
Identification of Compounds That Inhibit Estrogen-Related Receptor Alpha Signaling Using High-Throughput Screening Assays

Abstract
1. Introduction
2. Results
2.1. qHTS Performance and Reproducibility
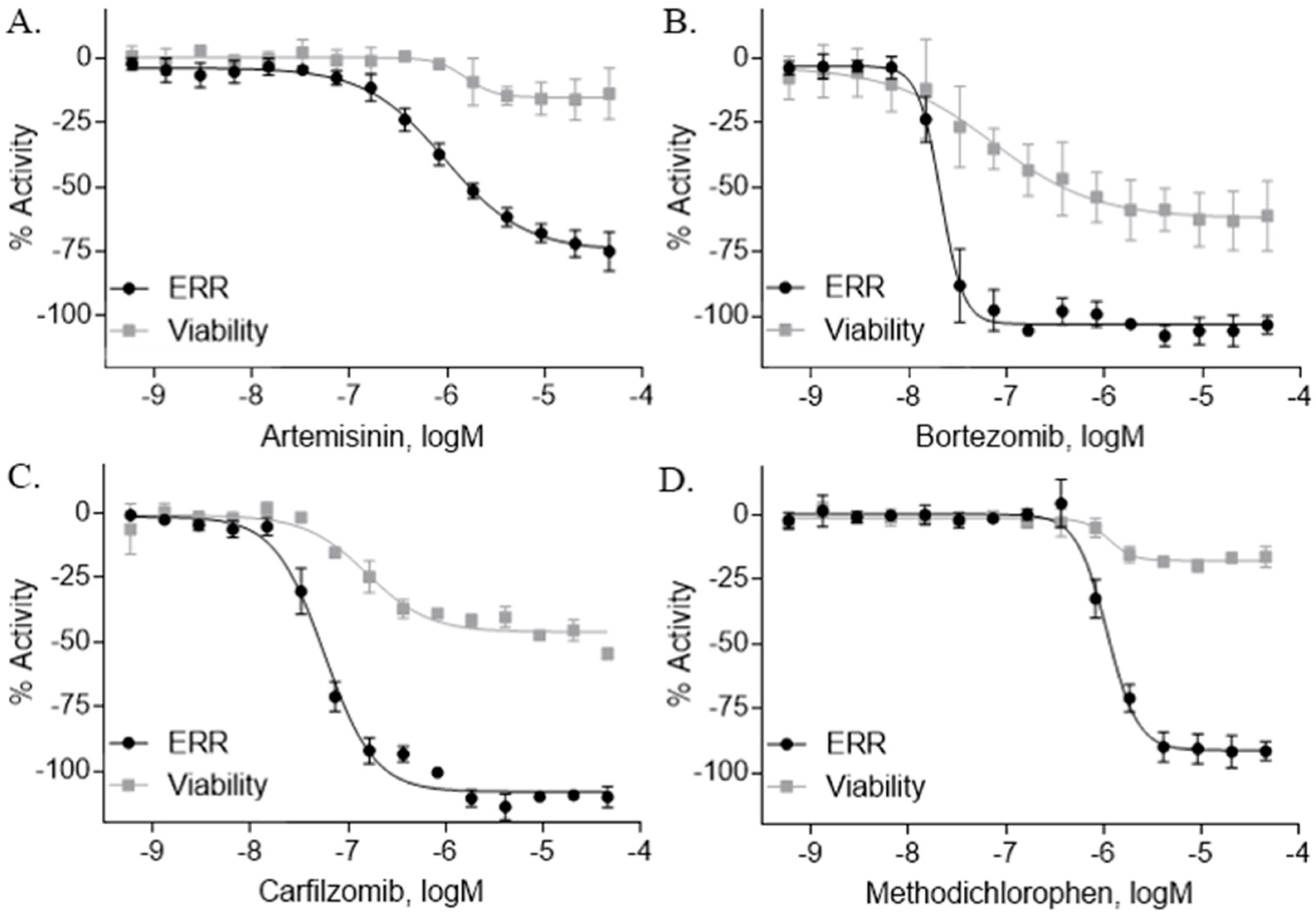
2.2. ERR Antagonists—Antineoplastic Agents
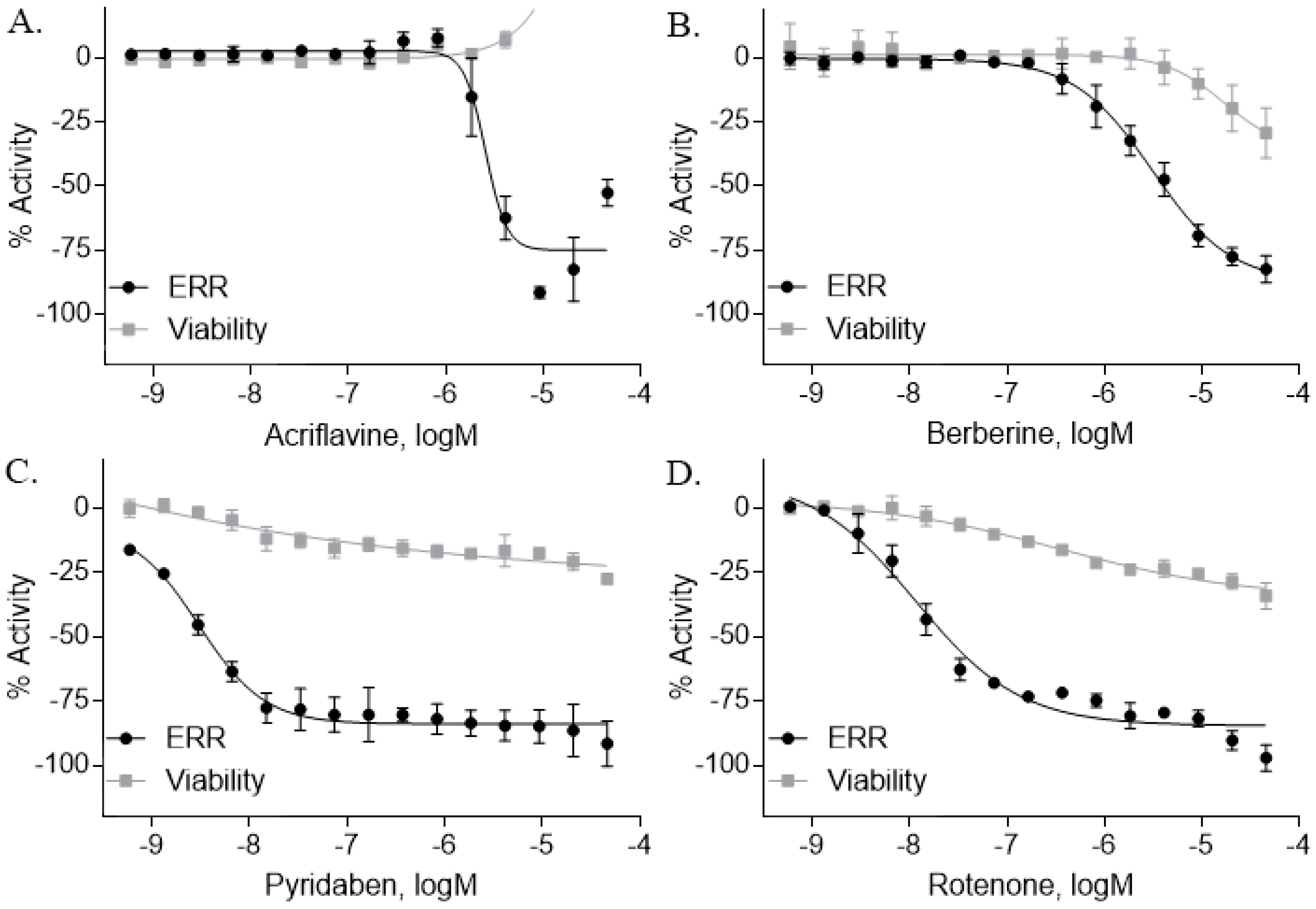
2.3. ERR Antagonists—Pesticides
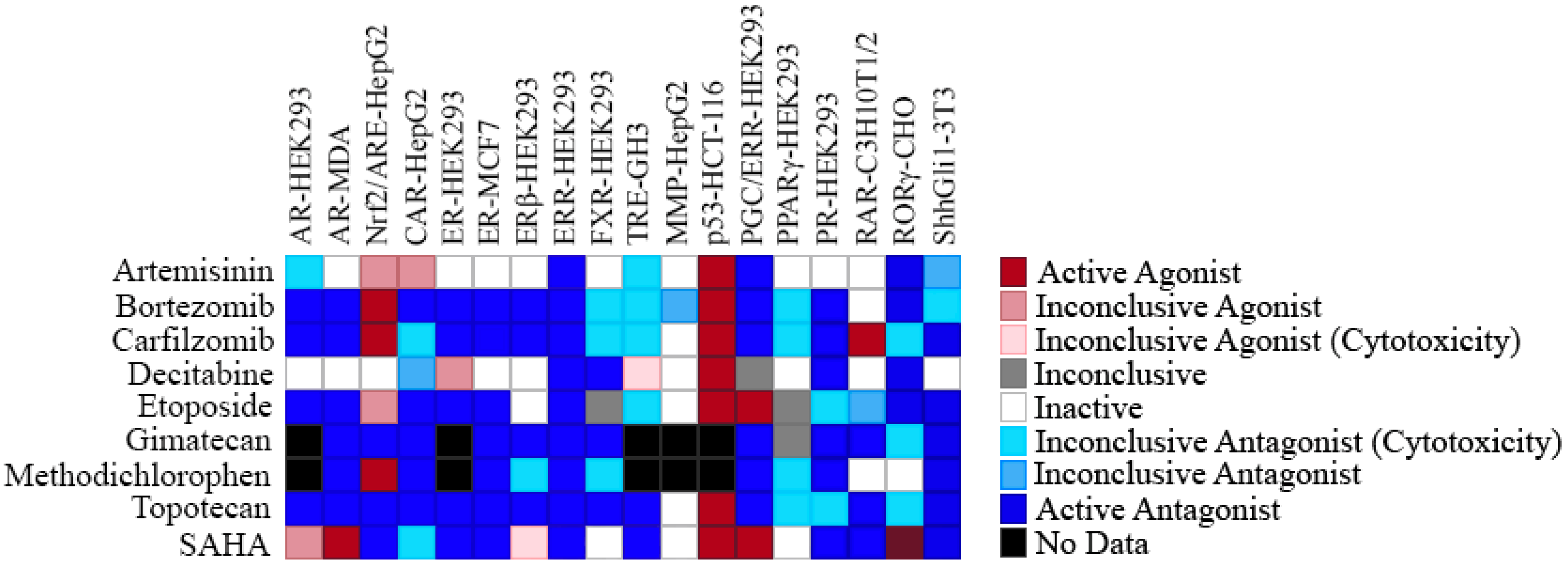
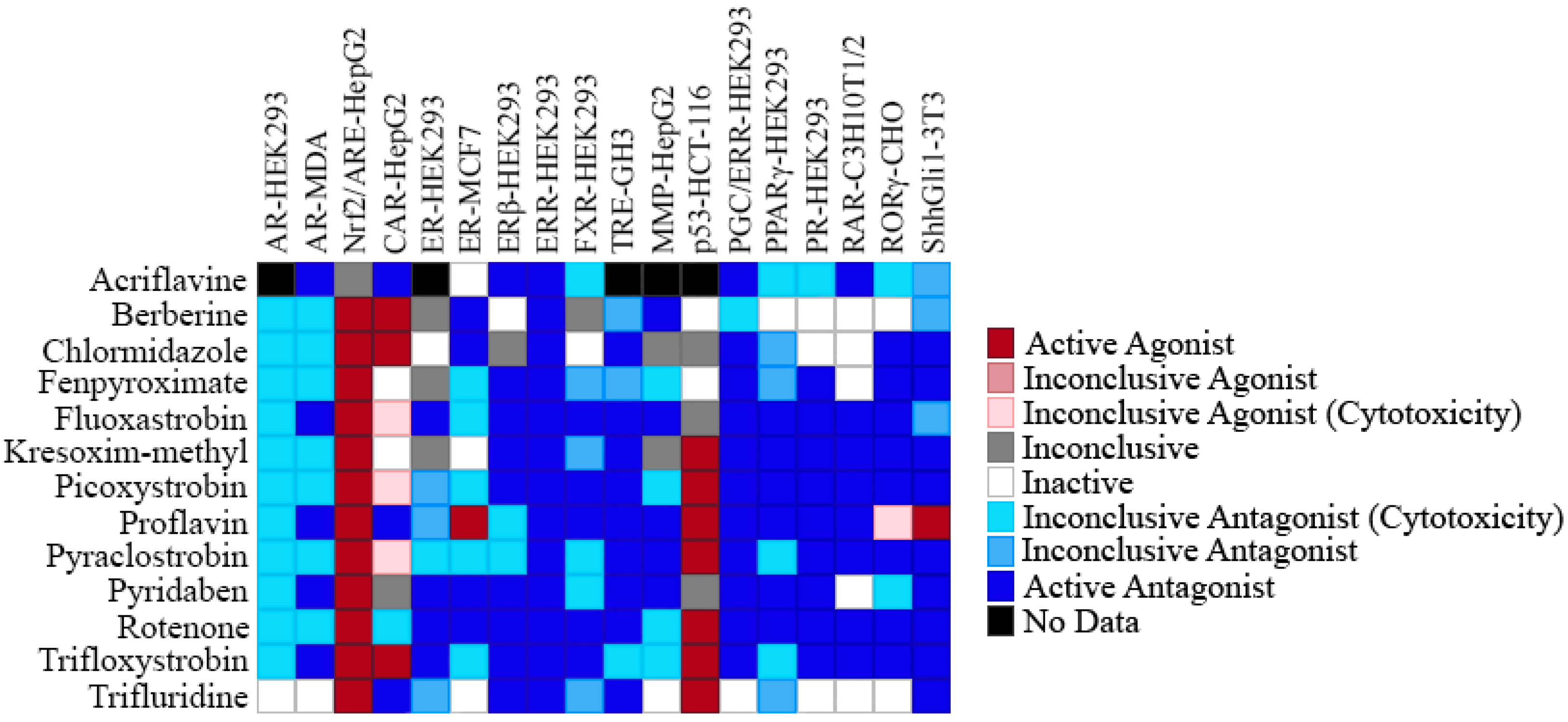
2.4. Selectivity of ERR Antagonists
2.5. Effects of the ERR Antineoplastic Agents on Gene Expression
2.6. Effects of the Pesticide Antagonists on Gene Expression
3. Discussion
4. Materials and Methods
4.1. Tox21 Chemical Library
4.2. Cell Lines and Culture Conditions
4.3. ERR and PGC/ERR Reporter Assays
4.4. AR-HEK293, ARE-HepG2, ER-HEK293, ERβ-HEK293, FXR-HEK293, p53-HCT116, PPARγ-HEK293, and PR-HEK293 β-Lactamase Reporter Gene Assays
4.5. Mitochondrial Membrane Potential (MMP) Assay
4.6. AR-MDA, CAR-HepG2, ER-MCF7, RAR-C3H10T1/2, RORγ-CHO, ShhGli1-3T3, and TRE-GH3 Luciferase Reporter Gene Assays
4.7. qHTS Data Analysis
4.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giguère, V.; Yang, N.; Segui, P.; Evans, R.M. Identification of a new class of steroid hormone receptors. Nature 1988, 331, 91. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V. To ERR in the estrogen pathway. Trends Endocrinol. Metab. 2002, 13, 220–225. [Google Scholar] [CrossRef]
- Shigeta, H.; Zuo, W.; Yang, N.; DiAugustine, R.; Teng, C.T. The mouse estrogen receptor-related orphan receptor alpha 1: Molecular cloning and estrogen responsiveness. J. Mol. Endocrinol. 1997, 19, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Shigeta, H.; Shi, H.; Teng, C.T. Estrogen-related receptor, hERR1, modulates estrogen receptor-mediated response of human lactoferrin gene promoter. J. Biol. Chem. 1996, 271, 5795–5804. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, Z.; Gladwell, W.; Teng, C.T. Estrogen stimulates estrogen-related receptor alpha gene expression through conserved hormone response elements. Endocrinology 2003, 144, 4894–4904. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr. Rev. 2008, 29, 677–696. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A.; Kralli, A. ERRalpha: A metabolic function for the oldest orphan. Trends Endocrinol. Metab. 2008, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Garbacz, W.G.; Xie, W. Constitutive activities of estrogen-related receptors: Transcriptional regulation of metabolism by the ERR pathways in health and disease. Biochim. Biophys. Acta 2015, 1852, 1912–1927. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sladek, R.; Carrier, J.; Bader, J.A.; Richard, D.; Giguere, V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol. Cell Biol. 2003, 23, 7947–7956. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Imahashi, K.; Dufour, C.R.; Weinheimer, C.J.; Courtois, M.; Kovacs, A.; Giguere, V.; Murphy, E.; Kelly, D.P. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007, 6, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Busch, B.B.; Stevens, W.C., Jr.; Martin, R.; Ordentlich, P.; Zhou, S.; Sapp, D.W.; Horlick, R.A.; Mohan, R. Identification of a selective inverse agonist for the orphan nuclear receptor estrogen-related receptor alpha. J. Med. Chem. 2004, 47, 5593–5596. [Google Scholar] [CrossRef] [PubMed]
- Chisamore, M.J.; Cunningham, M.E.; Flores, O.; Wilkinson, H.A.; Chen, J.D. Characterization of a novel small molecule subtype specific estrogen-related receptor alpha antagonist in MCF-7 breast cancer cells. PLoS ONE 2009, 4, e5624. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.T.; Beames, B.; Alex Merrick, B.; Martin, N.; Romeo, C.; Jetten, A.M. Development of a stable cell line with an intact PGC-1alpha/ERRalpha axis for screening environmental chemicals. Biochem. Biophys. Res. Commun. 2014, 444, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.T.; Hsieh, J.H.; Zhao, J.; Huang, R.; Xia, M.; Martin, N.; Gao, X.; Dixon, D.; Auerbach, S.S.; Witt, K.L.; et al. Development of Novel Cell Lines for High-Throughput Screening to Detect Estrogen-Related Receptor Alpha Modulators. SLAS Discov. 2017, 22, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Schwaid, A.G.; Wang, X.; Wang, X.; Chen, S.; Chu, Q.; Saghatelian, A.; Wan, Y. Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016, 23, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Willy, P.J.; Murray, I.R.; Qian, J.; Busch, B.B.; Stevens, W.C., Jr.; Martin, R.; Mohan, R.; Zhou, S.; Ordentlich, P.; Wei, P.; et al. Regulation of PPARgamma coactivator 1alpha (PGC-1alpha) signaling by an estrogen-related receptor alpha (ERRalpha) ligand. Proc. Natl. Acad. Sci. USA 2004, 101, 8912–8917. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Zhao, J.; Huang, R.; Kanaya, N.; Bernal, L.; Hsieh, J.H.; Auerbach, S.S.; Witt, K.L.; Merrick, B.A.; Chen, S.; et al. Identification of Estrogen-Related Receptor alpha Agonists in the Tox21 Compound Library. Endocrinology 2018, 159, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Clark, G.M.; Mertz, J.E. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res. 2002, 62, 6510–6518. [Google Scholar] [PubMed]
- Ariazi, E.A.; Kraus, R.J.; Farrell, M.L.; Jordan, V.C.; Mertz, J.E. Estrogen-related receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol. Cancer Res. 2007, 5, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Hall, J.A.; Perry, M.C.; Laganiere, J.; Ghahremani, M.; Park, M.; Hallett, M.; Giguere, V. Genome-wide identification of direct target genes implicates estrogen-related receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res. 2009, 69, 6149–6157. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Sailland, J.; Vanacker, J.M. ERRs and cancers: Effects on metabolism and on proliferation and migration capacities. J. Steroid Biochem. Mol. Biol. 2012, 130, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Handschin, C.; Arlow, D.; Xie, X.; St Pierre, J.; Sihag, S.; Yang, W.; Altshuler, D.; Puigserver, P.; Patterson, N.; et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. USA 2004, 101, 6570–6575. [Google Scholar] [CrossRef] [PubMed]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, e101. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.N.; Knutti, D.; Brogli, K.; Uhlmann, T.; Kralli, A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J. Biol. Chem. 2003, 278, 9013–9018. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Ohizumi, H.; Fujitani, Y.; Nemoto, T.; Tanaka, T.; Takahashi, N.; Kawada, T.; Miyoshi, M.; Ezaki, O.; Kakizuka, A. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc. Natl. Acad. Sci. USA 2003, 100, 12378–12383. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.A.; Gaillard, S.; McDonnell, D.P. Estrogen-related receptor alpha induces the expression of vascular endothelial growth factor in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2009, 114, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Fradet, A.; Sorel, H.; Bouazza, L.; Goehrig, D.; Depalle, B.; Bellahcene, A.; Castronovo, V.; Follet, H.; Descotes, F.; Aubin, J.E.; et al. Dual function of ERRalpha in breast cancer and bone metastasis formation: Implication of VEGF and osteoprotegerin. Cancer Res. 2011, 71, 5728–5738. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.A.; Chang, C.Y.; Kazmin, D.A.; Way, J.; Schroeder, T.; Wergin, M.; Dewhirst, M.W.; McDonnell, D.P. Estrogen-related receptor alpha is critical for the growth of estrogen receptor-negative breast cancer. Cancer Res. 2008, 68, 8805–8812. [Google Scholar] [CrossRef] [PubMed]
- Ao, A.; Wang, H.; Kamarajugadda, S.; Lu, J. Involvement of estrogen-related receptors in transcriptional response to hypoxia and growth of solid tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 7821–7826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, X.; Liang, N.; Li, S. Estrogen-related receptor alpha triggers the proliferation and migration of human non-small cell lung cancer via interleukin-6. Cell Biochem. Funct. 2018, 36, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Giguere, V. Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim. Biophys. Acta 2011, 1812, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.A.; Joseph, J.D.; Wade, H.E.; Eaton, M.L.; Kunder, R.S.; Kazmin, D.; Chang, C.Y.; McDonnell, D.P. WNT11 expression is induced by estrogen-related receptor alpha and beta-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res. 2010, 70, 9298–9308. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Zhao, J.; Huang, R.; Hsieh, J.H.; Hamm, J.; Chang, X.; Houck, K.; Xia, M. Quantitative high-throughput profiling of environmental chemicals and drugs that modulate farnesoid X receptor. Sci. Rep. 2014, 4, 6437. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Sakamuru, S.; Martin, M.T.; Reif, D.M.; Judson, R.S.; Houck, K.A.; Casey, W.; Hsieh, J.H.; Shockley, K.R.; Ceger, P.; et al. Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 2014, 4, 5664. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Zhao, J.; Huang, R.; Xiao, J.; Li, L.; Heyward, S.; Xia, M.; Wang, H. Quantitative high-throughput identification of drugs as modulators of human constitutive androstane receptor. Sci. Rep. 2015, 5, 10405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, Q.; Wang, Q.; Sun, W.; Pu, Z.; Wang, J.; Bao, Y. Pifithrin-alpha enhancing anticancer effect of topotecan on p53-expressing cancer cells. Eur. J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Chen, Z.J.; Jiang, G.M.; Zhang, K.S.; Liu, Q.; Liang, S.W.; Zhou, Y.; Huang, H.B.; Du, J.; Wang, H.S. Inverse agonist of estrogen-related receptor alpha suppresses the growth of triple negative breast cancer cells through ROS generation and interaction with multiple cell signaling pathways. Oncotarget 2016, 7, 12568–12581. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, H.; Duan, Z.; Zou, J.X.; Chen, H.; He, W.; Wang, J. Estrogen-related receptor alpha confers methotrexate resistance via attenuation of reactive oxygen species production and P53 mediated apoptosis in osteosarcoma cells. BioMed Res. Int. 2014, 2014, 616025. [Google Scholar] [CrossRef] [PubMed]
- Klayman, D.L. Qinghaosu (artemisinin): An antimalarial drug from China. Science 1985, 228, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Su, X.Z. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti-Infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Sasaki, T.; Singh, N.P. Targeted treatment of cancer with artemisinin and artemisinin-tagged iron-carrying compounds. Expert Opin. Ther. Targets 2005, 9, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Kumari, K.; Keshari, S.; Sengupta, D.; Sabat, S.C.; Mishra, S.K. Transcriptome analysis of genes associated with breast cancer cell motility in response to Artemisinin treatment. BMC Cancer 2017, 17, 858. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Liebes, L.; Ng, B.; Buckley, M.; Elliott, P.J.; Adams, J.; Jiang, J.D.; Muggia, F.M.; Perez-Soler, R. PS-341, a novel proteasome inhibitor, induces Bcl-2 phosphorylation and cleavage in association with G2-M phase arrest and apoptosis. Mol. Cancer Ther. 2002, 1, 841–849. [Google Scholar] [PubMed]
- Accardi, F.; Toscani, D.; Bolzoni, M.; Dalla Palma, B.; Aversa, F.; Giuliani, N. Mechanism of Action of Bortezomib and the New Proteasome Inhibitors on Myeloma Cells and the Bone Microenvironment: Impact on Myeloma-Induced Alterations of Bone Remodeling. BioMed Res. Int. 2015, 2015, 172458. [Google Scholar] [CrossRef] [PubMed]
- Price, L.A.; Goldie, J.H.; Hill, B.T. Methodichlorophen as anti-tumor drug. Br. Med. J. 1975, 2, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Okamura, K.; Kitaoka, C.; Kinoshita, F.; Namitome, R.; Nakamura, U.; Shiota, M.; Goto, K.; Ohtsubo, T.; Matsumura, K.; et al. Metyrapone-responsive ectopic ACTH-secreting pheochromocytoma with a vicious cycle via a glucocorticoid-driven positive-feedback mechanism. Endocr. J. 2018, 65, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Iwayama, H.; Hirase, S.; Nomura, Y.; Ito, T.; Morita, H.; Otake, K.; Okumura, A.; Takagi, J. Spontaneous adrenocorticotropic hormone (ACTH) normalisation due to tumour regression induced by metyrapone in a patient with ectopic ACTH syndrome: Case report and literature review. BMC Endocr. Disord. 2018, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, G.; Beretta, G.L.; Zunino, F. Gimatecan, a novel camptothecin with a promising preclinical profile. Anticancer Drugs 2004, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lau, L.F.; Dai, X.; Li, B. In Vitro and In Vivo Anticancer Activity of Gimatecan against Hepatocellular Carcinoma. Asian Pac. J. Cancer Prev. 2016, 17, 4853–4856. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, S.; Ray-Coquard, I.; Tredan, O.; Colombo, N.; Parma, G.; Tisi, G.; Katsaros, D.; Lhomme, C.; Lissoni, A.A.; Vermorken, J.B.; et al. Phase II of oral gimatecan in patients with recurrent epithelial ovarian, fallopian tube or peritoneal cancer, previously treated with platinum and taxanes. Ann. Oncol. 2010, 21, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Garcia-Vargas, J.; Hardwick, J.S. Development of vorinostat: Current applications and future perspectives for cancer therapy. Cancer Lett. 2009, 280, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Dequiedt, F.; Kasler, H.G. Class II histone deacetylases: Versatile regulators. Trends Genet. 2003, 19, 286–293. [Google Scholar] [CrossRef]
- Zhang, C.; Zhong, J.F.; Stucky, A.; Chen, X.L.; Press, M.F.; Zhang, X. Histone acetylation: Novel target for the treatment of acute lymphoblastic leukemia. Clin. Epigenet. 2015, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Wong, C. Oestrogen-related receptor alpha inverse agonist XCT-790 arrests A549 lung cancer cell population growth by inducing mitochondrial reactive oxygen species production. Cell Prolif. 2010, 43, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Mastropasqua, F.; Girolimetti, G.; Shoshan, M. PGC1alpha: Friend or Foe in Cancer? Genes 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E., Jr.; Lee, Y.; Gerhart-Hines, Z.; Puigserver, P. Nutrient-dependent regulation of PGC-1alpha’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim. Biophys. Acta 2010, 1804, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Fullgrabe, J.; Hermanson, O.; Joseph, B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide. Oncogene 2010, 29, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Ueha, T.; Kawamoto, T.; Hara, H.; Toda, M.; Harada, R.; Minoda, M.; Kurosaka, M.; Akisue, T. Regulation of mitochondrial proliferation by PGC-1alpha induces cellular apoptosis in musculoskeletal malignancies. Sci Rep. 2014, 4, 3916. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Wu, S.Y.; Ma, Q.; Shi, N. The pesticide deltamethrin increases free radical production and promotes nuclear translocation of the stress response transcription factor Nrf2 in rat brain. Toxicol. Ind. Health 2011, 27, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Lo, S.C.; Liu, J.H.; Hannink, M.; Lubahn, D.B. ERRbeta: A potent inhibitor of Nrf2 transcriptional activity. Mol. Cell Endocrinol. 2007, 278, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.C.; Anderson, W.A. Effects of acriflavine on the mitochondria and kinetoplast of Crithidia fasciculata. Correlation of fine structure changes with decreased mitochondrial enzyme activity. J. Cell Biol. 1969, 41, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.M.; Ahammadsahib, K.I.; Gadelhak, G.; McLaughlin, J.L. New inhibitors of complex I of the mitochondrial electron transport chain with activity as pesticides. Biochem. Soc. Trans. 1994, 22, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Affuso, F.; Mercurio, V.; Fazio, V.; Fazio, S. Cardiovascular and metabolic effects of Berberine. World J. Cardiol. 2010, 2, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R.; Kowalczyk, W. Azole antimycotics—A highway to new drugs or a dead end? Curr. Med. Chem. 2012, 19, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Stenrod, M.; Klemsdal, S.S.; Norli, H.R.; Eklo, O.M. Effects of picoxystrobin and 4-n-nonylphenol on soil microbial community structure and respiration activity. PLoS ONE 2013, 8, e66989. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A. Acridine derivatives as chemotherapeutic agents. Curr. Med. Chem. 2002, 9, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Demeunynck, M.; Charmantray, F.; Martelli, A. Interest of acridine derivatives in the anticancer chemotherapy. Curr. Pharm. Des. 2001, 7, 1703–1724. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Huang, R.; Shi, Q.; Boyd, W.A.; Zhao, J.; Sun, N.; Rice, J.R.; Dunlap, P.E.; Hackstadt, A.J.; Bridge, M.F.; et al. Comprehensive Analyses and Prioritization of Tox21 10K Chemicals Affecting Mitochondrial Function by in-Depth Mechanistic Studies. Environ. Health Perspect. 2018, 126, 077010. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, J.E.; Jeong, S.H.; Paik, M.K.; Kim, J.S.; Cho, M.H. Trifloxystrobin-induced mitophagy through mitochondrial damage in human skin keratinocytes. J. Toxicol. Sci. 2016, 41, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Balsa, E.; Thomas, A.; Hatting, M.; Jedrychowski, M.; Gygi, S.P.; Widlund, H.R.; Puigserver, P. ERRalpha Maintains Mitochondrial Oxidative Metabolism and Constitutes an Actionable Target in PGC1alpha-Elevated Melanomas. Mol. Cancer Res. 2017, 15, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Freitas, J.; Miller, N.; Mengeling, B.J.; Xia, M.; Huang, R.; Houck, K.; Rietjens, I.M.; Furlow, J.D.; Murk, A.J. Identification of thyroid hormone receptor active compounds using a quantitative high-throughput screening platform. Curr. Chem. Genom. Transl. Med. 2014, 8, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sakamuru, S.; Huang, R.; Reese, D.H.; Xia, M. Identification of compounds that modulate retinol signaling using a cell-based qHTS assay. Toxicol. In Vitro 2016, 32, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Huang, R. A Quantitative High-Throughput Screening Data Analysis Pipeline for Activity Profiling. Methods Mol. Biol. 2016, 1473, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, R. Correction of Microplate Data from High-Throughput Screening. In High-Throughput Screening Assays in Toxicology; Zhu, H., Xia, M., Eds.; Springer: New York, NY, USA, 2016; pp. 123–134. [Google Scholar]
- Wang, Y.; Jadhav, A.; Southal, N.; Huang, R.; Nguyen, D.T. A grid algorithm for high throughput fitting of dose-response curve data. Curr. Chem. Genom. 2010, 4, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Auld, D.S.; Jadhav, A.; Johnson, R.L.; Simeonov, A.; Yasgar, A.; Zheng, W.; Austin, C.P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. USA 2006, 103, 11473–11478. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xia, M.; Cho, M.H.; Sakamuru, S.; Shinn, P.; Houck, K.A.; Dix, D.J.; Judson, R.S.; Witt, K.L.; Kavlock, R.J.; et al. Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ. Health Perspect. 2011, 119, 1142–1148. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Name (CAS #, Source) [Purity] | Structure | ERR-HEK293 | PGC/ERR-HEK293 | ||
|---|---|---|---|---|---|
| Primary EC50 (µM) [Efficacy (%)] Viability | Follow-Up EC50 (µM) [Efficacy (%)] Viability | Primary EC50 (µM) [Efficacy (%)] Viability | Follow-Up EC50 (µM) [Efficacy (%)] Viability | ||
| Artemisinin (63968-64-9, Microsource) [A] |  | 1.01 ± 0.297 [−71.6 ± 5.57] Inactive | 2.44 ± 1.22 [−83.7 ± 14.6] Inactive | 1.78 ± 0.401 [−57.6 ± 2.03] Inactive | 14.5 ± 10.8 [−72.8 ± 22.0] Inactive |
| Bortezomib (179324-69-7, Selleck) [N/A] |  | 0.0170 ± 0.00781 [−92.2 ± 11.8] 0.0656 ± 0.0247 [−60.7 ± 7.59] | 0.0167 ± 0.00608 [−125 ± 11.0] 0.0780 ± 0.0140 [−61.3 ± 6.93] | 0.0169 ± 0.00554 [−92.2 ± 11.8] 0.0628 ± 0.0235 [−48.6 ± 1.34] | 0.00962 ± 0.00452 [−116 ± 12.7] 0.0643 ± 0.0110 [−61.1 ± 3.07] |
| Carfilzomib (868540-17-4, Sequoia) [A] |  | 0.0552 ± 0.00374 [−107 ± 2.77] 0.178 ± 0.0767 [−46.9 ± 4.15] | 0.0617 ± 0.00786 [−126 ± 10.9] 0.274 ± 0.0186 [−55.3 ± 0.231] | 0.0615 ± 0.0198 [−121 ± 7.36] 1.36 ± 1.73 [−61.8 ± 18.0] | 0.189 ± 0.0396 [−150 ± 8.02] 0.471 ± 0.0541 [−68.5 ± 2.09] |
| Decitabine (2353-33-5, Tocris) [A] |  | 1.52 ± 0.348 [−79.1 ± 6.79] Inactive | 3.10 ± 0.558 [−97.8 ± 15.4] Inactive | Inactive Inactive | Inactive Inactive |
| Etoposide (33419-42-0, Light Biologicals) [A] |  | 9.24 ± 8.56 [−115 ± 6.85] 56.3 ± 7.18 [−40.5 ± 13.0] | 8.12 ± 4.42 [−88.9 ± 6.62] Inactive | 2.10 ± 0.526 [62.9 ± 2.71] 56.3 ± 7.18 [−44.2 ± 8.70] | 10.7 ± 2.44 [89.3 ± 19.5] Inactive |
| Gimatecan (292618-32-7, GVK) [I] |  | 0.0497 ± 0.00894 [−117 ± 14.5] Inactive | 0.0442 ± 0.0204 [−145 ± 16.0] Inactive | 2.53 ± 1.92 [−115 ± 2.20] Inactive | 3.34 ± 2.29 [−130 ± 20.6] Inactive |
| Methodichlorophen (7761-45-7, GVK) [A] |  | 1.06 ± 0.122 [−90.9 ± 4.94] Inactive | 1.60 ± 0.104 [−104 ± 7.68] Inactive | 0.845 ± 0.0971 [−68.4 ± 7.64] Inactive | 1.57 ± 0.354 [−61.5 ± 12.6] Inactive |
| Topotecan hydrochloride (119413-54-6, Prestwick) [A] |  | 1.24 ± 0.0837 [−127 ± 11.8] Inactive | 0.722 ± 0.285 [−140 ± 24.4] Inactive | 14.1 ± 1.15 [−69.1 ± 5.82] Inactive | 5.26 ± 0.00 [−78.8 ± 7.20] Inactive |
| Vorinostat (SAHA) (149647-78-9, Prestwick) [A] |  | 0.162 ± 0.0105 [−54.7 ± 3.05] 5.16 ± 0.879 [−33.1 ± 1.56] | 0.359 ± 0.0234 [−55.0 ± 10.8] Inactive | 2.11 ± 0.00 [382 ± 23.2] Inactive | 2.44 ± 0.165 [479± 63.2] Inactive |
| Chemical Name (CAS #, Source) [Purity] | Structure | ERR-HEK293 | PGC/ERR-HEK293 | ||
|---|---|---|---|---|---|
| Primary EC50 (µM) [Efficacy (%)] Viability | Follow-Up EC50 (µM) [Efficacy (%)] Viability | Primary EC50 (µM) [Efficacy (%)] Viability | Follow-Up EC50 (µM) [Efficacy (%)] Viability | ||
| Acriflavine hydrochloride (69235-50-3, Microsource) [I] |  | 2.75 ± 0.885 [−83.8 ± 13.1] Inactive | 8.96 ± 2.65 [−153 ± 1.29] Inactive | 2.30 ± 0.316 [−50.7 ± 8.37] Inactive | 5.66 ± 3.31 [−132 ± 25.7] Inactive |
| Berberine (633-65-8, Labotest) [A] |  | 3.16 ± 0.713 [−88.4 ± 7.45] 12.8 ± 0.837 [−31.7 ± 10.2] | 3.29 ± 2.27 [−92.0 ± 16.0] 12.7 ± 5.33 [−54.1 ± 10.6] | 6.13 ± 1.71 [−77.3 ± 14.6] 17.3 ± 4.82 [−31.8 ± 3.57] | 8.93 ± 4.01 [−87.4 ± 20.8] 17.3 ± 1.17 [−43.4 ± 9.71] |
| Chlormidazole (3689-76-7, Vitas) [A] |  | 0.784 ± 0.0999 [−64.3 ± 5.95] Inactive | 2.31 ± 2.55 [−96.2 ± 32.3] 19.3 ± 6.20 [−43.6 ± 15.3] | 1.48 ± 1.02 [−42.2 ± 2.79] Inactive | 0.565 ± 0.352 [−56.9 ± 16.8] Inactive |
| Fenpyroximate (111812-58-9, Sigma) [N/A] |  | 0.00452 ± 0.000577 [−67.1 ± 9.31] Inactive | 0.00699 ± 0.00328 [−89.6 ± 6.08] 51.4 ± 87.2 [−36.9 ± 12.9] | 0.00875 ± 0.000572 [−36.8 ± 2.17] Inactive | 0.0186 ± 0.0210 [−56.2 ± 10.5] Inactive |
| Fluoxastrobin (361377-29-9, Sigma) [N/A] |  | 2.05 ± 0.893 [−90.0 ± 16.2] 22.5 ± 11.2 [−49.1 ± 7.27] | 7.19 ± 3.10 [−109 ± 19.7] 24.1 ± 12.2 [−46.2 ± 5.55] | 14.0 ± 12.0 [−77.2 ± 11.6] 38.5 ± 13.8 [−46.4 ± 6.61] | 7.62 ± 2.72 [−90.2 ± 12.1] 41.3 ± 2.69 [−43.4 ± 6.25] |
| Kresoxim-methyl (143390-89-0, Light Biologicals) [A] |  | 5.65 ± 6.96 [−86.6 ± 21.1] Inactive | 8.31 ± 3.29 [−102 ± 1.40] Inactive | 7.66 ± 6.87 [−56.7 ± 8.51] Inactive | 17.7 ± 6.04 [−61.1 ± 10.0] Inactive |
| Picoxystrobin (117428-22-5, Sigma) [N/A] |  | 0.807 ± 0.279 [−83.9 ± 10.6] Inactive | 3.42 ± 0.393 [−86.7 ± 15.8] 32.1 ± 14.8 [−30.1 ± 2.49] | 2.79 ± 2.40 [−44.2 ± 4.37] Inactive | 4.19 ± 2.97 [−43.6 ± 14.2] Inactive |
| Proflavin hemisulfate (1811-28-5, Vitas1) [A] |  | 2.89 ± 0.398 [−95.0 ± 10.9] Inactive | 3.38 ± 0.772 [−86.8 ± 8.82] Inactive | 20.9 ± 16.2 [−94.9 ± 12.3] Inactive | 1.88 ± 2.61 [−39.0 ± 79.4] Inactive |
| Pyraclostrobin (175013-18-0, Sigma) [A] |  | 0.239 ± 0.130 [−90.7 ± 11.9] 40.9 ± 17.8 [−31.0 ± 2.62] | 0.762 ± 0.00 [−80.0 ± 5.67] Inactive | 0.838 ± 0.771 [−58.2 ± 4.17] 74.5 ± 10.3 [−31.3 ± 5.61] | 0.539 ± 0.760 [−42.1 ± 9.82] Inactive |
| Pyridaben (96489-71-3, Sigma) [A] |  | 0.00592 ± 0.000815 [−77.3 ± 4.10] Inactive | 0.0245 ± 0.00942 [−94.1 ± 13.4] Inactive | 0.00781 ± 0.00301 [−57.6 ± 8.49] Inactive | 0.0434 ± 0.0200 [−63.1 ± 29.7] Inactive |
| Rotenone (83-79-4, Microsource) [B] |  | 0.0133 ± 0.00476 [−92.4 ± 3.03] Inactive | 0.0160 ± 0.00671 [−95.8 ± 17.8] 0.429 ± 0.168 [−50.4 ± 7.00] | 0.00742 ± 0.00207 [−31.0 ± 4.14] 1.01 ± 0.454 [−35.8 ± 12.3] | 0.0618 ± 0.0610 [−41.9 ± 8.70] 2.97 ± 0.341 [−52.4 ± 5.35] |
| Trifloxystrobin (141517-21-7, Sigma) [A] |  | 0.801 ± 0.0542 [−88.1 ± 18.7] Inactive | 4.36 ± 0.998 [−100 ± 4.87] Inactive | 2.30 ± 0.187 [−45.2 ± 1.61] Inactive | 5.86 ± 0.807 [−56.7 ± 10.7] Inactive |
| Trifluridine (70-00-8, Prestwick) [A] |  | 0.875 ± 0.0593 [−51.8 ± 5.77] Inactive | 1.75 ± 0.314 [−67.7 ± 6.20] Inactive | Inactive Inactive | Inactive Inactive |
| COX8α | IDH3α | PPARα | COX4I1 | Cytochrome C | |
|---|---|---|---|---|---|
| XCT790 (12 µM) | 65.0 ± 8.41 | 42.6 ± 6.59 | 16.0 ± 1.70 | 46.0 ± 2.16 | 60.8 ± 2.16 |
| Artemisinin (°: 2 µM)(*: 7 µM) | 39.0° ± 15.1 | 30.2* ± 4.28 | 26.2* ± 8.02 | 20.5* ± 4.06 | 36.2* ± 3.38 |
| Bortezomib (20 µM) | 37.7 ± 5.50 | 17.6 ± 4.66 | 14.0 ± 5.17 | 35.3 ± 3.49 | 48.3 ± 1.92 |
| Carfilzomib (60 nM) | 39.6 ± 1.81 | 1.73 ± 0.76 | 7.39 ± 4.61 | 37.8 ± 5.12 | 44.2 ± 0.70 |
| Decitabine (2 µM) | 5.79 ± 45.6 | ||||
| Etoposide (18 µM) | 7.19 ± 23.5 | 6.55 ± 3.18 | |||
| Gimatecan (°: 0.2 µM)(*: 1 µM) | 9.84° ± 0.47 | 7.43* ± 4.35 | 31.5* ± 5.35 | ||
| Methodichlorophen (2 µM) | 17.1 ± 29.5 | 50.8 ± 8.69 | 12.5 ± 2.60 | 47.6 ± 7.45 | |
| Topotecan (°: 1.5 µM)(*: 7 µM) | 23.6* ± 13.6 | 18.1° ± 9.85 | |||
| SAHA (300 nM) | 33.4 ± 22.5 | 26.1 ± 32.9 |
| COX8α | IDH3α | PPARα | COX4I1 | Cytochrome C | |
|---|---|---|---|---|---|
| XCT790 (12 µM) | 65.0 ± 8.41 | 42.6 ± 6.59 | 16.0 ± 1.70 | 46.0 ± 2.16 | 60.8 ± 2.16 |
| Acriflavine (3 µM) | 30.2 ± 18.2 | 74.2 ± 9.92 | 4.76 ± 8.24 | 41.8 ± 3.72 | |
| Berberine (20 µM) | 5.15 ± 0.17 | 15.1 ± 2.97 | 20.5 ± 4.06 | 46.2 ± 2.61 | |
| Chlormidazole (5 µM) | 23.8 ± 3.17 | 4.71 ± 5.09 | 35.8 ± 3.92 | ||
| Fenpyroximate (7.5 nM) | 5.28 ± 4.56 | ||||
| Fluoxastrobin (7 µM) | 13.3 ± 2.44 | 25.5 ± 7.81 | |||
| Kresoxim-methyl (2 µM) | 9.70 ± 12.8 | 8.26 ± 19.3 | 9.31 ± 0.48 | ||
| Picoxystrobin (°: 1 µM)(*: 1.5 µM) | 28.4° ± 6.07 | 17.7° ± 34.4 | 3.06* ± 0.28 | 31.6* ± 3.25 | |
| Proflavin (3 µM) | 34.8 ± 26.7 | 24.9 ± 20.6 | 20.6 ± 8.69 | ||
| Pyraclostrobin (1 µM) | 5.06 ± 5.03 | 11.0 ± 8.54 | |||
| Pyridaben (°: 5 nM)(*: 15 nM) | 26.8° ± 23.4 | 14.6* ± 8.15 | 20.3* ± 2.05 | ||
| Rotenone (175 nM) | 22.4 ± 3.14 | 2.34 ± 3.78 | 27.3 ± 7.33 | ||
| Trifloxystrobin (0.5 µM) | 38.4 ± 17.2 | 10.4 ± 0.21 | |||
| Trifluridine (1 µM) | 43.2 ± 21.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynch, C.; Zhao, J.; Sakamuru, S.; Zhang, L.; Huang, R.; Witt, K.L.; Merrick, B.A.; Teng, C.T.; Xia, M. Identification of Compounds That Inhibit Estrogen-Related Receptor Alpha Signaling Using High-Throughput Screening Assays. Molecules 2019, 24, 841. https://doi.org/10.3390/molecules24050841
Lynch C, Zhao J, Sakamuru S, Zhang L, Huang R, Witt KL, Merrick BA, Teng CT, Xia M. Identification of Compounds That Inhibit Estrogen-Related Receptor Alpha Signaling Using High-Throughput Screening Assays. Molecules. 2019; 24(5):841. https://doi.org/10.3390/molecules24050841
Chicago/Turabian StyleLynch, Caitlin, Jinghua Zhao, Srilatha Sakamuru, Li Zhang, Ruili Huang, Kristine L. Witt, B. Alex Merrick, Christina T. Teng, and Menghang Xia. 2019. "Identification of Compounds That Inhibit Estrogen-Related Receptor Alpha Signaling Using High-Throughput Screening Assays" Molecules 24, no. 5: 841. https://doi.org/10.3390/molecules24050841
APA StyleLynch, C., Zhao, J., Sakamuru, S., Zhang, L., Huang, R., Witt, K. L., Merrick, B. A., Teng, C. T., & Xia, M. (2019). Identification of Compounds That Inhibit Estrogen-Related Receptor Alpha Signaling Using High-Throughput Screening Assays. Molecules, 24(5), 841. https://doi.org/10.3390/molecules24050841