase, a novel allergen of Octopus fangsiao. J. Agric. Food Chem. 2012, 60, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Fukuta, H.; Nagato, H.; Umekawa, M. Arginine kinase from Nautilus pompilius, a living fossil. Site-directed mutagenesis studies on the role of amino acid residues in the Guanidino specificity region. J. Biol. Chem. 2000, 275, 23884–23890. [Google Scholar] [CrossRef]
- Storey, K.B. Purification and characterization of arginine kinase from the mantle muscle of the squid, Symplectoteuthis oualaniensis. Role of the phosphagen/phosphagen kinase system in a highly aerobic muscle. Arch. Biochem. Biophys. 1977, 179, 518–529. [Google Scholar] [CrossRef]
- Jarilla, B.R.; Uda, K.; Suzuki, T.; Acosta, L.P.; Urabe, M.; Agatsuma, T. Characterization of arginine kinase from the caenogastropod Semisulcospira libertina, an intermediate host of Paragonimus westermani. J. Molluscan Stud. 2014, 80, 444–451. [Google Scholar] [CrossRef]
- Agatsuma, T.; Fukunaga, S.; Jarilla, B.R.; Nagataki, M.; Tokuhiro, S.; Xiao, J.-Y.; Devi, K.R.; Nomura, H.; Shimada, M.; Uda, K. Molecular characterization of a cDNA-derived phosphagen kinase from Biomphalaria glabrata, the intermediate host of Schistosoma mansoni. Med. Entomol. Zool. 2011, 62, 1–11. [Google Scholar] [CrossRef]
- Uda, K.; Ellington, W.R.; Suzuki, T. A diverse array of creatine kinase and arginine kinase isoform genes is present in the starlet sea anemone Nematostella vectensis, a cnidarian model system for studying developmental evolution. Gene 2012, 497, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kawasaki, Y.; Furukohri, T. Evolution of phosphagen kinase. Isolation, characterization and cDNA-derived amino acid sequence of two-domain arginine kinase from the sea anemone Anthopleura japonicus. Biochem. J. 1997, 328, 301–306. [Google Scholar] [CrossRef]
- Matsuo, T.; Yano, D.; Uda, K.; Iwasaki, N.; Suzuki, T. Arginine Kinases from the Precious Corals Corallium rubrum and Paracorallium japonicum: Presence of Two Distinct Arginine Kinase Gene Lineages in Cnidarians. Protein, J. 2017, 36, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Yano, D.; Mimura, S.; Uda, K.; Suzuki, T. Arginine kinase from Myzostoma cirriferum, a basal member of annelids. Comp. Biochem. Physiol. Part B 2016, 198, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.-X.; Wong, Y.-H.; Qian, P.-Y. The regulatory role of arginine kinase during larval settlement of the bryozoan Bugula neritina. Mar. Biol. 2018, 165, 52. [Google Scholar] [CrossRef]
- Guo, S.-Y.; Guo, Z.; Guo, Q.; Chen, B.-Y.; Wang, X.-C. Expression, purification, and characterization of arginine kinase from the sea cucumber Stichopus japonicus. Protein Expr. Purif. 2003, 29, 230–234. [Google Scholar] [CrossRef]
- Held, B.C.; Wright-Weber, B.; Grossman, S.H. Kinetic analysis of two purified forms of arginine kinase: Absence of cooperativity in substrate binding of dimeric phosphagen kinase. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2007, 148, 6–13. [Google Scholar] [CrossRef]
- Seals, J.D.; Grossman, S.H. Purification and characterization of arginine kinase from the sea cucumber Caudina arenicola. Comp. Biochem. Physiol. Part B Comp. Biochem. 1988, 89, 701–707. [Google Scholar] [CrossRef]
- Fujimaki, H.; Yanagisawa, T. Changes in activities of creatine kinase, arginine kinase and their multienzyme forms during embryonic and larval development of sea urchins. Dev. Growth Differ. 1978, 20, 125–131. [Google Scholar] [CrossRef]
- Ratto, A.; Christen, R. Purification and characterization of arginine kinase from sea-urchin eggs. Eur. J. Biochem. 1988, 173, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Chouno, K.; Yano, D.; Uda, K.; Fujita, T.; Iwasaki, N.; Suzuki, T. Arginine kinases from the marine feather star Tropiometra afra macrodiscus: The first finding of a prenylation signal sequence in metazoan phosphagen kinases. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2015, 187, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Perovic-Ottstadt, S.; Wiens, M.; Schroder, H.-C.; Batel, R.; Giovine, M.; Krasko, A.; Muller, I.M.; Muller, W.E.G. Arginine kinase in the demosponge Suberites domuncula: Regulation of its expression and catalytic activity by silicic acid. J. Exp. Biol. 2005, 208, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.F.; MacDonald, M.H.; Thai, V.K.; Tucker, M.L. Molecular characterization of arginine kinases in the soybean cyst nematode (Heterodera glycines). J. Nematol. 2003, 35, 252. [Google Scholar] [PubMed]
- Umair, S.; Knight, J.S.; Bland, R.J.; Simpson, H. V Molecular and biochemical characterisation of arginine kinases in Haemonchus contortus and Teladorsagia circumcincta. Exp. Parasitol. 2013, 134, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Platzer, E.G.; Wang, W.; Thompson, S.N.; Borchardt, D.B. Arginine kinase and phosphoarginine, a functional phosphagen, in the rhabditoid nematode steinernema carpocapsae. J. Parasitol. 1999, 85, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.; Yatawara, L.; Nagataki, M.; Agatsuma, T. Arginine kinase in Toxocara canis: Exon–intron organization, functional analysis of site-directed mutants and evaluation of putative enzyme inhibitors. Asian Pac. J. Trop. Med. 2016, 9, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, M.; Gao, W.; Gadahi, J.A.; Lu, M.; Liu, X.; Wang, Y.; Yan, R.; Xu, L.; Song, X.; Li, X. Arginine kinase from Haemonchus contortus decreased the proliferation and increased the apoptosis of goat PBMCs in vitro. Parasit. Vectors 2017, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Kulathunga, D.G.R.S.; Wickramasinghe, S.; Rajapakse, R.P.V.J.; Yatawara, L.; Jayaweera, W.R.; Agatsuma, T. Immunolocalization of arginine kinase (AK) in Toxocara canis, Toxocara vitulorum, and Ascaris lumbricoides. Parasitol. Res. 2012, 111, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Nagataki, M.; Uda, K.; Jarilla, B.R.; Tokuhiro, S.; Wickramasinghe, S.; Suzuki, T.; Blair, D.; Agatsuma, T. Molecular and catalytic properties of an arginine kinase from the nematode Ascaris suum. J. Helminthol. 2012, 86, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Fraga, D.; Aryal, M.; Hall, J.E.; Rae, E.; Snider, M. Characterization of the arginine kinase isoforms in Caenorhabditis elegans. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2015, 187, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.-P.; Rotureau, B.; Gribaldo, S.; Georgikou, C.; Julkowska, D.; Blisnick, T.; Perrot, S.; Subota, I.; Bastin, P. The Flagellar Arginine Kinase in Trypanosoma brucei Is Important for Infection in Tsetse Flies. PLoS ONE 2015, 10, e0133676. [Google Scholar] [CrossRef] [PubMed]
- Voncken, F.; Gao, F.; Wadforth, C.; Harley, M.; Colasante, C. The Phosphoarginine Energy-Buffering System of Trypanosoma brucei Involves Multiple Arginine Kinase Isoforms with Different Subcellular Locations. PLoS ONE 2013, 8, e65908. [Google Scholar] [CrossRef] [PubMed]
- Canepa, G.E.; Carrillo, C.; Miranda, M.R.; Saye, M.; Pereira, C.A. Arginine kinase in Phytomonas, a trypanosomatid parasite of plants. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2011, 160, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.A.; Alonso, G.D.; Paveto, M.C.; Iribarren, A.; Cabanas, M.L.; Torres, H.N.; Flawiá, M.M. Trypanosoma cruzi arginine kinase characterization and cloning a novel energetic pathway in protozoan parasites. J. Biol. Chem. 2000, 275, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.A.; Alonso, G.D.; Ivaldi, S.; Silber, A.M.; Alves, M.J.M.; Torres, H.N.; Flawiá, M.M. Arginine kinase overexpression improves Trypanosoma cruzi survival capability. FEBS Lett. 2003, 554, 201–205. [Google Scholar] [CrossRef]
- Noguchi, M.; Sawada, T.; Akazawa, T. ATP-regenerating system in the cilia of Paramecium caudatum. J. Exp. Biol. 2001, 204, 1063 LP–1071 LP. [Google Scholar]
- Bragg, J.; Rajkovic, A.; Anderson, C.; Curtis, R.; Van Houten, J.; Begres, B.; Naples, C.; Snider, M.; Fraga, D.; Singer, M. Identification and Characterization of a Putative Arginine Kinase Homolog from Myxococcus xanthus Required for Fruiting Body Formation and Cell Different. J. Bacteriol. 2012, 194, 2668 LP–2676 LP. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Uda, K.; Ishida, K.; Kokufuta, C.; Iwasaki, N.; Suzuki, T. Comparison of kinetic constants of creatine kinase isoforms. Int. J. Biol. Macromol. 2006, 38, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Winnard Jr, P.; Cashon, R.E.; Sidell, B.D.; Vayda, M.E. Isolation, characterization and nucleotide sequence of the muscle isoforms of creatine kinase from the Antarctic teleost Chaenocephalus aceratus. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2003, 134, 651–667. [Google Scholar] [CrossRef]
- Fisher, S.E.; Whitt, G.S. Purification of the creatine kinase isozymes of the green sunfish (Lepomis cyanellus) with Blue Sepharose CL-6B. Anal. Biochem. 1979, 94, 89–95. [Google Scholar] [CrossRef]
- Grzyb, K.; Skorkowski, E.F. Purification and some properties of two creatine kinase isoforms from herring (Clupea harengus) spermatozoa. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2006, 144, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Grzyb, K.; Rychłowski, M.; Biegniewska, A.; Skorkowski, E.F. Quantitative determination of creatine kinase release from herring (Clupea harengus) spermatozoa induced by tributyltin. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2003, 134, 207–213. [Google Scholar] [CrossRef]
- Grzyb, K.; Skorkowski, E.F. Characterization of creatine kinase isoforms in herring (Clupea harengus) skeletal muscle. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2005, 140, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecka, N.; Grzyb, K.; Nona-Moldawa, A.; Gronczewska, J.; Skorkowski, E.F. Purification and stability of octameric mitochondrial creatine kinase isoform from herring (Clupea harengus) organ of vision. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2015, 185, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Gosselin-Rey, C.; Gerday, C. Isolation and molecular properties of creatine kinase from carp white muscle. Biochim. Biophys. Acta 1970, 221, 241–254. [Google Scholar] [CrossRef]
- Nakagawa, T.; Nagayama, F. Enzymatic properties of fish muscle creatine kinase. Comp. Biochem. Physiol. Part B Comp. Biochem. 1991, 98, 349–354. [Google Scholar] [CrossRef]
- Wu, C.-L.; Li, B.-Y.; Wu, J.-L.; Hui, C.-F. The activity of carp muscle-specific creatine kinase at low temperature is enhanced by decreased hydrophobicity of residue 268. Physiol. Biochem. Zool. 2014, 87, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Birkedal, R.; Gesser, H. Creatine kinase and mitochondrial respiration in hearts of trout, cod and freshwater turtle. J. Comp. Physiol. B. 2003, 173, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Wang, P.-F.; Babbitt, P.C.; McLeish, M.J.; Kenyon, G.L.; Allen, K.N. The 2.1 Å Structure of Torpedo californica Creatine Kinase Complexed with the ADP-Mg2+− NO3-− Creatine Transition-State Analogue Complex. Biochemistry 2002, 41, 13861–13867. [Google Scholar] [CrossRef]
- Barrantes, F.J.; Braceras, A.; Caldironi, H.A.; Mieskes, G.; Moser, H.; Toren, E.C.J.; Roque, M.E.; Wallimann, T.; Zechel, A. Isolation and characterization of acetylcholine receptor membrane-associated (nonreceptor v2-protein) and soluble electrocyte creatine kinases. J. Biol. Chem. 1985, 260, 3024–3034. [Google Scholar] [PubMed]
- Gray, K.A.; Grossman, S.H.; Summers, D.D. Purification and characterization of creatine kinase isozymes from the nurse shark Ginglymostoma cirratum. Comp. Biochem. Physiol. B. 1986, 83, 613–620. [Google Scholar] [CrossRef]
- Simonarson, B.; Watts, D.C. Purification and properties of adenosine triphosphate-creatine phosphotransferase from muscle of the dogfish Scylliorhinus canicula. Biochem. J. 1972, 128, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Kobel, H.R. Purification and characterization of cytoplasmic creatine kinase isozymes of Xenopus laevis. Biochem. Genet. 1988, 26, 543–555. [Google Scholar] [CrossRef]
- Wang, W.; Lee, J.; Hao, H.; Park, Y.-D.; Qian, G.-Y. Hydrogen peroxide (H2O2) irreversibly inactivates creatine kinase from Pelodiscus sinensis by targeting the active site cysteine. Int. J. Biol. Macromol. 2017, 105, 1595–1601. [Google Scholar] [CrossRef]
- Lipskaya, T.Y. Mitochondrial creatine kinase: Properties and function. Biochemistry. 2001, 66, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.; Schlegel, J.; James, P.; Eppenberger, H.M.; Wallimann, T. Mitochondrial creatine kinase from chicken brain. Purification, biophysical characterization, and generation of heterodimeric and heterooctameric molecules with subunits of other creatine kinase isoenzymes. J. Biol. Chem. 1990, 265, 15900–15908. [Google Scholar] [PubMed]
- Abnous, K.; Storey, K.B. Regulation of skeletal muscle creatine kinase from a hibernating mammal. Arch. Biochem. Biophys. 2007, 467, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Tisi, D.; Bax, B.; Loew, A. The three-dimensional structure of cytosolic bovine retinal creatine kinase. Acta Crystallogr. Sect. D 2001, 57, 187–193. [Google Scholar] [CrossRef]
- Bong, S.M.; Moon, J.H.; Nam, K.H.; Lee, K.S.; Chi, Y.M.; Hwang, K.Y. Structural studies of human brain-type creatine kinase complexed with the ADP–Mg2+–NO3−–creatine transition-state analogue complex. FEBS Lett. 2008, 582, 3959–3965. [Google Scholar] [CrossRef] [PubMed]
- Kouttinen, A. Purification of human and canine creatine kinase isozymes. Acta Med, Scand Suppl 1978, 623, 115–117. [Google Scholar]
- Roberts, R.; Grace, A.M. Purification of mitochondrial creatine kinase. Biochemical and immunological characterization. J. Biol. Chem. 1980, 255, 2870–2877. [Google Scholar] [PubMed]
- Couthon, F.; Clottes, E.; Vial, C. High salt concentrations induce dissociation of dimeric rabbit muscle creatine kinase. Physico-chemical characterization of the monomeric species. Biochim. Biophys. Acta 1997, 1339, 277–288. [Google Scholar] [CrossRef]
- Iwanami, K.; Uda, K.; Tada, H.; Suzuki, T. Cytoplasmic and mitochondrial creatine kinases from the skeletal muscle of sperm whale (Physeter macrocephalus). Molecular cloning and enzyme characterization. Protein J. 2008, 27, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Terblanche, S.E.; Masondo, T.C.; Nel, W. Effects of cold acclimation on the activity levels of creatine kinase, lactate dehydrogenase and lactate dehydrogenase isoenzymes in various tissues of the rat. Cell Biol. Int. 1998, 22, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, C.; Tanaka, K.; Suzuki, T. Isolation, characterization, and cDNA-derived amino acid sequence of glycocyamine kinase from the tropical marine worm Namalycastis sp. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2005, 140, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.G.; Sona, S.; Bertin, M.; Ellington, W.R. The role of an absolutely conserved tryptophan residue in octamer formation and stability in mitochondrial creatine kinases. Biochim. Biophys. Acta 2006, 1764, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Tombes, R.M.; Shapiro, B.M. Metabolite channeling: A phosphorylcreatine shuttle to mediate high energy phosphate transport between sperm mitochondrion and tail. Cell 1985, 41, 325–334. [Google Scholar] [CrossRef]
- Tombes, R.M.; Brokaw, C.J.; Shapiro, B.M. Creatine kinase-dependent energy transport in sea urchin spermatozoa. Flagellar wave attenuation and theoretical analysis of high energy phosphate diffusion. Biophys. J. 1987, 52, 75–86. [Google Scholar] [CrossRef]
- Tombes, R.M. Isolation and characterization of sea urchin flagellar creatine kinase. Methods Cell Biol. 1995, 47, 467–472. [Google Scholar] [PubMed]
- Sona, S.; Suzuki, T.; Ellington, W.R. Cloning and expression of mitochondrial and protoflagellar creatine kinases from a marine sponge: Implications for the origin of intracellular energy transport systems. Biochem. Biophys. Res. Commun. 2004, 317, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Kagda, M.S.; Vu, A.L.; Ah-Fong, A.M.V.; Judelson, H.S. Phosphagen kinase function in flagellated spores of the oomycete Phytophthora infestans integrates transcriptional regulation, metabolic dynamics and protein retargeting. Mol. Microbiol. 2018, 110, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Uda, K.; Hoshijima, M.; Suzuki, T. A novel taurocyamine kinase found in the protist Phytophthora infestans. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 2013, 165, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.; Begres, B.N.; Van Houten, J.M.; Snider, M.J.; Fraga, D. Characterization of a putative oomycete taurocyamine kinase: Implications for the evolution of the phosphagen kinase family. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2013, 166, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Schiedek, D. Nephtys hombergii, a free-living predator in marine sediments: Energy production under environmental stress. Mar. Biol. 1997, 129, 643–650. [Google Scholar] [CrossRef]
- Pradel, L.A.; Kassab, R.; Conlay, C.; Thoai, N. Van Properties and amino acid composition of purified ATP: Guanidinoacetate phosphotransferase. Biochim. Biophys. Acta 1968, 154, 305–314. [Google Scholar] [CrossRef]
- Furukohri, T.; Suzuki, T. Preparation of Glycocyamine Kinase from Polychaete, Perinereis Brevicirrus; Reports of the Usa Marine Biological Institute-Kochi University; Usa Marine Biological Institute: Tosa, Japan, 1987. [Google Scholar]
- Shirokane, Y.; Nakajima, M.; Mizusawa, K. Purification and Properties of Guanidinoacetate Kinase from a Polychaete, Perinereis sp. Agric. Biol. Chem. 1991, 55, 2235–2242. [Google Scholar] [CrossRef]
- Virden, R.; Watts, D.C. The distribution of guanidine-adenosine triphosphate phosphotransferases and adenosine triphosphatase in animals from several phyla. Comp. Biochem. Physiol. 1964, 13, 161–177. [Google Scholar] [CrossRef]
- Ellington, W.R.; Bush, J. Cloning and expression of a lombricine kinase from an echiuroid worm: Insights into structural correlates of substrate specificity. Biochem. Biophys. Res. Commun. 2002, 291, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Uda, K.; Shimada, M.; Takahashi, K.; Gamou, S.; Ellington, W.R.; Suzuki, T. Evolution of the Cytoplasmic and Mitochondrial Phosphagen Kinases Unique to Annelid Groups. J. Mol. Evol. 2007, 65, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, T.J.; Rosenberg, H.; Ennor, A.H. The purification and properties of adenosine triphosphate-lombricine phosphotransferase. Biochem. J. 1964, 90, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Florkin, M. Chemical Zoology V4: Annelida, Echiuria, and Sipuncula; Elsevier: Amsterdam, The Netherlands, 2012; ISBN 0323145876. [Google Scholar]
- Gercken, G.; Döring, V. Inhibition of creatine kinase by creatinine phosphate. FEBS Lett. 1974, 46, 87–91. [Google Scholar] [CrossRef]
- Iyengar, M.R.; Coleman, D.W.; Butler, T.M. Phosphocreatinine, a high-energy phosphate in muscle, spontaneously forms phosphocreatine and creatinine under physiological conditions. J. Biol. Chem. 1985, 260, 7562–7567. [Google Scholar] [PubMed]
- VanWagenen, B.C.; Larsen, R.; Cardellina, J.H.; Randazzo, D.; Lidert, Z.C.; Swithenbank, C. Ulosantoin, a potent insecticide from the sponge Ulosa ruetzleri. J. Org. Chem. 1993, 58, 335–337. [Google Scholar] [CrossRef]
- Fankhauser, H.; Schiff, J.A.; Garber, L.J. Purification and properties of adenylyl sulphate:ammonia adenylyltransferase from Chlorella catalysing the formation of adenosine 5′-phosphoramidate from adenosine 5′-phosphosulphate and ammonia. Biochem. J. 1981, 195, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, H.; Berkowitz, G.A.; Schiff, J.A. A nucleotide with the properties of adenosine 5′ phosphoramidate from Chlorella cells. Biochem. Biophys. Res. Commun. 1981, 101, 524–532. [Google Scholar] [CrossRef]
- Bretes, E.; Wojdyla-Mamon, A.M.; Kowalska, J.; Jemielity, J.; Kaczmarek, R.; Baraniak, J.; Guranowski, A. Hint2, the mitochondrial nucleoside 5′-phosphoramidate hydrolase; properties of the homogeneous protein from sheep (Ovis aries) liver. Acta Biochim. Pol. 2013, 60, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Guranowski, A.; Wojdyla, A.M.; Rydzik, A.M.; Stepinski, J.; Jemielity, J. Plant nucleoside 5’-phosphoramidate hydrolase; simple purification from yellow lupin (Lupinus luteus) seeds and properties of homogeneous enzyme. Acta Biochim. Pol. 2011, 58, 131–136. [Google Scholar] [PubMed]
- Wojdyla-Mamon, A.M.; Guranowski, A. Adenylylsulfate-ammonia adenylyltransferase activity is another inherent property of Fhit proteins. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Rossomando, E.F.; Crean, E.V.; Kestler, D.P. Isolation and characterization of an adenylyl-protein complex formed during the incubation of membranes from Dictyostelium discoideum with ATP. Biochim. Biophys. Acta (BBA) General Subj. 1981, 675, 386–391. [Google Scholar] [CrossRef]
- Hadjimichael, J.; Rossomando, E.F. Isolation and characterization of the protein phosphoamidates formed by a membrane bound adenylyl transferase reaction in Dictyostelium discoideum. Int. J. Biochem. 1991, 23, 535–539. [Google Scholar] [CrossRef]
- Lerbs, W.; Luckner, M. Cyclopeptine synthetase activity in surface cultures of Penicillium cyclopium. J. Basic Microbiol. 1985, 25, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Schwelle, N.; Lerbs, W.; Luckner, M. Enzymatic synthesis of cyclopeptine intermediates in Penicillium cyclopium. Phytochemistry 1985, 24, 1935–1939. [Google Scholar] [CrossRef]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system–From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, S.O.; Larghi, E.L.; Kaufman, T.S. The 3,4-dioxygenated 5-hydroxy-4-aryl-quinolin-2(1H)-one alkaloids. Results of 20 years of research, uncovering a new family of natural products. Nat. Prod. Rep. 2016, 33, 1425–1446. [Google Scholar] [CrossRef] [PubMed]
- Purich, D.L. Enzyme Kinetics: Catalysis and Control: A Reference of Theory and Best-Practice Methods; Elsevier: Amsterdam, The Netherlands, 2010; ISBN 0123809258. [Google Scholar]
- Martin, J.; Magnino, F.; Schmidt, K.; Piguet, A.; Lee, J.; Semela, D.; St–Pierre, M.V.; Ziemiecki, A.; Cassio, D.; Brenner, C.; et al. Hint2, A Mitochondrial Apoptotic Sensitizer Down-Regulated in Hepatocellular Carcinoma. Gastroenterology 2006, 130, 2179–2188. [Google Scholar] [CrossRef]
- Seidle, H.F.; Bieganowski, P.; Brenner, C. Disease-associated mutations inactivate AMP-lysine hydrolase activity of Aprataxin. J. Biol. Chem. 2005, 280, 20927–20931. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, S.; Antigny, F.; Vetterli, L.; Dufour, J.-F.; Rossier, M.F. Hint2 Is Expressed in the Mitochondria of H295R Cells and Is Involved in Steroidogenesis. Endocrinology 2008, 149, 5461–5469. [Google Scholar] [CrossRef] [PubMed]
- Maize, K.M.; Wagner, C.R.; Finzel, B.C. Structural characterization of human histidine triad nucleotide-binding protein 2, a member of the histidine triad superfamily. FEBS J. 2013, 280, 3389–3398. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-F.; Bieganowski, P.; Shilinski, K.; Cheng, J.; Brenner, C.; Wagner, C.R. 31P NMR and genetic analysis establish hinT as the only Escherchia coli purine nucleoside phosphoramidase and as essential for growth under high salt conditions. J. Biol. Chem. 2005, 280, 15356–15361. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, A.; Pace, H.C.; Blackburn, G.M.; Adams, M.; Mekhalfia, A.; Kaczmarek, R.; Baraniak, J.; Stec, W.J.; Brenner, C. Biochemical, crystallographic, and mutagenic characterization of hint, the AMP-lysine hydrolase, with novel substrates and inhibitors. J. Biol. Chem. 2004, 279, 18711–18716. [Google Scholar] [CrossRef] [PubMed]
- Bieganowski, P.; Garrison, P.N.; Hodawadekar, S.C.; Faye, G.; Barnes, L.D.; Brenner, C. Adenosine monophosphoramidase activity of Hint and Hnt1 supports function of Kin28, Ccl1, and Tfb3. J. Biol. Chem. 2002, 277, 10852–10860. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C. Hint, Fhit, and GalT: Function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry 2002, 41, 9003–9014. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kawasaki, M.; Hatano, M. Occurrence of a toxic phospholipid in cabezon roe. Toxicon 1976, 14, 141–143. [Google Scholar] [CrossRef]
- Hatano, M.; Hashimoto, Y. Properties of a toxic phospholipid in the northern blenny roe. Toxicon 1974, 12, 231–236. [Google Scholar] [CrossRef]
- Hatano, M.; Marumoto, R.; Hashimoto, Y. Structure of a Toxic Phospholipid in the Northern Blenny Roe BT-Animal, Plant, and Microbial Toxins: Volume 2 Chemistry, Pharmacology, and Immunology; Ohsaka, A., Hayashi, K., Sawai, Y., Murata, R., Funatsu, M., Tamiya, N., Eds.; Springer: Boston, MA, USA, 1976; pp. 145–151. ISBN 978-1-4684-0889-8. [Google Scholar]
- Matsuura, N.; Onose, R.; Osada, H. Morphology reversion activity of phosmidosine and phosmidosine B, a newly isolated derivative, on src transformed NRK cells. J. Antibiot. 1996, 49, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.R.; Uramoto, M.; Isono, K.; McCloskey, J.A. Structure of the antifungal nucleotide antibiotic phosmidosine. J. Org. Chem. 1993, 58, 854–859. [Google Scholar] [CrossRef]
- Matsunaga, S.; Takahashi, N.; Fusetani, N. Dinogunellins AD: Putative ichthyootoxic phospholipids of northern blenny Stichaeus grigorjewi eggs. Pure Appl. Chem. 2009, 81, 1001–1008. [Google Scholar] [CrossRef]
- Heip, J.; Chatterjee, G.C.; Vandekerckhove, J.; Van, M.M.; Schell, J. Purification of the Agrobacterium radiobacter 84 agrocin. Arch. Int. Physiol. Biochim. 1975, 83, 974–975. [Google Scholar] [PubMed]
- McCardell, B.A.; Pootjes, C.F. Chemical Nature of Agrocin 84 and Its Effect on a Virulent Strain of Agrobacterium tumefaciens. Antimicrob. Agents Chemother. 1976, 10, 498 LP–502 LP. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.P.; Tate, M.E.; Kerr, A. Agrocin 84 is a 6-N-phosphoramidate of an adenine nucleotide analogue. Nature 1977, 265, 379. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Basu, M.; Chatterjee, G.C. Studies on the mode of action of agrocin 84. J. Antibiot. 1978, 31, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.J.; Hamilton, R.H.; Pootjes, C.F. Purification and characterization of agrocin 84. Antimicrob. Agents Chemother. 1979, 16, 293 LP–296 LP. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.E.; Murphy, P.J.; Roberts, W.P.; Kerr, A. Adenine N6-substituent of agrocin 84 determines its bacteriocin-like specificity. Nature 1979, 280, 697. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.S.; Ordoukhanian, P.T.; Kim, J.-G.; de Crécy-Lagard, V.; Hwang, I.; Farrand, S.; Schimmel, P. Major Biocontrol of Plant Tumors Targets tRNA Synthetase. Science 2005, 309, 1533 LP–1553 LP. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Palencia, A.; Virus, C.; Schulwitz, S.; Temple, B.R.; Cusack, S.; Reader, J. Structural characterization of antibiotic self-immunity tRNA synthetase in plant tumour biocontrol agent. Nat. Commun. 2016, 7, 12928. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Palencia, A.; Virus, C.; Tripathy, A.; Temple, B.R.; Velazquez-Campoy, A.; Cusack, S.; Reader, J.S. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat. Commun. 2013, 4, 1417. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Tate, M.E.; Kerr, A. Substituents at N6 and C-5′ control selective uptake and toxicity of the adenine-nucleotide bacteriocin, agrocin 84, in Agrobacteria. Eur. J. Biochem. 1981, 115, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Farrand, S.K.; Wang, C.-L.; Hong, S.-B.; O’Morchoe, S.B.; Slota, J.E. Deletion derivatives of pAgK84 and their use in the analysis of Agrobacterium plasmid functions. Plasmid 1992, 28, 201–212. [Google Scholar] [CrossRef]
- Wang, C.-L.; Farrand, S.K.; Hwang, I. Organization and expression of the genes on pAgK84 that encode production of agrocin 84. Mol. plant-microbe Interact. MPMI 1994. [Google Scholar]
- Ryder, M.H.; Slota, J.E.; Scarim, A.; Farrand, S.K. Genetic analysis of agrocin 84 production and immunity in Agrobacterium spp. J. Bacteriol. 1987, 169, 4184–4189. [Google Scholar] [CrossRef] [PubMed]
- Farrand, S.K.; Slota, J.E.; Shim, J.-S.; Kerr, A. Tn5 insertions in the agrocin 84 plasmid: The conjugal nature of pAgK84 and the locations of determinants for transfer and agrocin 84 production. Plasmid 1985, 13, 106–117. [Google Scholar] [CrossRef]
- Slota, J.E.; Farrand, S.K. Genetic isolation and physical characterization of pAgK84, the plasmid responsible for agrocin 84 production. Plasmid 1982, 8, 175–186. [Google Scholar] [CrossRef]
- Ellis, J.G.; Kerr, A.; Van Montagu, M.; Schell, J. Agrobacterium: Genetic studies on agrocin 84 production and the biological control of crown gall. Physiol. Plant Pathol. 1979, 15, 311–319. [Google Scholar] [CrossRef]
- Kim, J.-G.; Park, B.K.; Kim, S.-U.; Choi, D.; Nahm, B.H.; Moon, J.S.; Reader, J.S.; Farrand, S.K.; Hwang, I. Bases of biocontrol: Sequence predicts synthesis and mode of action of agrocin 84, the Trojan horse antibiotic that controls crown gall. Proc. Natl. Acad. Sci. USA 2006, 103, 8846–8851. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Roberts, W.P. A basis for agrocin 84 sensitivity in Agrobacterium radiobacter. Microbiology 1979, 114, 207–213. [Google Scholar] [CrossRef]
- Gelvin, S.B. Agrobacterium and plant genes involved in T-DNA transfer and integration. Annu. Rev. Plant Biol. 2000, 51, 223–256. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.G.; Murphy, P.J. Four new opines from crown gall tumours—their detection and properties. Mol. Gen. Genet. MGG 1981, 181, 36–43. [Google Scholar] [CrossRef]
- Hayman, G.T.; Farrand, S.K. Characterization and mapping of the agrocinopine-agrocin 84 locus on the nopaline Ti plasmid pTiC58. J. Bacteriol. 1988, 170, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Farrand, S.K. Characterization of the acc operon from the nopaline-type Ti plasmid pTiC58, which encodes utilization of agrocinopines A and B and susceptibility to agrocin 84. J. Bacteriol. 1997, 179, 7559–7572. [Google Scholar] [CrossRef] [PubMed]
- Von Bodman, S.B.; Hayman, G.T.; Farrand, S.K. Opine catabolism and conjugal transfer of the nopaline Ti plasmid pTiC58 are coordinately regulated by a single repressor. Proc. Natl. Acad. Sci. USA 1992, 89, 643–647. [Google Scholar] [CrossRef]
- Uramoto, M.; Kim, C.-J.; Shin-Ya, K.; Kusakabe, H.; Isono, K.; Phillips, D.R.; McCloskey, J.A. Isolation and characterization of phosmidosine a new antifungal nucleotide antibiotic. J. Antibiot. (Tokyo) 1991, 44, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Asai, N.; Okada, K.; Seio, K.; Sasaki, T.; Sekine, M. First synthesis and anticancer activity of phosmidosine and its related compounds. J. Org. Chem. 2002, 67, 3290–3300. [Google Scholar] [CrossRef] [PubMed]
- Sekine, M.; Moriguchi, T.; Wada, T.; Seio, K. Total synthesis of agrocin 84 and phosmidosine as naturally occurring nucleotidic antibiotics having PN bond linkages. J. Synth. Org. Chem. Japan 2001, 59, 1109–1120. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Kakeya, H.; Osada, H.; Obata, T.; Sasaki, T. Synthesis of Chemically Stabilized Phosmidosine Analogues and the Structure-Activity Relationship of Phosmidosine. J. Org. Chem. 2004, 69, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Bantysh, O.; Serebryakova, M.; Makarova, K.S.; Dubiley, S.; Datsenko, K.A.; Severinov, K. Enzymatic synthesis of bioinformatically predicted microcin C-like compounds encoded by diverse bacteria. MBio 2014, 5, e01059-14. [Google Scholar] [CrossRef] [PubMed]
- Paz-Yepes, J.; Brahamsha, B.; Palenik, B. Role of a microcin-C-like biosynthetic gene cluster in allelopathic interactions in marine Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 12030–12035. [Google Scholar] [CrossRef] [PubMed]
- Kulikovsky, A.; Tsibulskaya, D.; Piskunova, J.; Severinov, K.; Serebryakva, M.; Dubiley, S. Microcin C-like peptide-adenylate antibiotic from Rubidus massiliensis. In FEBS Open Bio; WILEY: Hoboken, NJ, USA, 2018; Volume 8, p. 280. [Google Scholar]
- Severinov, K.; Semenova, E.; Kazakov, A.; Kazakov, T.; Gelfand, M.S. Low-molecular-weight post-translationally modified microcins. Mol. Microbiol. 2007, 65, 1380–1394. [Google Scholar] [CrossRef] [PubMed]
- Serebryakova, M.; Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Nautiyal, M.; Van Aerschot, A.; Severinov, K.; Dubiley, S. A Trojan-Horse Peptide-Carboxymethyl-Cytidine Antibiotic from Bacillus amyloliquefaciens. J. Am. Chem. Soc. 2016, 138, 15690–15698. [Google Scholar] [CrossRef] [PubMed]
- Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Piskunova, J.; Severinov, K.; Serebryakova, M.; Dubiley, S. The Product of Yersinia pseudotuberculosis mcc Operon Is a Peptide-Cytidine Antibiotic Activated Inside Producing Cells by the TldD/E Protease. J. Am. Chem. Soc. 2017, 139, 16178–16187. [Google Scholar] [CrossRef]
- Garcia-Bustos, J.F.; Pezzi, N.; Mendez, E. Structure and mode of action of microcin 7, an antibacterial peptide produced by Escherichia coli. Antimicrob. Agents Chemother. 1985, 27, 791 LP–797 LP. [Google Scholar] [CrossRef] [PubMed]
- Metlitskaya, A.Z.; Katrukha, G.S.; Shashkov, A.S.; Zaitsev, D.A.; Egorov, T.A.; Khmel, I.A. Structure of microcin C51, a new antibiotic with a broad spectrum of activity. FEBS Lett. 1995, 357, 235–238. [Google Scholar] [CrossRef]
- Fomenko, D.E.; Metlitskaya, A.Z.; Péduzzi, J.; Goulard, C.; Katrukha, G.S.; Gening, L.V.; Rebuffat, S.; Khmel, I.A. Microcin C51 Plasmid Genes: Possible Source of Horizontal Gene Transfer. Antimicrob. Agents Chemother. 2003, 47, 2868 LP–2874 LP. [Google Scholar] [CrossRef] [PubMed]
- Regni, C.A.; Roush, R.F.; Miller, D.J.; Nourse, A.; Walsh, C.T.; Schulman, B.A. How the MccB bacterial ancestor of ubiquitin E1 initiates biosynthesis of the microcin C7 antibiotic. EMBO J. 2009, 28, 1953 LP–1964 LP. [Google Scholar] [CrossRef] [PubMed]
- Kulikovsky, A.; Serebryakova, M.; Bantysh, O.; Metlitskaya, A.; Borukhov, S.; Severinov, K.; Dubiley, S. The molecular mechanism of aminopropylation of peptide-nucleotide antibiotic microcin C. J. Am. Chem. Soc. 2014, 136, 11168–11175. [Google Scholar] [CrossRef] [PubMed]
- Zukher, I.; Novikova, M.; Tikhonov, A.; Nesterchuk, M.V.; Osterman, I.A.; Djordjevic, M.; Sergiev, P.V.; Sharma, C.M.; Severinov, K. Ribosome-controlled transcription termination is essential for the production of antibiotic microcin C. Nucleic Acids Res. 2014, 42, 11891–11902. [Google Scholar] [CrossRef] [PubMed]
- Severinov, K.; Nair, S.K. Microcin C: Biosynthesis and mechanisms of bacterial resistance. Future Microbiol. 2012, 7, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, T.; Vondenhoff, G.H.; Datsenko, K.A.; Novikova, M.; Metlitskaya, A.; Wanner, B.L.; Severinov, K. Escherichia coli Peptidase A, B, or N Can Process Translation Inhibitor Microcin C. J. Bacteriol. 2008, 190, 2607 LP–2610 LP. [Google Scholar] [CrossRef] [PubMed]
- Vondenhoff, G.H.M.; Van Aerschot, A. Microcin C: Biosynthesis, Mode of Action, and Potential as a Lead in Antibiotics Development. Nucleosides, Nucleotides and Nucleic Acids 2011, 30, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Metlytskaya, A.; Severinov, K.V.; Nair, S.K. Structural basis for Microcin C7 inactivation by the MccE acetyltransferase. J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Kazakov, T.; Vondenhoff, G.H.; Semenova, E.; Rozenski, J.; Metlytskaya, A.; Zukher, I.; Tikhonov, A.; Van Aerschot, A.; Severinov, K. MccE provides resistance to protein synthesis inhibitor Microcin C by acetylating the processed form of the antibiotic. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [PubMed]
- Piskunova, J.; Maisonneuve, E.; Germain, E.; Gerdes, K.; Severinov, K. Peptide-nucleotide antibiotic Microcin C is a potent inducer of stringent response and persistence in both sensitive and producing cells. Mol. Microbiol. 2017, 104, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, A.; Kazakov, T.; Semenova, E.; Serebryakova, M.; Vondenhoff, G.; Van Aerschot, A.; Reader, J.S.; Govorun, V.M.; Severinov, K. The mechanism of microcin C resistance provided by the MccF peptidase. J. Biol. Chem. 2010, 285, 37944–37952. [Google Scholar] [CrossRef] [PubMed]
- Duquesne, S.; Destoumieux-Garzón, D.; Peduzzi, J.; Rebuffat, S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 2007, 24, 708–734. [Google Scholar] [CrossRef] [PubMed]
- Rebuffat, S. Chapter 20—Microcins. In Handbook of Biologically Active Peptides: Bacterial/Antibiotic Peptides, 2nd ed.; Kastin, A., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 129–137. ISBN 978-0-12-385095-9. [Google Scholar]
- Takeuchi, H.; Asai, N.; Tanabe, K.; Kozaki, T.; Fujita, M.; Sakai, T.; Okuda, A.; Naruse, N.; Yamamoto, S.; Sameshima, T. EM2487, a Novel Anti-HIV-1 Antibiotic, Produced by Streptomyces sp. Mer-2487. J. Antibiot. 1999, 52, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Okamoto, M.; Takeuchi, H. Inhibition of human immunodeficiency virus type 1 replication in acutely and chronically infected cells by EM2487, a novel substance produced by a Streptomyces species. Antimicrob. Agents Chemother. 1999, 43, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, T.; Horiuchi, N.; Ishikawa, M.; Wanibuchi, K.; Moriguchi, T.; Takahashi, S. 1100-50, a novel nematocide from Streptomyces lavendulae SANK 64297. J. Antibiot. 2003, 56, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, B.W.; Payne, D.J. Dictionary of Antibiotics and Related Substances: With CD-ROM.; CRC Press: Boca Rato, FL, USA, 2013; ISBN 1482282151. [Google Scholar]
- Wada, T.; Moriguchi, T.; Sekine, M. Synthesis and Properties of N-Phosphorylated Ribonucleosides. J. Am. Chem. Soc. 1994, 116, 9901–9911. [Google Scholar] [CrossRef]
- Poudyal, R.R.; Nguyen, P.D.M.; Lokugamage, M.P.; Callaway, M.K.; Gavette, J.V.; Krishnamurthy, R.; Burke, D.H. Nucleobase modification by an RNA enzyme. Nucleic Acids Res. 2017, 45, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Besant, P.G.; Attwood, P.V.; Piggott, M.J. Focus on phosphoarginine and phospholysine. Curr Protein Pept Sci 2009, 10, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Attwood, P.V.; Piggott, M.J.; Zu, X.L.; Besant, P.G. Focus on phosphohistidine. Amino Acids 2007, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Piggott, M.J.; Attwood, P. V Focus on O-phosphohydroxylysine, O-phosphohydroxyproline, N 1-phosphotryptophan and S-phosphocysteine. Amino Acids 2017, 49, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, J.; Fraczyk, T.; Rode, W. Phosphorylation of basic amino acid residues in proteins: Important but easily missed. Acta Biochim Pol 2011, 58, 137–148. [Google Scholar] [PubMed]
- Azevedo, C.; Saiardi, A. Why always lysine? The ongoing tale of one of the most modified amino acids. Adv. Biol. Regul. 2016, 60, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.R. Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: A possible regulator of the mitogen-activated protein kinase cascade. Pharmacol. Ther. 1995, 67, 323–350. [Google Scholar] [CrossRef]
- Klumpp, S.; Krieglstein, J. Phosphorylation and dephosphorylation of histidine residues in proteins. Eur. J. Biochem. 2002, 269, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Besant, P.G.; Attwood, P.V. Detection and analysis of protein histidine phosphorylation. Mol. Cell. Biochem. 2009, 329, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Wakim, B.T.; Aswad, G.D. Ca (2+)-calmodulin-dependent phosphorylation of arginine in histone 3 by a nuclear kinase from mouse leukemia cells. J. Biol. Chem. 1994, 269, 2722–2727. [Google Scholar] [PubMed]
- Wakim, B.T.; Grutkoski, P.S.; Vaughan, A.T.M.; Engelmann, G.L. Stimulation of a Ca2+-Calmodulin-activated Histone 3 Arginine Kinase in Quiescent Rat Heart Endothelial Cells Compared to Actively Dividing Cells. J. Biol. Chem. 1995, 270, 23155–23158. [Google Scholar] [CrossRef] [PubMed]
- Levy-Favatier, F.; Delpech, M.; Kruh, J. Characterization of an arginine-specific protein kinase tightly bound to rat liver DNA. Eur. J. Biochem. 1987, 166, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E.; Consigli, R.A. Characterization of a protein kinase activity associated with purified capsids of the granulosis virus infecting Plodia interpunctella. Virology 1985, 143, 516–525. [Google Scholar] [CrossRef]
- Wilson, M.E.; Consigli, R.A. Functions of a protein kinase activity associated with purified capsids of the granulosis virus infecting Plodia interpunctella. Virology 1985, 143, 526–535. [Google Scholar] [CrossRef]
- Junker, S.; Maaß, S.; Otto, A.; Michalik, S.; Morgenroth, F.; Gerth, U.; Hecker, M.; Becher, D. Spectral library based analysis of arginine phosphorylations in Staphylococcus aureus. Mol. Cell. Proteomics 2017. [Google Scholar] [CrossRef]
- Junker, S.; Maaß, S.; Otto, A.; Hecker, M.; Becher, D. Toward the Quantitative Characterization of Arginine Phosphorylations in Staphylococcus aureus. J. Proteome Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, J.; Subramanian, V.; Kojetin, D.J.; Thompson, P.R. Activity-Based Profiling Reveals a Regulatory Link between Oxidative Stress and Protein Arginine Phosphorylation. Cell Chem. Biol. 2016, 23, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Trentini, D.B.; Suskiewicz, M.J.; Heuck, A.; Kurzbauer, R.; Deszcz, L.; Mechtler, K.; Clausen, T. Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature 2016, 539, 48. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.J.; Clausen, T. Chemical Biology Interrogates Protein Arginine Phosphorylation. Cell Chem. Biol. 2016, 23, 888–890. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, J.; Mierzwa, B.; Trentini, D.B.; Spiess, S.; Lehner, A.; Charpentier, E.; Clausen, T. Structural Basis for Recognizing Phosphoarginine and Evolving Residue-Specific Protein Phosphatases in Gram-Positive Bacteria. Cell Rep. 2013, 3, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, J.; Schmidt, A.; Spiess, S.; Lehner, A.; Turgay, K.; Mechtler, K.; Charpentier, E.; Clausen, T. McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 2009, 324, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Elsholz, A.K.W.; Turgay, K.; Michalik, S.; Hessling, B.; Gronau, K.; Oertel, D.; Mader, U.; Bernhardt, J.; Becher, D.; Hecker, M.; et al. Global impact of protein arginine phosphorylation on the physiology of Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2012, 109, 7451–7456. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Ammerer, G.; Mechtler, K. Studying the fragmentation behavior of peptides with arginine phosphorylation and its influence on phospho-site localization. Proteomics 2013, 13, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Černý, J.; Svozil, D.; Čech, P.; Gelly, J.-C.; de Brevern, A.G. Bioinformatic analysis of the protein/DNA interface. Nucleic Acids Res. 2014, 42, 3381–3394. [Google Scholar] [CrossRef] [PubMed]
- Slade, D.J.; Subramanian, V.; Fuhrmann, J.; Thompson, P.R. Chemical and biological methods to detect post-translational modifications of arginine. Biopolymers 2014, 101, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Zetterqvist, Ö. Further studies on acid-labile [32P]phosphate bound to high-molecular weight material from rat-liver cell sap after incubation with [32P]adenosine triphosphate. Biochim. Biophys. Acta (BBA) Gen. Subj. 1967, 141, 533–539. [Google Scholar] [CrossRef]
- Zetterqvist, Ö.; Engström, L. Isolation of N-ε-[32P]phosphoryl-lysine from rat-liver cell sap after incubation with [32P]Adenosine triphosphate. Biochim. Biophys. Acta (BBA) Gen. Subj. 1967, 141, 523–532. [Google Scholar] [CrossRef]
- Chen, C.-C.; Smith, D.L.; Bruegger, B.B.; Halpern, R.M.; Smith, R.A. Occurrence and distribution of acid-labile histone phosphates in regenerating rat liver. Biochemistry 1974, 13, 3785–3789. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Bruegger, B.B.; Kern, C.W.; Lin, Y.C.; Halpern, R.M.; Smith, R.A. Phosphorylation of nuclear proteins in rat regenerating liver. Biochemistry 1977, 16, 4852–4855. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Chen, C.-C.; Bruegger, B.B.; Holtz, S.L.; Halpern, R.M.; Smith, R.A. Characterization of protein kinases forming acid-labile histone phosphates in Walker-256 carcinosarcoma cell nuclei. Biochemistry 1974, 13, 3780–3785. [Google Scholar] [CrossRef] [PubMed]
- Hiraishi, H.; Yokoi, F.; Kumon, A. 3-Phosphohistidine and 6-Phospholysine Are Substrates of a 56-kDa Inorganic Pyrophosphatase from Bovine Liver. Arch. Biochem. Biophys. 1998, 349, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Hiraishi, H.; Yokoi, F.; Kumon, A. Bovine Liver Phosphoamidase as a Protein Histidine/Lysine Phosphatase. J. Biochem. 1999, 126, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, F.; Hiraishi, H.; Izuhara, K. Molecular Cloning of a cDNA for the Human Phospholysine Phosphohistidine Inorganic Pyrophosphate Phosphatase. J. Biochem. 2003, 133, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Attwood, P. V PN bond protein phosphatases. Biochim. Biophys. Acta (BBA) Proteins Proteomics 2013, 1834, 470–478. [Google Scholar] [CrossRef]
- Ek, P.; Ek, B.; Zetterqvist, O. Phosphohistidine phosphatase 1 (PHPT1) also dephosphorylates phospholysine of chemically phosphorylated histone H1 and polylysine. Ups. J. Med. Sci. 2015, 120, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Bertran-Vicente, J.; Serwa, R.A.; Schümann, M.; Schmieder, P.; Krause, E.; Hackenberger, C.P.R. Site-Specifically Phosphorylated Lysine Peptides. J. Am. Chem. Soc. 2014, 136, 13622–13628. [Google Scholar] [CrossRef] [PubMed]
- Bertran-Vicente, J.; Schümann, M.; Schmieder, P.; Krause, E.; Hackenberger, C.P.R. Direct access to site-specifically phosphorylated-lysine peptides from a solid-support. Org. Biomol. Chem. 2015, 13, 6839–6843. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Penkert, M.; Hackenberger, C.P.R. Chemical Approaches to Investigate Labile Peptide and Protein Phosphorylation. Acc. Chem. Res. 2017, 50, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-Y.; Chai, Y.-R.; Jia, Y.-L.; Gao, J.-H.; Peng, X.-J.; Han, H.-F. Crosstalk among the proteome, lysine phosphorylation, and acetylation in romidepsin-treated colon cancer cells. Oncotarget 2016, 7, 53471–53501. [Google Scholar] [CrossRef] [PubMed]
- Hardman, G.; Perkins, S.; Ruan, Z.; Kannan, N.; Brownridge, P.; Byrne, D.P.; Eyers, P.A.; Jones, A.R.; Eyers, C.E. Extensive non-canonical phosphorylation in human cells revealed using strong-anion exchange-mediated phosphoproteomics. bioRxiv 2017. [Google Scholar]
- Schwartz, D. Prediction of lysine post-translational modifications using bioinformatic tools. Essays Biochem. 2012, 52, 165 LP–177 LP. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Livermore, T.; Saiardi, A. Protein Polyphosphorylation of Lysine Residues by Inorganic Polyphosphate. Mol. Cell 2015, 58, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Saiardi, A. The new world of inorganic polyphosphates. Biochem. Soc. Trans. 2016, 44, 13. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Saiardi, A. Functions of inorganic polyphosphates in eukaryotic cells: A coat of many colours. 2014. [Google Scholar]
- Saiardi, A. How inositol pyrophosphates control cellular phosphate homeostasis? Adv. Biol. Regul. 2012, 52, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Bentley-DeSousa, A.; Holinier, C.; Moteshareie, H.; Tseng, Y.-C.; Kajjo, S.; Nwosu, C.; Amodeo, G.F.; Bondy-Chorney, E.; Sai, Y.; Rudner, A.; et al. A Screen for Candidate Targets of Lysine Polyphosphorylation Uncovers a Conserved Network Implicated in Ribosome Biogenesis. Cell Rep. 2018, 22, 3427–3439. [Google Scholar] [CrossRef] [PubMed]
- Bru, S.; Samper-Martín, B.; Quandt, E.; Hernández-Ortega, S.; Martínez-Laínez, J.M.; Garí, E.; Rafel, M.; Torres-Torronteras, J.; Martí, R.; Ribeiro, M.P.C.; et al. Polyphosphate is a key factor for cell survival after DNA damage in eukaryotic cells. DNA Repair 2017, 57, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Nevitt, C.; Tooley, J.G.; Schaner Tooley, C.E. N-terminal acetylation and methylation differentially affect the function of MYL9. Biochem. J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Petkowski, J.J.; Schaner Tooley, C.E.; Anderson, L.C.; Shumilin, I.A.; Balsbaugh, J.L.; Shabanowitz, J.; Hunt, D.F.; Minor, W.; Macara, I.G. Substrate specificity of mammalian N-terminal alpha-amino methyltransferase NRMT. Biochemistry 2012, 51, 5942–5950. [Google Scholar] [CrossRef] [PubMed]
- Szijgyarto, Z.; Garedew, A.; Azevedo, C.; Saiardi, A. Influence of Inositol Pyrophosphates on Cellular Energy Dynamics. Science 2011, 334, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Shears, S.B. Inositol pyrophosphates: Why so many phosphates? Adv. Biol. Regul. 2015, 57, 203–216. [Google Scholar] [CrossRef]
- Wilson, M.S.C.; Livermore, T.M.; Saiardi, A. Inositol pyrophosphates: Between signalling and metabolism. Biochem. J. 2013, 452, 369 LP–379 LP. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Szijgyarto, Z.; Bosch, D.; Loss, O.; Azevedo, C.; Saiardi, A. Identification of an Evolutionarily Conserved Family of Inorganic Polyphosphate Endopolyphosphatases. J. Biol. Chem. 2011, 286, 31966–31974. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Saiardi, A.; Ahmadibeni, Y.; Snowman, A.M.; Resnick, A.C.; Kristiansen, T.Z.; Molina, H.; Pandey, A.; Werner, J.K.; Juluri, K.R.; et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc. Natl. Acad. Sci. USA 2007, 104, 15305. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Burton, A.; Ruiz-Mateos, E.; Marsh, M.; Saiardi, A. Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proc. Natl. Acad. Sci. USA 2009, 106, 21161 LP–21166 LP. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Bhandari, R.; Resnick, A.C.; Snowman, A.M.; Snyder, S.H. Phosphorylation of Proteins by Inositol Pyrophosphates. Science 2004, 306, 2101 LP–2105 LP. [Google Scholar] [CrossRef] [PubMed]
- Besant, P.G.; Attwood, P. V Chapter 21—Histidine Phosphorylation in Histones and in Other Mammalian Proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2010; Volume 471, pp. 403–426. ISBN 0076-6879. [Google Scholar]
- Zu, X.-L.; Besant, P.G.; Attwood, P. V Chapter 14 Protein Histidine Phosphorylation. In Comprehensive Analytical Chemistry; Whitelegge, J.P., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 52, pp. 315–352. ISBN 0166-526X. [Google Scholar]
- Puttick, J.; Baker, E.N.; Delbaere, L.T.J. Histidine phosphorylation in biological systems. Biochim. Biophys. Acta - Proteins Proteomics 2008, 1784, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Fuhs, S.R.; Hunter, T. pHisphorylation: The emergence of histidine phosphorylation as a reversible regulatory modification. Curr. Opin. Cell Biol. 2017, 45, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Kee, J.-M.; Muir, T.W. Chasing Phosphohistidine, an Elusive Sibling in the Phosphoamino Acid Family. ACS Chem. Biol. 2012, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Makwana, M.V.; Muimo, R.; Jackson, R.F.W. Advances in development of new tools for the study of phosphohistidine. Lab. Investig. 2017, 98, 291. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, U.M.; Ludwig, K.; Krieglstein, J.; König, S. Stepchild phosphohistidine: Acid-labile phosphorylation becomes accessible by functional proteomics. Anal. Bioanal. Chem. 2010, 397, 3209–3212. [Google Scholar] [CrossRef] [PubMed]
- Besant, P.G.; Attwood, P.V. Histone H4 histidine phosphorylation: Kinases, phosphatases, liver regeneration and cancer. Biochem. Soc. Trans. 2012, 40, 290. [Google Scholar] [CrossRef] [PubMed]
- Attwood, P.V.; Wieland, T. Nucleoside diphosphate kinase as protein histidine kinase. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 153–160. [Google Scholar] [CrossRef]
- Wieland, T.; Attwood, P. V Alterations in reversible protein histidine phosphorylation as intracellular signals in cardiovascular disease. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Attwood, P.V.; Muimo, R. The actions of NME1/NDPK-A and NME2/NDPK-B as protein kinases. Lab. Investig. 2017, 98, 283. [Google Scholar] [CrossRef] [PubMed]
- Attwood, P.V. Histidine kinases from bacteria to humans. Biochem. Soc. Trans. 2013, 41, 1023 LP–1028 LP. [Google Scholar] [CrossRef] [PubMed]
- Walton, T.; Szostak, J.W. A Highly Reactive Imidazolium-Bridged Dinucleotide Intermediate in Nonenzymatic RNA Primer Extension. J. Am. Chem. Soc. 2016, 138, 11996–12002. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Joyce, M.A.; Ryan, D.G.; Wolodko, W.T. Two glutamate residues, Glu 208α and Glu 197β, are crucial for phosphorylation and dephosphorylation of the active-site histidine residue in succinyl-CoA synthetase. Biochemistry 2002, 41, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Rose, Z.B. Evidence for a phosphohistidine protein intermediate in the phosphoglycerate mutase reaction. Arch. Biochem. Biophys. 1970, 140, 508–513. [Google Scholar] [CrossRef]
- Walinder, O. Evidence of presence of 1-phosphohistidine as main phosphorylated component at active site of bovine liver nucleoside diphosphate kinase. Acta Chem Scand 1969, 23, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular Mechanisms of Two-Component Signal Transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Srivastava, S.; Li, Z.; Vaeth, M.; Fuhs, S.R.; Hunter, T.; Skolnik, E.Y. Identification of PGAM5 as a Mammalian Protein Histidine Phosphatase that Plays a Central Role to Negatively Regulate CD4+ T Cells. Mol. Cell 2016, 63, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Panda, S.; Li, Z.; Fuhs, S.R.; Hunter, T.; Thiele, D.J.; Hubbard, S.R.; Skolnik, E.Y. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife 2016, 5, e16093. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.R. Limiting T cell histidine phosphorylation. Sci. Signal. 2016, 9, ec185. [Google Scholar] [CrossRef]
- Shen, H.; Yang, P.; Liu, Q.; Tian, Y. Nuclear expression and clinical significance of phosphohistidine phosphatase 1 in clear-cell renal cell carcinoma. J. Int. Med. Res. 2015, 43, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Hao, J.; Zhang, Z.; Tian, T.; Jiang, S.; Hao, J.; Liu, C.; Huang, L.; Xiao, X.; He, D. 14-kDa phosphohistidine phosphatase and its role in human lung cancer cell migration and invasion. Lung Cancer 2010, 67, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Potel, C.M.; Lin, M.-H.; Heck, A.J.R.; Lemeer, S. Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nat. Methods 2018, 15, 187. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.R.; Dutta, P.; Mahajan, S.; Varma, S.; Stevens Jr., S. M. Structural and activity characterization of human PHPT1 after oxidative modification. Sci. Rep. 2016, 6, 23658. [Google Scholar] [CrossRef] [PubMed]
- Ek, P.; Pettersson, G.; Ek, B.; Gong, F.; Li, J.-P.; Zetterqvist, O. Identification and characterization of a mammalian 14-kDa phosphohistidine phosphatase. Eur. J. Biochem. 2002, 269, 5016–5023. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.K.Y.; Betz, C.; et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature 2018, 555, 678. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, H.A.; Perich, J.W.; Wade, J.D.; Tregear, G.W.; Johns, R.B. Synthesis of an N(1)-phosphotryptophan-containing tripeptide: Glutamyl-N(1)-phosphotryptophylleucine. J. Org. Chem. 1989, 54, 5731–5736. [Google Scholar] [CrossRef]
- Guillaume, H.A.; Perich, J.W.; Johns, R.B.; Tregear, G.W. Synthesis of N(1)-phosphorylated tryptophan derivatives. J. Org. Chem. 1989, 54, 1664–1668. [Google Scholar] [CrossRef]
- Tooley, J.G.; Schaner Tooley, C.E. New roles for old modifications: Emerging roles of N-terminal post-translational modifications in development and disease. Protein Sci. 2014, 23, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Varland, S.; Osberg, C.; Arnesen, T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 2015, 15, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Tooley, C.E.S.; Petkowski, J.J.; Muratore-Schroeder, T.L.; Balsbaugh, J.L.; Shabanowitz, J.; Sabat, M.; Minor, W.; Hunt, D.F.; Macara, I.G. NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 2010, 466, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Petkowski, J.J. NRMT: An α-N-methyl transferase. University of Virginia: Charlottesville, VA, USA, 2012; ISBN 1267866160. [Google Scholar]
- Ree, R.; Varland, S.; Arnesen, T. Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 2018, 50, 90. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta - Proteins Proteomics 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed]
- Foyn, H.; Van Damme, P.; Støve, S.I.; Glomnes, N.; Evjenth, R.; Gevaert, K.; Arnesen, T. Protein N-terminal acetyltransferases act as N-terminal propionyltransferases in vitro and in vivo. Mol. Cell. Proteomics 2013, 12, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Fatty Acylation of Proteins: The Long and the Short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Lee, M.C.; Cui, M.; Liu, T.; Chen, Z.-Z.; Li, Y.-M.; Ju, Y.; Zhao, Y.-F.; Chen, K.; Jiang, H. A common intermediate for prebiotic synthesis of proteins and nucleosides: A density functional theory (DFT) study on the formation of penta-coordinate phosphorus carboxylic–phosphoric mixed anhydride from N-phosphoryl amino acids. J. Mol. Struct. THEOCHEM 2004, 672, 51–60. [Google Scholar] [CrossRef]
- Karki, M.; Gibard, C.; Bhowmik, S.; Krishnamurthy, R. Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water. Life 2017, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.M.; Liu, X.H.; Li, Y.M.; Ma, Y.; Tan, B.; Wan, R.; Zhao, Y.F. N-Phosphoryl Amino Acids and Biomolecular Origins. Review Paper in Honor of the 50th Anniversary of the Publication of “A Production of Amino Acids under Possible Primitive Earth Conditions” (Miller, 1953). Orig. Life Evol. Biosph. 2004, 34, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tong, Y.; Chen, S.; Li, Y.; Chen, Y.; Zhao, Y.; Wang, J. Orientation of the peptide formation of N-phosphoryl amino acids in solution. Chin. Sci. Bull. 2002, 47, 1866. [Google Scholar] [CrossRef]
- Qiang, L.-M.; Cao, S.-X.; Zhao, X.-Y.; Liu, R.-Y.; Liu, J.-H.; Lu, J.-S.; Zhao, Y.-F. Hydrolysis Reaction of N-Phosphoryl-α-, β- and γ-amino Acids Studied by HPLC. Chin. J. Chem. 2007, 25, 1559–1562. [Google Scholar] [CrossRef]
- Yu, Y.; Shu, W.; Liu, Y.; Zhao, Y. Co-origin of Oligopeptide/Oligonucleotide/Membrane with an N-phosphoryl Amino Acid Model in Origin of Life. In Proceedings of the XVIIIth International Conference on the Origin of Life, San Diego, CA, USA, 16–21 July 2017; Volume 1967. [Google Scholar]
- Wang, J.-N.; Liu, Y.; He, D.-C.; Zhao, Y.-F. Studies on the Reaction between Peptides or Proteins with N-Phosphoryl Amino Acids in Aqueous Solution. Phosphorus. Sulfur. Silicon Relat. Elem. 2008, 183, 764–772. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Cao, P.-S. N-Phospho-α-Amino Acids and Co-Evolution of Nucleic Acid and Protein. Phosphorus. Sulfur. Silicon Relat. Elem. 1999, 144, 757–760. [Google Scholar] [CrossRef]
- Chen, Z.-Z.; Tan, B.; Li, Y.-M.; Zhao, Y.-F.; Tong, Y.-F.; Wang, J.-F. Activity Difference between α-COOH and β-COOH in N-Phosphorylaspartic Acids. J. Org. Chem. 2003, 68, 4052–4058. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Fu, C.; Gao, X.; Liu, Y.; Xu, P.; Liu, L.; Lv, Y.; Fu, S.; Sun, Y.; Han, D.; et al. N-phosphoryl amino acid models for P–N bonds in prebiotic chemical evolution. Sci. China Chem. 2015, 58, 374–382. [Google Scholar] [CrossRef]
- Li, Y.; Yin, Y.; Zhao, Y. Phosphoryl group participation leads to peptide formation from N-phosphorylamino acids. Int. J. Pept. Protein Res. 1992, 39, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Deng, H.; Tang, G.; Liu, Y.; Xu, P.; Zhao, Y. Intermolecular Phosphoryl Transfer of N-Phosphoryl Amino Acids. European J. Org. Chem. 2011, 2011, 3220–3228. [Google Scholar] [CrossRef]
- Bertran-Vicente, J.; Penkert, M.; Nieto-Garcia, O.; Jeckelmann, J.-M.; Schmieder, P.; Krause, E.; Hackenberger, C.P.R. Chemoselective synthesis and analysis of naturally occurring phosphorylated cysteine peptides. Nat. Commun. 2016, 7, 12703. [Google Scholar] [CrossRef] [PubMed]
- Kegley, S.; Hill, B.; Orme, S.; Choi, A. PAN Pesticide Database; Pesticide Action Network: San Francisco, CA, USA, 2016. [Google Scholar]
- Alam, M.; Sanduja, R.; Hossain, M.B.; Van der Helm, D. Gymnodinium breve toxins. 1. Isolation and X-ray structure of O,O-dipropyl (E)-2-(1-methyl-2-oxopropylidene)phosphorohydrazidothioate (E)-oxime from the red tide dinoflagellate Gymnodinium breve. J. Am. Chem. Soc. 1982, 104, 5232–5234. [Google Scholar] [CrossRef]
- Husain, K.; Singh, R.; Kaushik, M.P.; Gupta, A.K. Acute Toxicity of SyntheticGymnodinium breveToxin Metabolite and Its Analogues in Mice. Ecotoxicol. Environ. Saf. 1996, 35, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.N.; Gupta, S.D.; Gupta, A.K.; Dube, S.N.; Deshpande, S.B. Relative potency of synthetic analogs of Ptychodiscus brevis toxin in depressing synaptic transmission evoked in neonatal rat spinal cord in vitro. Toxicol. Lett. 2002, 128, 177–183. [Google Scholar] [CrossRef]
- Abraham, S.P.; Hoang, T.D.; Alam, M.; Jones, E.B.G. Chemistry of the cytotoxic principles of the ma rine fungus Lignincola laevis. Pure Appl. Chem. 1994, 66, 2391–2394. [Google Scholar] [CrossRef]
- Buchowiecka, A.K. Puzzling over protein cysteine phosphorylation—assessment of proteomic tools for S-phosphorylation profiling. Analyst 2014, 139, 4118–4123. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.P.; Zhou, M.-M.; Van Etten, R.L. Kinetic and site-directed mutagenesis studies of the cysteine residues of bovine low molecular weight phosphotyrosyl protein phosphatase. J. Biol. Chem. 1994, 269, 8734–8740. [Google Scholar] [PubMed]
- Pannifer, A.D.B.; Flint, A.J.; Tonks, N.K.; Barford, D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by X-ray crystallography. J. Biol. Chem. 1998, 273, 10454–10462. [Google Scholar] [CrossRef] [PubMed]
- Deshimaru, S.; Miyake, Y.; Ohmiya, T.; Tatsu, Y.; Endo, Y.; Yumoto, N.; Toraya, T. Heterologous expression and catalytic properties of the C-terminal domain of starfish cdc25 dual-specificity phosphatase, a cell cycle regulator. J. Biochem. 2002, 131, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Brandão, T.A.S.; Hengge, A.C.; Johnson, S.J. Insights into the reaction of protein tyrosine phosphatase 1B. Crystal structures for transition-state analogs of both catalytic steps. J. Biol. Chem. 2010, 285, 15874–15883. [Google Scholar]
- Guan, K.L.; Dixon, J.E. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 1991, 266, 17026–17030. [Google Scholar] [PubMed]
- Pas, H.H.; Robillard, G.T. S-phosphocysteine and phosphohistidine are intermediates in the phosphoenolpyruvate-dependent mannitol transport catalyzed by Escherichia coli EIIMtl. Biochemistry 1988, 27, 5835–5839. [Google Scholar] [CrossRef] [PubMed]
- Pas, H.H.; Meyer, G.H.; Kruizinga, W.H.; Tamminga, K.S.; Van Weeghel, R.P.; Robillard, G.T. 31phospho-NMR demonstration of phosphocysteine as a catalytic intermediate on the Escherichia coli phosphotransferase system EIIMtl. J. Biol. Chem. 1991, 266, 6690–6692. [Google Scholar] [PubMed]
- Meins, M.; Jenö, P.; Müller, D.; Richter, W.J.; Rosenbusch, J.P.; Erni, B. Cysteine phosphorylation of the glucose transporter of Escherichia coli. J. Biol. Chem. 1993, 268, 11604–11609. [Google Scholar] [PubMed]
- Gemmecker, G.; Eberstadt, M.; Buhr, A.; Lanz, R.; Golic Grdadolnik, S.; Kessler, H.; Erni, B. Glucose transporter of Escherichia coli: NMR characterization of the phosphocysteine form of the IIBGlc domain and its binding interface with the IIAGlc subunit. Biochemistry 1997, 36, 7408–7417. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Ding, Y.; Ji, Q.; Liang, Z.; Deng, X.; Wong, C.C.L.; Yi, C.; Zhang, L.; Xie, S.; Alvarez, S.; et al. Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 15461–15466. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.R.; Brugarolas, P.; He, C. Redox signaling in human pathogens. Antioxid. Redox Signal. 2011, 14, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhu, M.; Schaub, M.C.; Gehrig, P.; Roschitzki, B.; Lucchinetti, E.; Zaugg, M. Phosphoproteome analysis of isoflurane-protected heart mitochondria: Phosphorylation of adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial function. Cardiovasc. Res. 2008, 80, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Chase, M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, S.; Xu, T.; Taghizadeh, K.; Wishnok, J.S.; Zhou, X.; You, D.; Deng, Z.; Dedon, P.C. Phosphorothioation of DNA in bacteria by dnd genes. Nat Chem Biol 2007, 3, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, S.; Deng, Z.; Dedon, P.C.; Chen, S. DNA phosphorothioate modification—A new multi-functional epigenetic system in bacteria. FEMS Microbiol. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; He, X.; Liang, J.; Li, A.; Xu, T.; Kieser, T.; Helmann, J.D.; Deng, Z. A novel DNA modification by sulphur. Mol. Microbiol. 2005, 57, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Liang, J.; Chen, S.; Wang, L.; He, X.; You, D.; Wang, Z.; Li, A.; Xu, Z.; Zhou, X. DNA phosphorothioation in Streptomyces lividans: Mutational analysis of the dnd locus. BMC Microbiol. 2009, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, S.; Vergin, K.L.; Giovannoni, S.J.; Chan, S.W.; DeMott, M.S.; Taghizadeh, K.; Cordero, O.X.; Cutler, M.; Timberlake, S.; et al. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc. Natl. Acad. Sci. USA 2011, 108, 2963–2968. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, S.; Deng, Z. Phosphorothioation: An Unusual Post-Replicative Modification on the DNA Backbone. In DNA Replication-Current Advances; Seligmann, H., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Xu, T.; Yao, F.; Zhou, X.; Deng, Z.; You, D. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010, 38, 7133–7141. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ou, H.-Y.; Wang, T.; Li, L.; Tan, H.; Zhou, X.; Rajakumar, K.; Deng, Z.; He, X. Cleavage of phosphorothioated DNA and methylated DNA by the type IV restriction endonuclease ScoMcrA. PLoS Genet. 2010, 6, e1001253. [Google Scholar] [CrossRef]
- Dai, D.; Du, A.; Xiong, K.; Pu, T.; Zhou, X.; Deng, Z.; Liang, J.; He, X.; Wang, Z. DNA Phosphorothioate Modification Plays a Role in Peroxides Resistance in Streptomyces lividans. Front. Microbiol. 2016, 7, 1380. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liang, J.; Pu, T.; Xu, F.; Yao, F.; Yang, Y.; Zhao, Y.-L.; You, D.; Zhou, X.; Deng, Z.; et al. Phosphorothioate DNA as an antioxidant in bacteria. Nucleic Acids Res. 2012, 40, 9115–9124. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.; Chen, S.; Wang, L.; Tang, Y.; Ryu, J.Y.; Jiang, S.; Wu, X.; Chen, C.; Luo, J.; Deng, Z.; et al. Occurrence, evolution, and functions of DNA phosphorothioate epigenetics in bacteria. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, L.; Chen, S.; Wu, X.; Gu, M.; Chen, X.; Jiang, S.; Wang, Y.; Deng, Z.; Dedon, P.C.; et al. Convergence of DNA methylation and phosphorothioation epigenetics in bacterial genomes. Proc. Natl. Acad. Sci. USA 2017, 114, 4501–4506. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Wu, X.; He, W.; Liu, Z.; Wu, S.; Chen, C.; Chen, S.; Xiang, Q.; Deng, Z.; Liang, D.; et al. DNA phosphorothioate modifications influence the global transcriptional response and protect DNA from double-stranded breaks. Sci. Rep. 2014, 4, 6642. [Google Scholar] [CrossRef] [PubMed]
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, W.W.; van der Donk, W.A. Biosynthesis of phosphonic and phosphinic acid natural products. Annu. Rev. Biochem. 2009, 78, 65–94. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.W.; Chin, J.P.; Quinn, J.P. Organophosphonates revealed: New insights into the microbial metabolism of ancient molecules. Nat. Rev. Microbiol. 2013, 11, 412. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.L.; Ingall, E.D.; Benner, R. Marine phosphorus is selectively remineralized. Nature 1998, 393, 426. [Google Scholar] [CrossRef]
- Kolowith, L.C.; Ingall, E.D.; Benner, R. Composition and cycling of marine organic phosphorus. Limnol. Oceanogr. 2001, 46, 309–320. [Google Scholar] [CrossRef]
- Dyhrman, S.T.; Benitez-Nelson, C.R.; Orchard, E.D.; Haley, S.T.; Pellechia, P.J. A microbial source of phosphonates in oligotrophic marine systems. Nat. Geosci. 2009, 2, 696. [Google Scholar] [CrossRef]
- Villarreal-Chiu, J.F.; Quinn, J.P.; McGrath, J.W. The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 2012, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- White, A.K.; Metcalf, W.W. Microbial Metabolism of Reduced Phosphorus Compounds. Annu. Rev. Microbiol. 2007, 61, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Peck, S.C.; Gao, J.; Van Der Donk, W.A. Discovery and biosynthesis of phosphonate and phosphinate natural products. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 516, pp. 101–123. ISBN 0076-6879. [Google Scholar]
- Ruth, J.H. Odor Thresholds and Irritation Levels of Several Chemical Substances: A Review. Am. Ind. Hyg. Assoc. J. 1986, 47, A-142–A-151. [Google Scholar] [CrossRef]
- Morton, S.C.; Edwards, M. Reduced Phosphorus Compounds in the Environment. Crit. Rev. Environ. Sci. Technol. 2005, 35, 333–364. [Google Scholar] [CrossRef]
- Devai, I.; Felfoldy, L.; Wittner, I.; Plosz, S. Detection of phosphine: New aspects of the phosphorus cycle in the hydrosphere. Nature 1988, 333, 343–345. [Google Scholar] [CrossRef]
- Glindemann, D.; Stottmeister, U.; Bergmann, A. Free phosphine from the anaerobic biosphere. Environ. Sci. Pollut. Res. 1996, 3, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, G. Phosphine in the fluvial and marine hydrosphere. Mar. Chem. 1994, 45, 197–205. [Google Scholar] [CrossRef]
- Han, C.; Geng, J.; Hong, Y.; Zhang, R.; Gu, X.; Wang, X.; Gao, S.; Glindemann, D. Free atmospheric phosphine concentrations and fluxes in different wetland ecosystems, China. Environ. Pollut. 2011, 159, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Geng, J.; Zhang, J.; Wang, X.; Gao, S. Phosphine migration at the water–air interface in Lake Taihu, China. Chemosphere 2011, 82, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liang, H.; Zhu, Y.; Mo, W.; Wang, Q.; Ren, H.; Wang, X.; Edwards, M.; Glindemann, D. Sources of matrix-bound phosphine in advanced wastewater treatment system. Chin. Sci. Bull. 2005, 50, 1274–1276. [Google Scholar] [CrossRef]
- Roels, J.; Verstraete, W. Occurrence and origin of phosphine in landfill gas. Sci. Total Environ. 2004, 327, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Bains, W.; Petkowski, J.J.; Sousa-Silva, C.; Seager, S. New environmental model for thermodynamic ecology of biological phosphine production. Sci. Total Environ. 2019, 658, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Pech, H.; Vazquez, M.G.; Van Buren, J.; Foster, K.L.; Shi, L.; Salmassi, T.M.; Ivey, M.M.; Pasek, M.A. Elucidating the Redox Cycle of Environmental Phosphorus Using Ion Chromatography. J. Chromatogr. Sci. 2011, 49, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Wang, Q.; Ding, W.; Wang, C.; Hou, L.; Ma, D. Penguins significantly increased phosphine formation and phosphorus contribution in maritime Antarctic soils. Sci. Rep. 2014, 4, 7055. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, G.; Glindemann, D. Phosphane (PH3) in the Biosphere. Angew. Chemie Int. Ed. English 1993, 32, 761–763. [Google Scholar] [CrossRef]
- Chughtai, M.; Pridham, J.B. Determination of phosphine by packed column gas chromatography with alkali flame ionisation detection. Anal. Comm. 1998, 35, 109–111. [Google Scholar] [CrossRef]
- Cahill, M.M. Bacterial flora of fishes: A review. Microb. Ecol. 1990, 19, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Guarner, F.; Malagelada, J.-R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Sissons, J.W. Potential of probiotic organisms to prevent diarrhoea and promote digestion in farm animals—A review. J. Sci. Food Agric. 1989, 49, 1–13. [Google Scholar] [CrossRef]
- Schmidt, O.; Wüst, P.K.; Hellmuth, S.; Borst, K.; Horn, M.A.; Drake, H.L. Novel [NiFe]- and [FeFe]-Hydrogenase Gene Transcripts Indicative of Active Facultative Aerobes and Obligate Anaerobes in Earthworm Gut Contents. Appl. Environ. Microbiol. 2011, 77, 5842–5850. [Google Scholar] [CrossRef] [PubMed]
- Brune, A.; Emerson, D.; Breznak, J.A. The Termite Gut Microflora as an Oxygen Sink: Microelectrode Determination of Oxygen and pH Gradients in Guts of Lower and Higher Termites. Appl. Environ. Microbiol. 1995, 61, 2681–2687. [Google Scholar] [PubMed]
- Jenkins, R.O.; Morris, T.A.; Craig, P.J.; Ritchie, A.W.; Ostah, N. Phosphine generation by mixed- and monoseptic-cultures of anaerobic bacteria. Sci. Total Environ. 2000, 250, 73–81. [Google Scholar] [CrossRef]
- Rutishauser, B.V.; Bachofen, R. Phosphine Formation from Sewage Sludge Cultures. Anaerobe 1999, 5, 525–531. [Google Scholar] [CrossRef]
- Bains, W.; Petkowski, J.J.; Sousa-Silva, C.; Seager, S. Trivalent Phosphorus and Phosphines as Components of Biochemistry in Anoxic Environments. Astrobiology 2019. in review. [Google Scholar]
- Sousa-Silva, C.; Seager, S.; Petkowski, J.J.; Ranjan, S.; Hu, R.; Zhan, Z.; Bains, W. On Phosphine as a Biosignature Gas in Exoplanet Atmospheres. Astrobiology 2019. (In review) [Google Scholar]
- Davies, M.J.; Hardege, J. Chemical Communication in the European Otter, Lutra lutra, University of Hull. Ph.D. Thesis, University of Hull, Hull, UK, 2008. [Google Scholar]
- Behnken, S.; Hertweck, C. Anaerobic bacteria as producers of antibiotics. Appl. Microbiol. Biotechnol. 2012, 96, 61–67. [Google Scholar] [CrossRef] [PubMed]






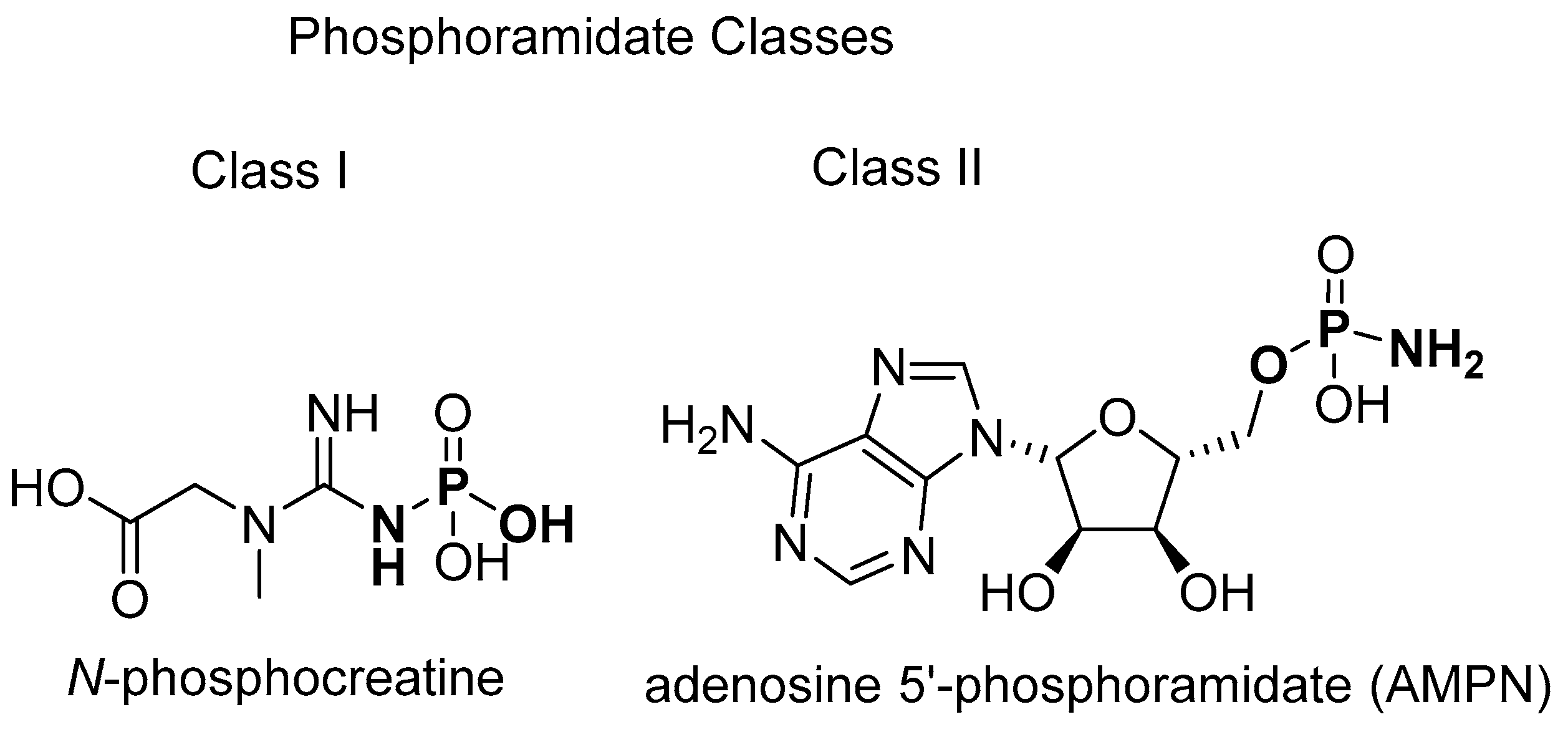{kind=link}
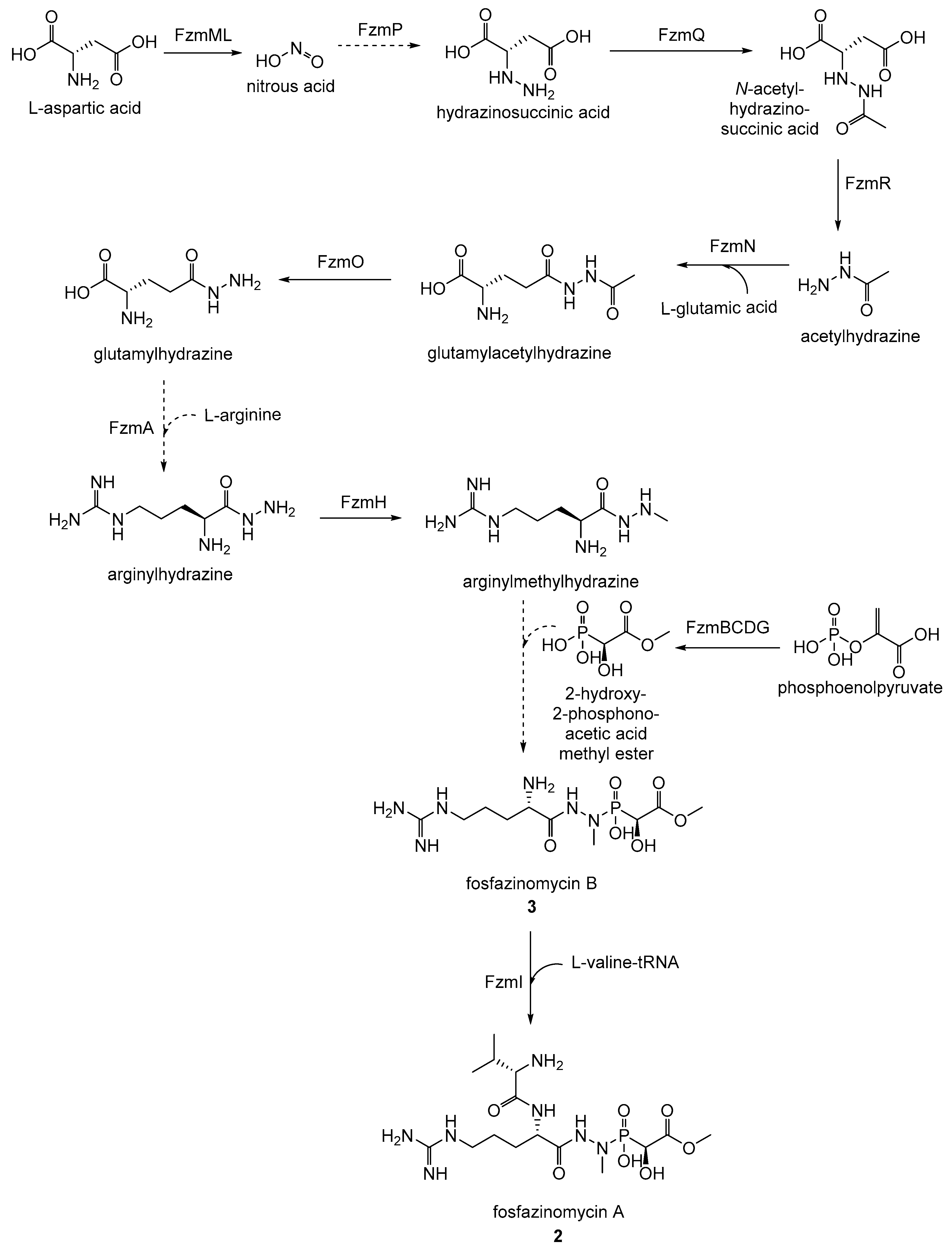{kind=link}
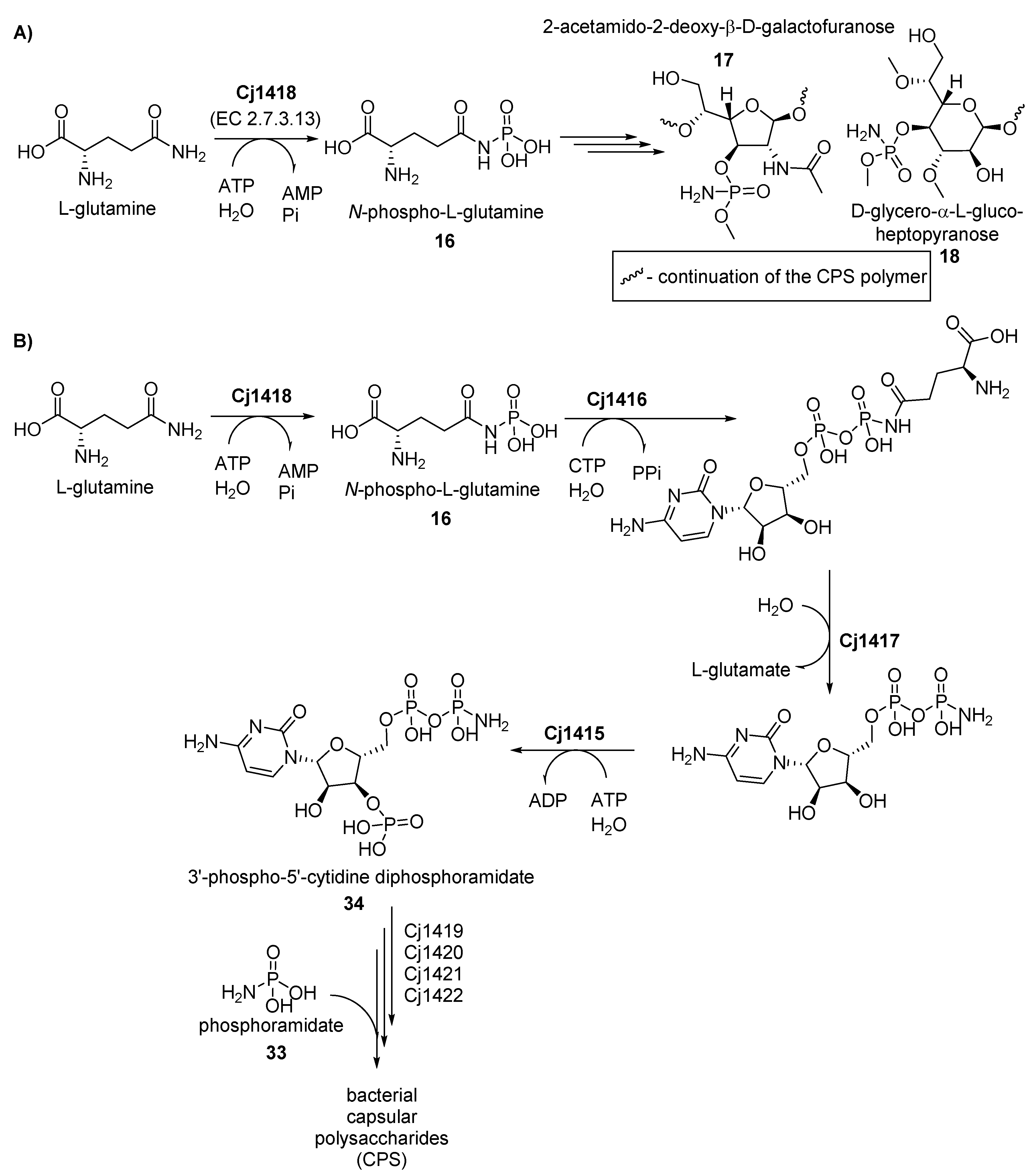{kind=link}
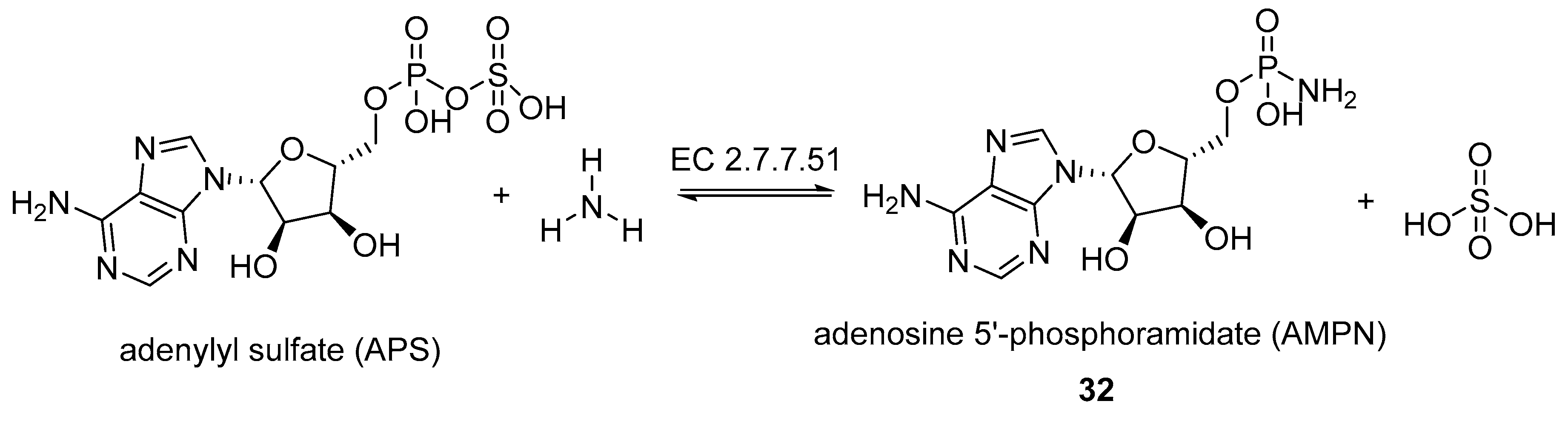{kind=link}
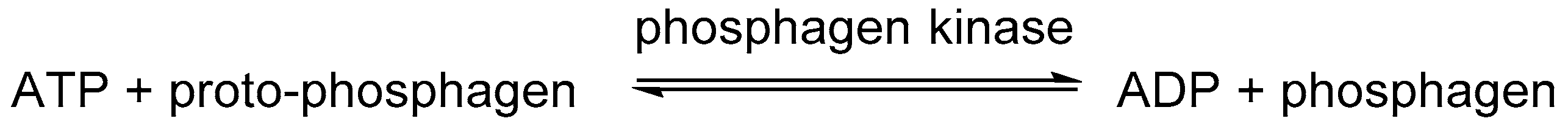{kind=link}
| Phosphagen | Natural Occurrence and Biochemical and Structural Characterization of the Phosphagen Kinases | ||||
|---|---|---|---|---|---|
| Domain, Kingdom (or other) | Phylum | Class | Species * | Reference | |
| N-phospho-arginine (19) | Animalia | Arthropoda | Insecta | Anasa tristis, Periplaneta americana, Solenopsis invicta, Cissites cephalotes, Plodia interpunctella, Manduca sexta, Locusta migratoria, Ctenocephalides felis, Lucilia cuprina, Apis mellifera, Bombyx mori, Ctenocephalides felis, Drosophila melanogaster, Musca domestica, Phormia regina, Frankliniella occidentalis | [119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135] |
| Arachnida | Polybetes pythagoricus, Holocnemus pluchei, Palamneus phipsoni | [70,136,137] | |||
| Branchiopoda | Daphnia magna | [138] | |||
| Chelicerata | Limulus polyphemus | [66,139,140] | |||
| Maxillopoda | Amphibalanus amphitrite | [141] | |||
| Malacostraca | Litopenaeus vannamei, Metapenaeus ensis, Neocaridina denticulata, Euphausia superba, Macrobrachium rosenbergii, Marsupenaeus japonicus, Pleocyemata sp., Portunus trituberculatus, Procambarus clarkii, Scylla serrata | [68,142,143,144,145,146,147,148,149,150,151,152,153] | |||
| Mollusca | Bivalvia | Crassostrea gigas, Chlamys farreri, Ensis directus, Calyptogena kaikoi, Corbicula japonica, Solen strictus, Pecten maximus, Archivesica packardana, Scapharca broughtonii | [154,155,156,157,158,159,160,161,162,163,164] | ||
| Cephalopoda | Sepia pharaonis, Amphioctopus fangsiao, Nautilus pompilius, Octopus vulgaris, Sepioteuthis lessoniana, Sthenoteuthis oualaniensis | [165,166,167,168,169] | |||
| Gastropoda | Semisulcospira libertina, Biomphalaria glabrata | [170,171] | |||
| Cnidaria | Anthozoa | Nematostella vectensis, Anthopleura japonicus, Paracorallium japonicum, Corallium rubrum | [67,172,173,174] | ||
| Annelida | Polychaeta | Myzostoma cirriferum, Sabellastarte indica | [109,110,175] | ||
| Bryozoa | Gymnolaemata | Bugula neritina | [176] | ||
| Echinodermata | Holothuroidea | Stichopus japonicus, Isostychopus badonotus, Molpadia arenicola | [64,177,178,179] | ||
| Echinoidea | Strongylocentrotus purpuratus, Heliocidaris crassispina, Hemicentrotus pulcherrimus, Paracentrotus lividus, Pseudocentrotus depressus | [178,180,181] | |||
| Crinoidea | Tropiometra afra Macrodiscus | [182] | |||
| Porifera | Demospongiae | Suberites domuncula, Clathria prolifera,Hartaetosiga gracilis, Suberites ficus | [58,183] | ||
| Hexactinellida | Aphrocallistes beatrix | [58] | |||
| Nematoda | Chromadorea | Heterodera glycines, Teladorsagia circumcincta, Steinernema carpocapsae,Haemonchus contortus, Toxocara canis, Toxocara vitulorum, Ascaris lumbricoides, Ascaris suum, Caenorhabditis elegans | [184,185,186,187,188,189,190,191] | ||
| Unranked | Unranked | Choanoflagellida | Hartaetosiga gracilis, Monosiga brevicollis, Monosiga ovata | [58] | |
| Excavata | Euglenozoa | Kinetoplastea | Trypanosoma brucei, Trypanosoma cruzi, Phytomonas Jma | [69,192,193,194,195,196] | |
| Alveolata | Ciliophora | Oligohymenophorea | Paramecium caudatum, Paramecium tetraurelia, Tetrahymena pyriformis | [56,76,197] | |
| Bacteria | Proteobacteria | Deltaproteobacteria | Desulfotalea psychrophila, Myxococcus xanthus | [53,55,198] | |
| Epsilonproteobacteria | Sulfurovum lithotrophicum | [54] | |||
| N-phospho-creatine (20) | Animalia | Chordata | Cephalaspidomorphi | Lampetra japonica | [199] |
| Actinopterygii | Danio rerio, Chaenocephalus aceratus, Clupea harengus, Cyprinus carpio, Gadus morhua, Lepomis cyanellus, Oncorhynchus mykiss, Pagrus major, Scomber japonicus | [74,200,201,202,203,204,205,206,207,208,209] | |||
| Chondrichthyes | Torpedo californica, Discopyge tschudii, Ginglymostoma cirratum, Scylliorhinus canicula | [210,211,212,213] | |||
| Amphibia | Xenopus laevis | [214] | |||
| Reptilia | Pelodiscus sinensis, Trachemys scripta | [209,215] | |||
| Aves | Columba livia, Gallus gallus | [216,217] | |||
| Mammalia | Bos taurus, Canis lupus familiaris, Homo sapiens, Mus Musculus, Eidolon helvum, Equus caballus, Oryctolagus cuniculus, Physeter catodon, Rattus norvegicus, Urocitellus richardsonii | [117,199,211,218,219,220,221,222,223,224,225] | |||
| Annelida | Polychaeta | Namalycastis sp., Neanthes diversicolor, Chaetopterus variopedatus, | [199,226,227] | ||
| Cnidaria | Anthozoa | Nematostella vectensis, Dendronephthya gigantea | [172,199] | ||
| Echinodermata | Echinoidea | Strongylocentrotus purpuratus,Heliocidaris crassispina, Hemicentrotus pulcherrimus, Paracentrotus lividus, Pseudocentrotus depressus | [180,181,228,229,230] | ||
| Porifera | Demospongiae | Tethya aurantia | [231] | ||
| N-phospho-taurocyamine (21) | Animalia | Platyhelminthes | Termatoda | Schistosoma mansoni, Schistosoma japonicum | [59,86,88] |
| Rhabditophora | Paragonimus westermani, Clonorchis sinensis | [82,83,87,89] | |||
| Annelida | Polychaeta | Arenicola brasiliensis, Riftia pachyptila,Arenicola marina | [81,84,85] | ||
| Chromista | Heterokontophyta | Oomycota | Phytophthora infestans, Phytophthora sojae | [232,233,234] | |
| N-phospho-hypotaurocyamine (22) | Animalia | Sipuncula | Sipunculidea | Golfingia vulgaris, Golfingia elongata, Siphonosoma cumanense | [77,78,104] |
| N-phospho-glycocyamine (23) | Animalia | Annelida | Polychaeta | Namalycastis sp., Nephtys hombergii, Neanthes diversicolor, Myxicola infundibulum, Nephtys caeca, Perinereis brevicirrus | [60,102,226,235,236,237,238,239] |
| Platyhelminthes | Rhabditophora | Polycelis cornuta | [239] | ||
| N-phospho-agmatine (24) | Chromista | Ochrophyta | Chrysophyceae | Ochromonas danica | [90] |
| Excavata | Euglenozoa | Euglenoidea | Euglena gracilis | [90] | |
| N-phospho-lombricine (25) | Animalia | Annelida | Echiura | Urechis caupo | [63,240] |
| Clitellata | Octolasium cyaneum, Allolobophora caliginosa, Lumbricus terrestris, Eisenia fetida, Enchytraeus sp., Stylaria sp., Lumbriculus variegatus, Tubifex tubifex, Megascolides cameroni | [75,95,96,99,100,101,241,242] | |||
| N-phospho-opheline (26) | Animalia | Annelida | Polychaeta | Ophelia neglecta | [94] |
| N-phospho-thalassemine (27) | Animalia | Annelida | Clitellata | Lumbricus terrestris | [103] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkowski, J.J.; Bains, W.; Seager, S. Natural Products Containing ‘Rare’ Organophosphorus Functional Groups. Molecules 2019, 24, 866. https://doi.org/10.3390/molecules24050866
Petkowski JJ, Bains W, Seager S. Natural Products Containing ‘Rare’ Organophosphorus Functional Groups. Molecules. 2019; 24(5):866. https://doi.org/10.3390/molecules24050866
Chicago/Turabian StylePetkowski, Janusz J., William Bains, and Sara Seager. 2019. "Natural Products Containing ‘Rare’ Organophosphorus Functional Groups" Molecules 24, no. 5: 866. https://doi.org/10.3390/molecules24050866
APA StylePetkowski, J. J., Bains, W., & Seager, S. (2019). Natural Products Containing ‘Rare’ Organophosphorus Functional Groups. Molecules, 24(5), 866. https://doi.org/10.3390/molecules24050866