
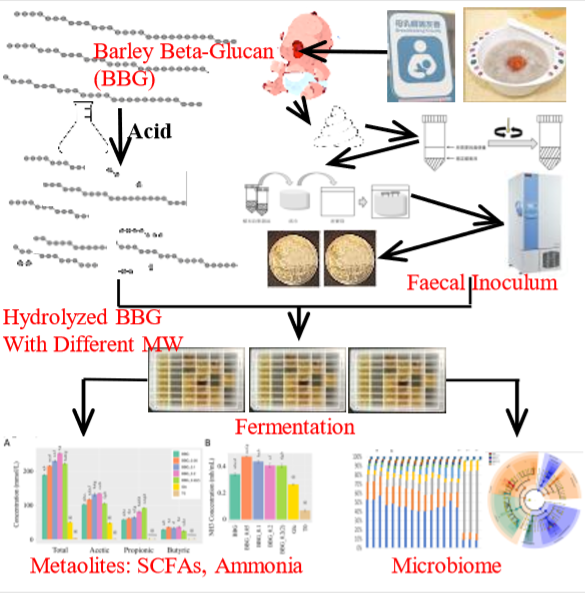
In Vitro Infant Faecal Fermentation of Low Viscosity Barley β-Glucan and Its Acid Hydrolyzed Derivatives: Evaluation of Their Potential as Novel Prebiotics

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results
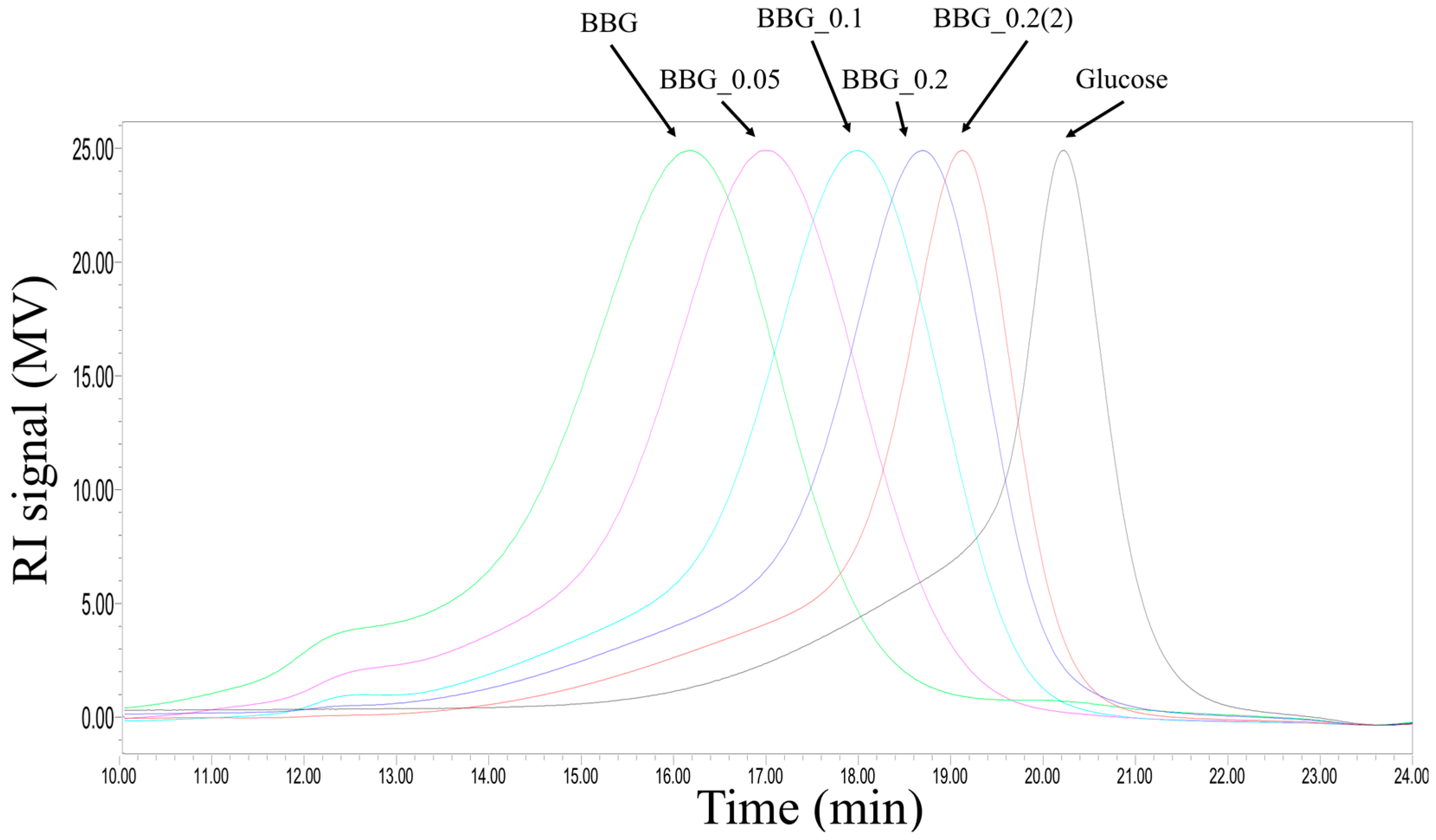
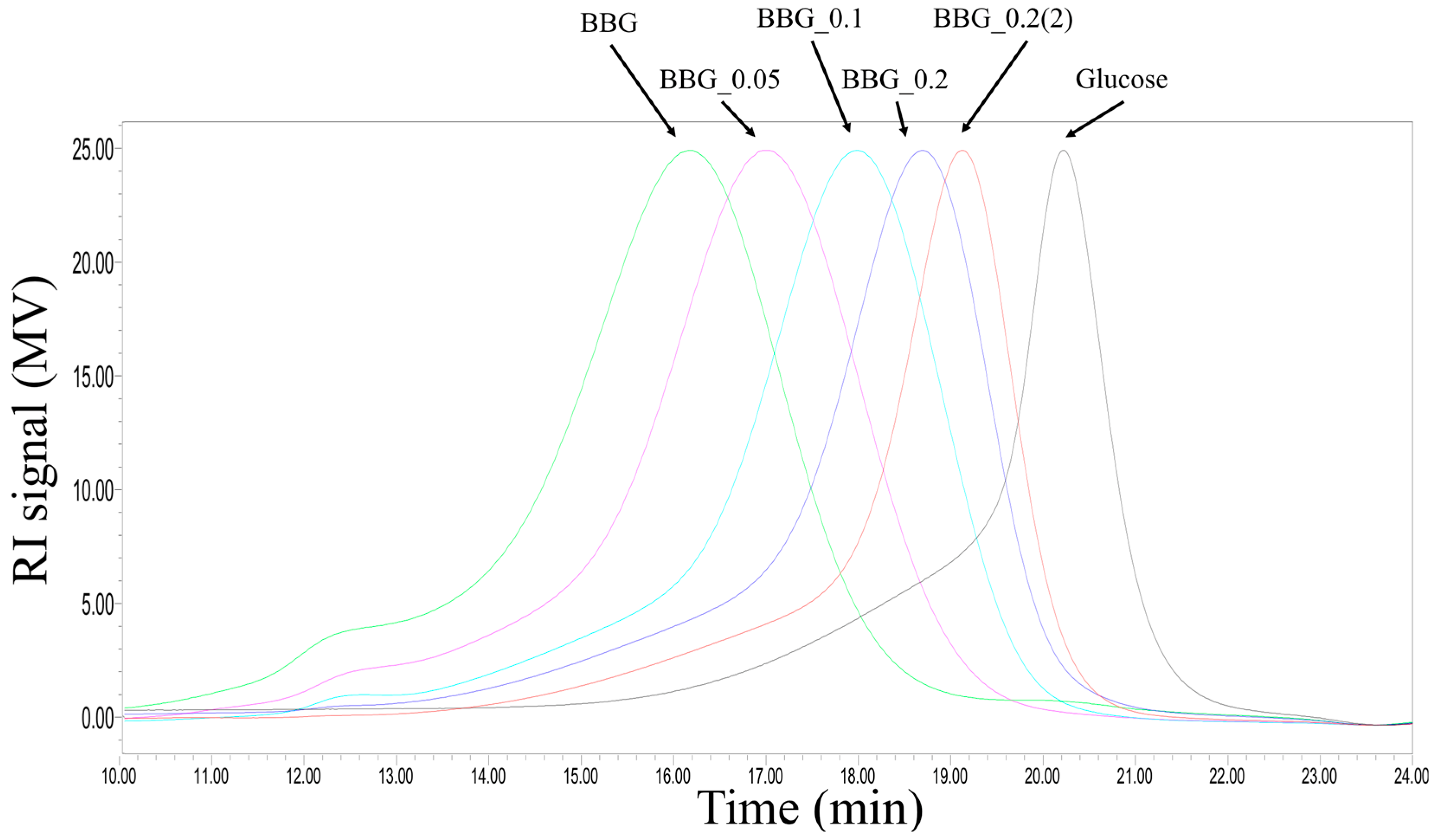
2.1. Chemical Composition and Structual Characterization of BBG Samples
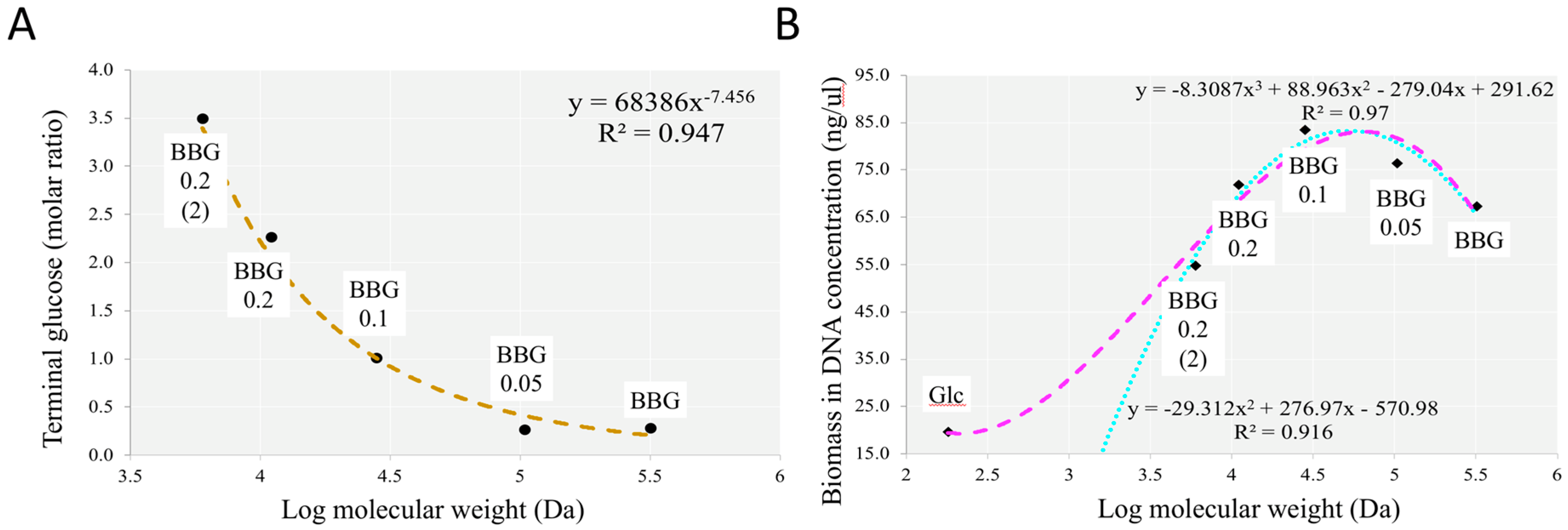
2.2. BBG In Vitro Fermentation Using Infant Faecal Inocula
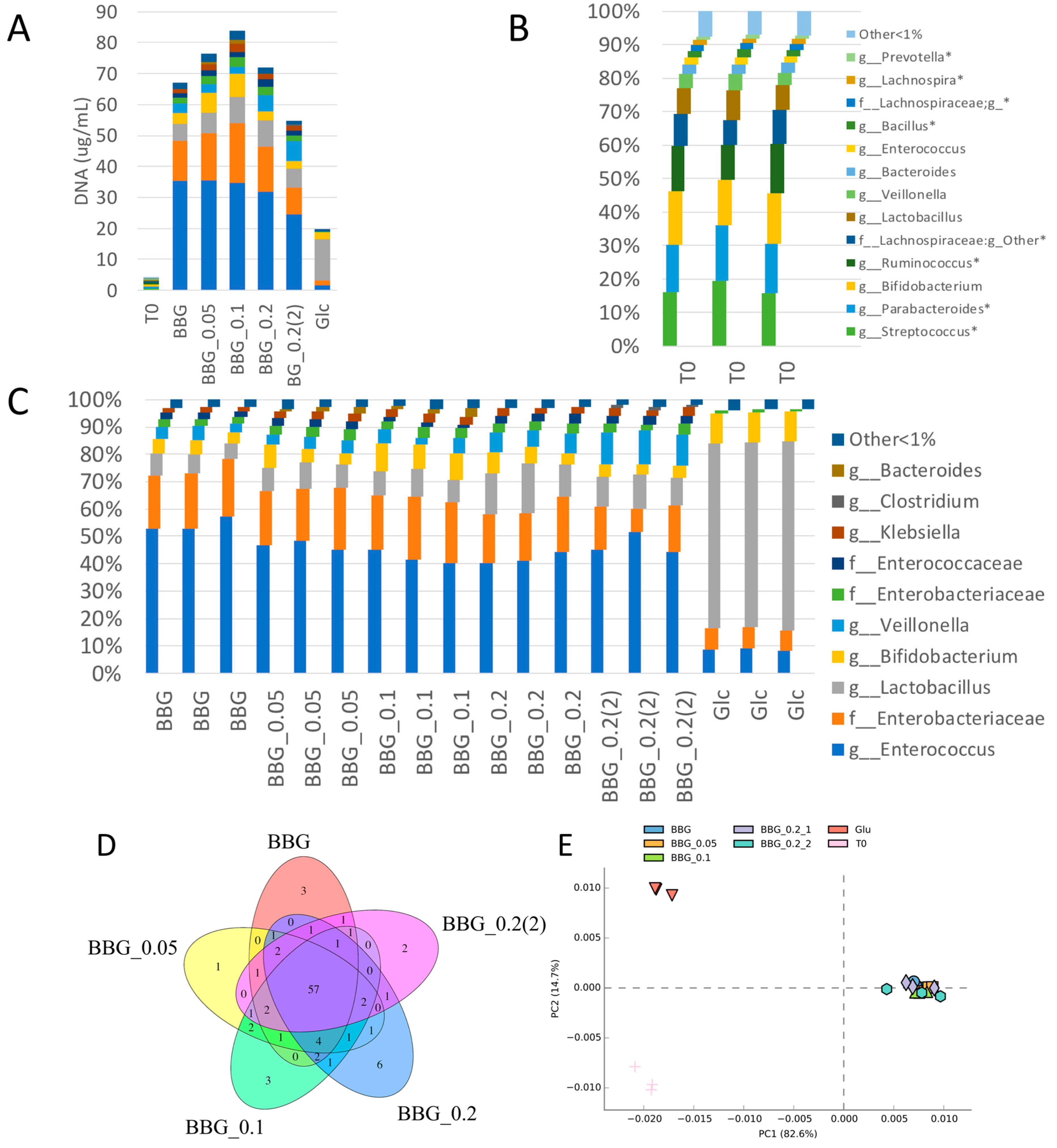
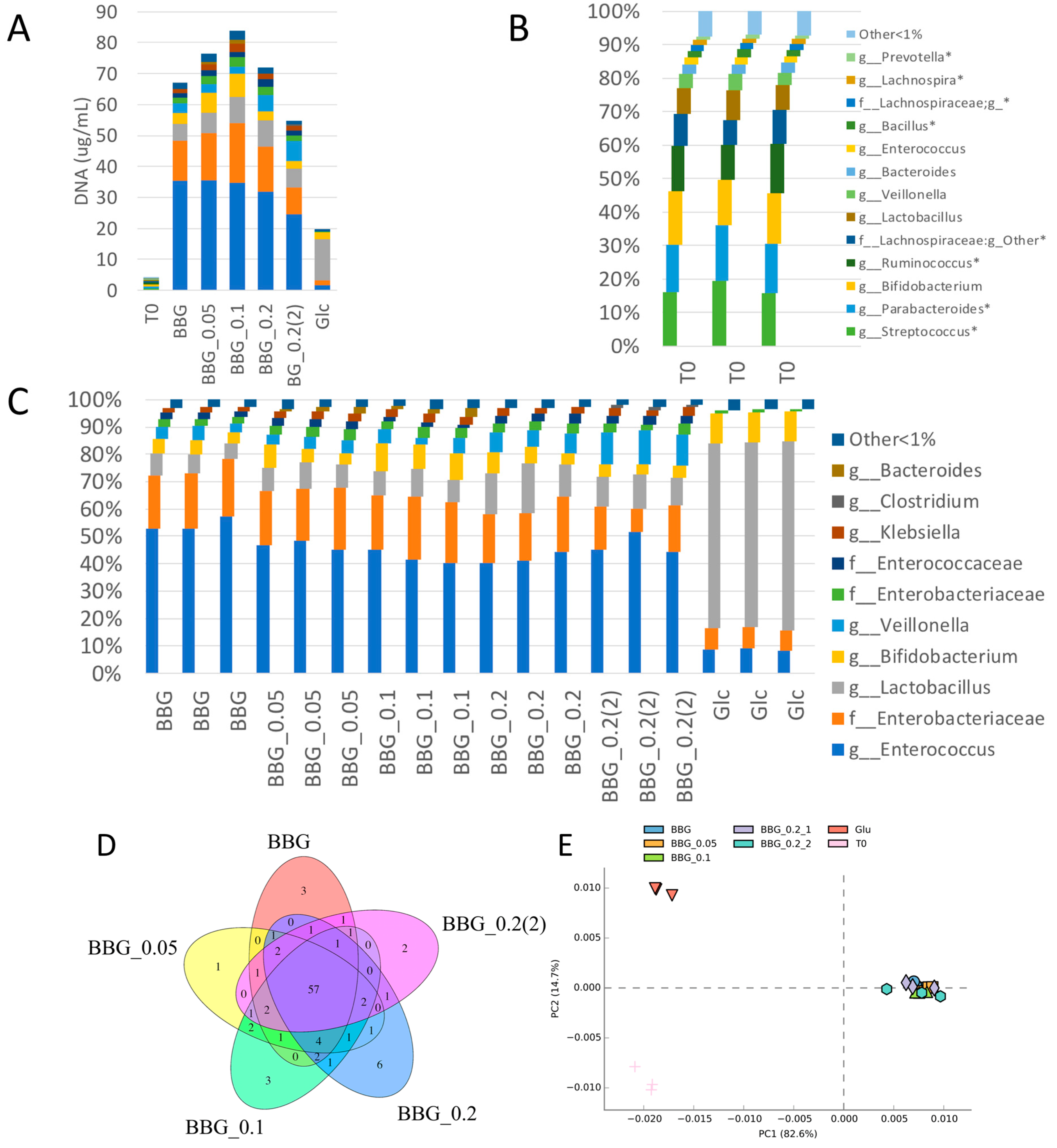
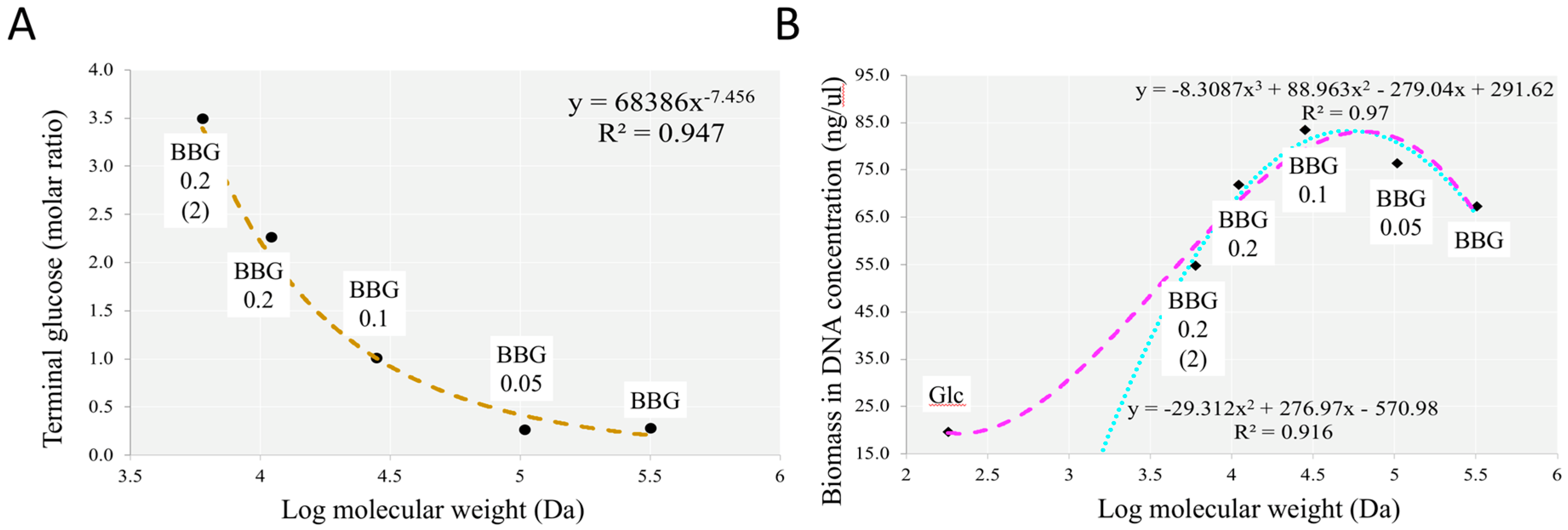
2.2.1. Fermentation of BBG Samples Evaluated by Total Bacterial Count and Total DNA Extracted
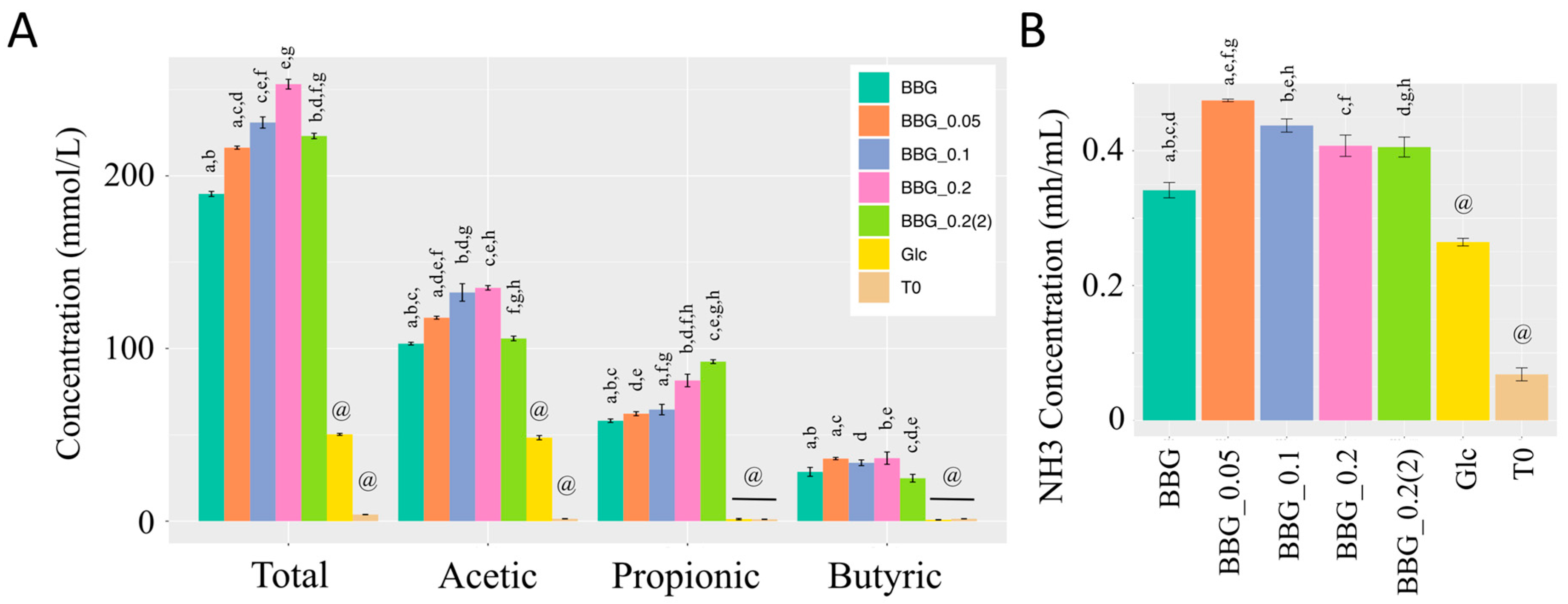
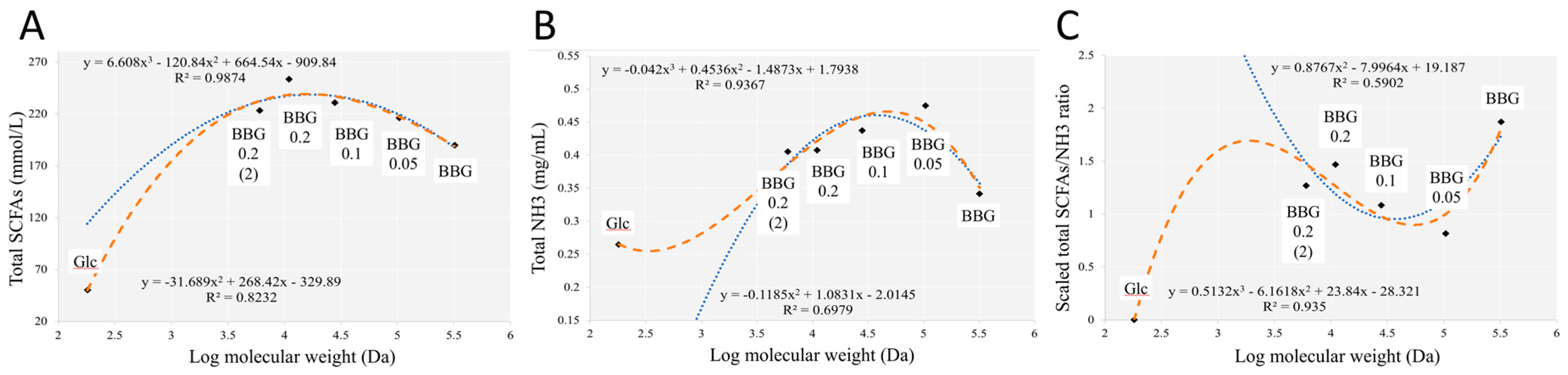
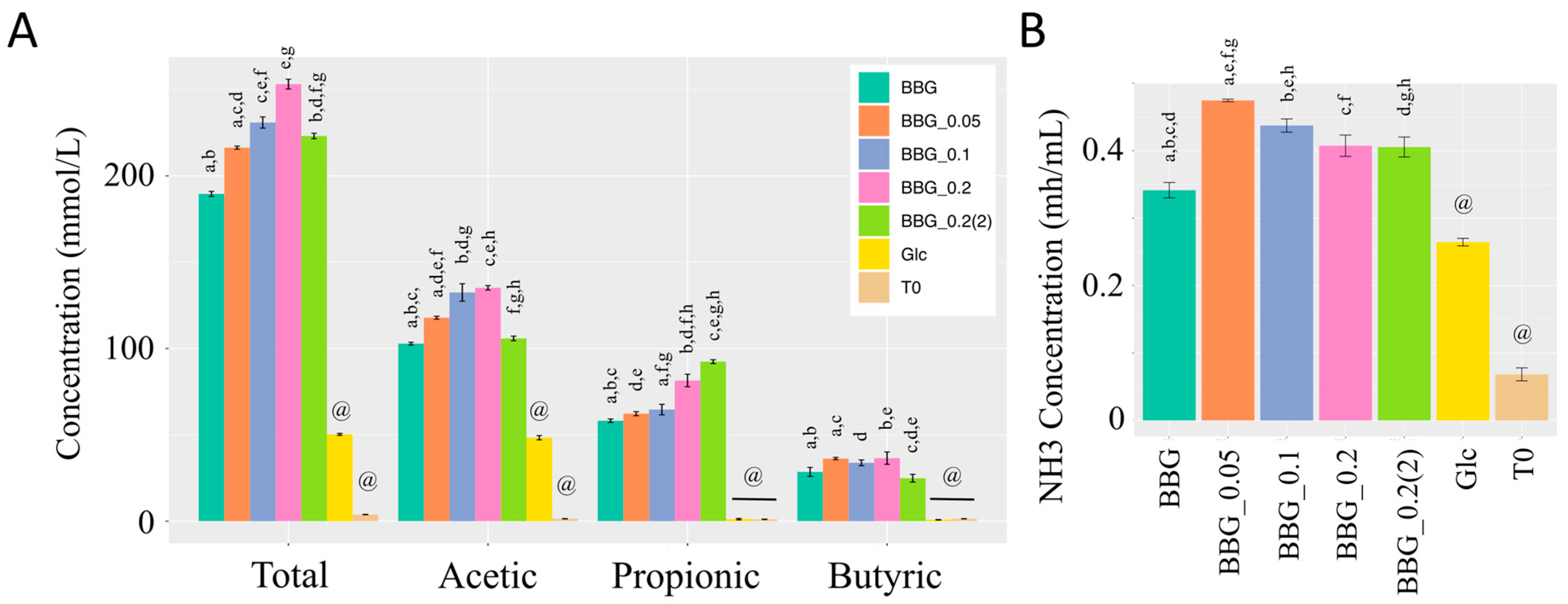
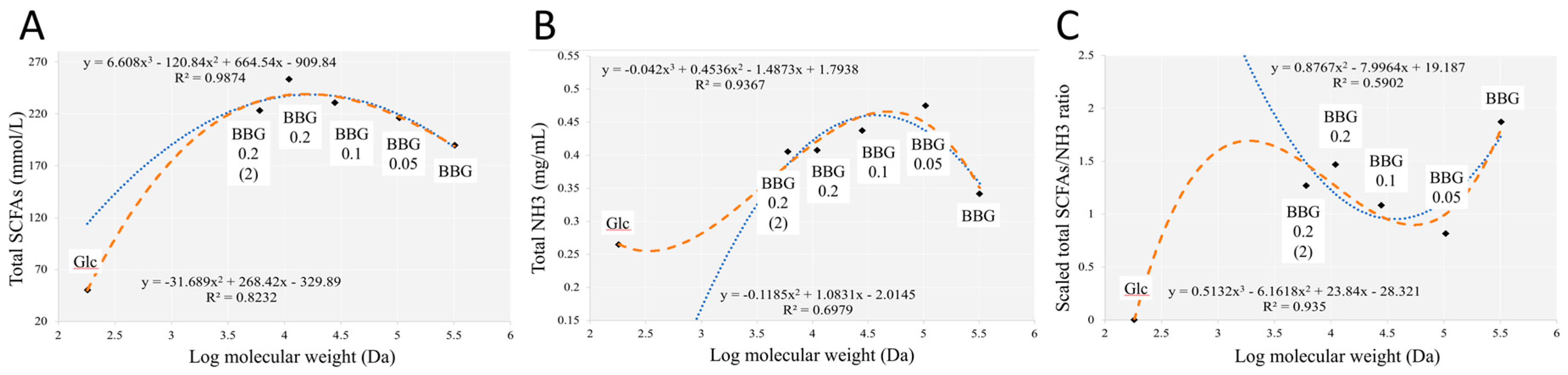
2.2.2. Short Chain Fatty Acids and Dissolved Ammonia Content after In Vitro Fermentation
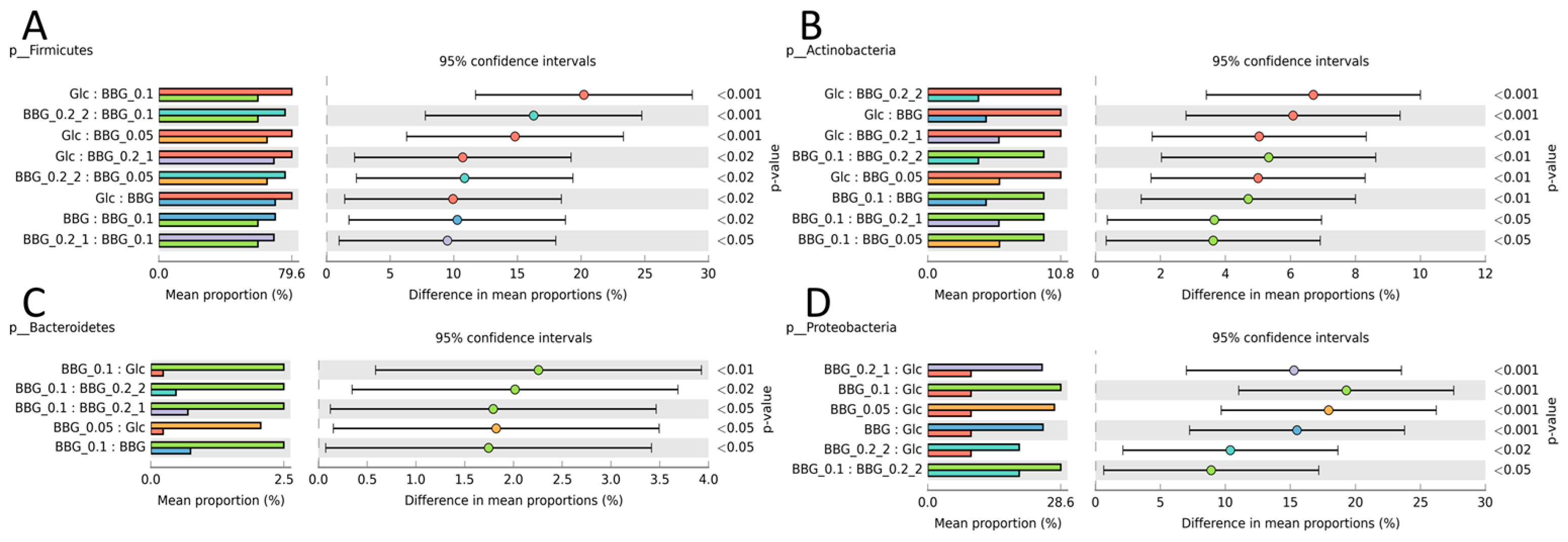
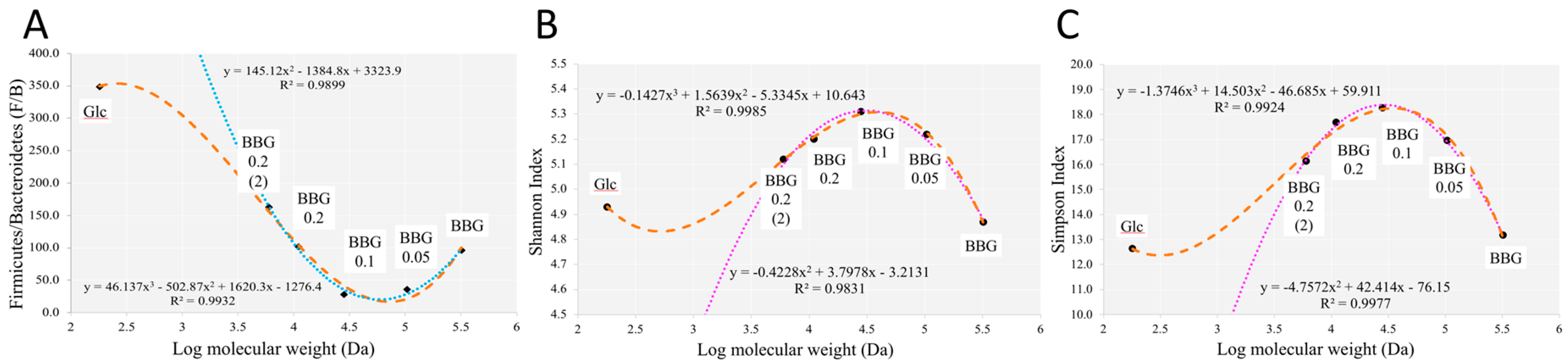
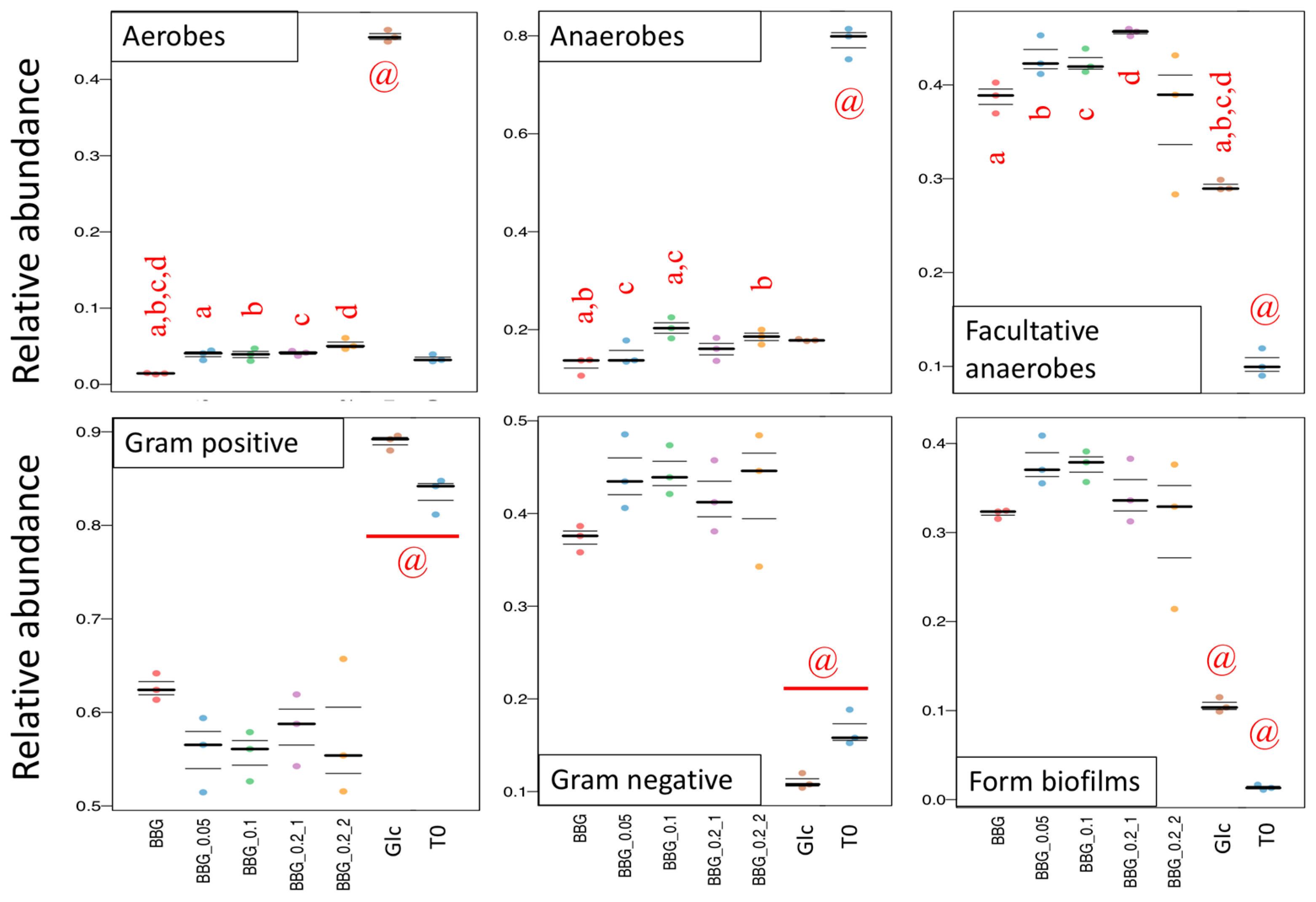
2.3. Microbiome Profile Changes after BBG Fermentation
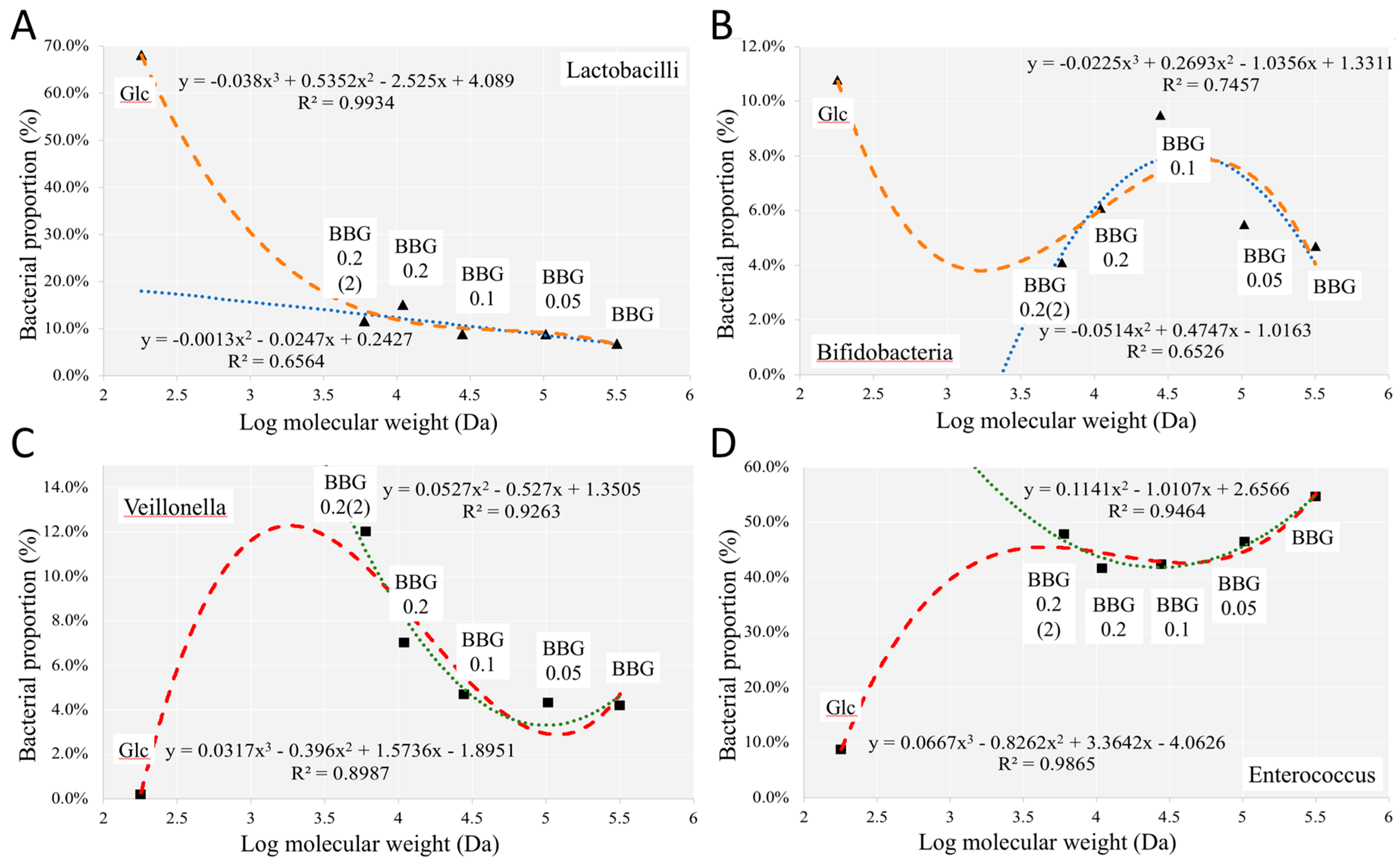
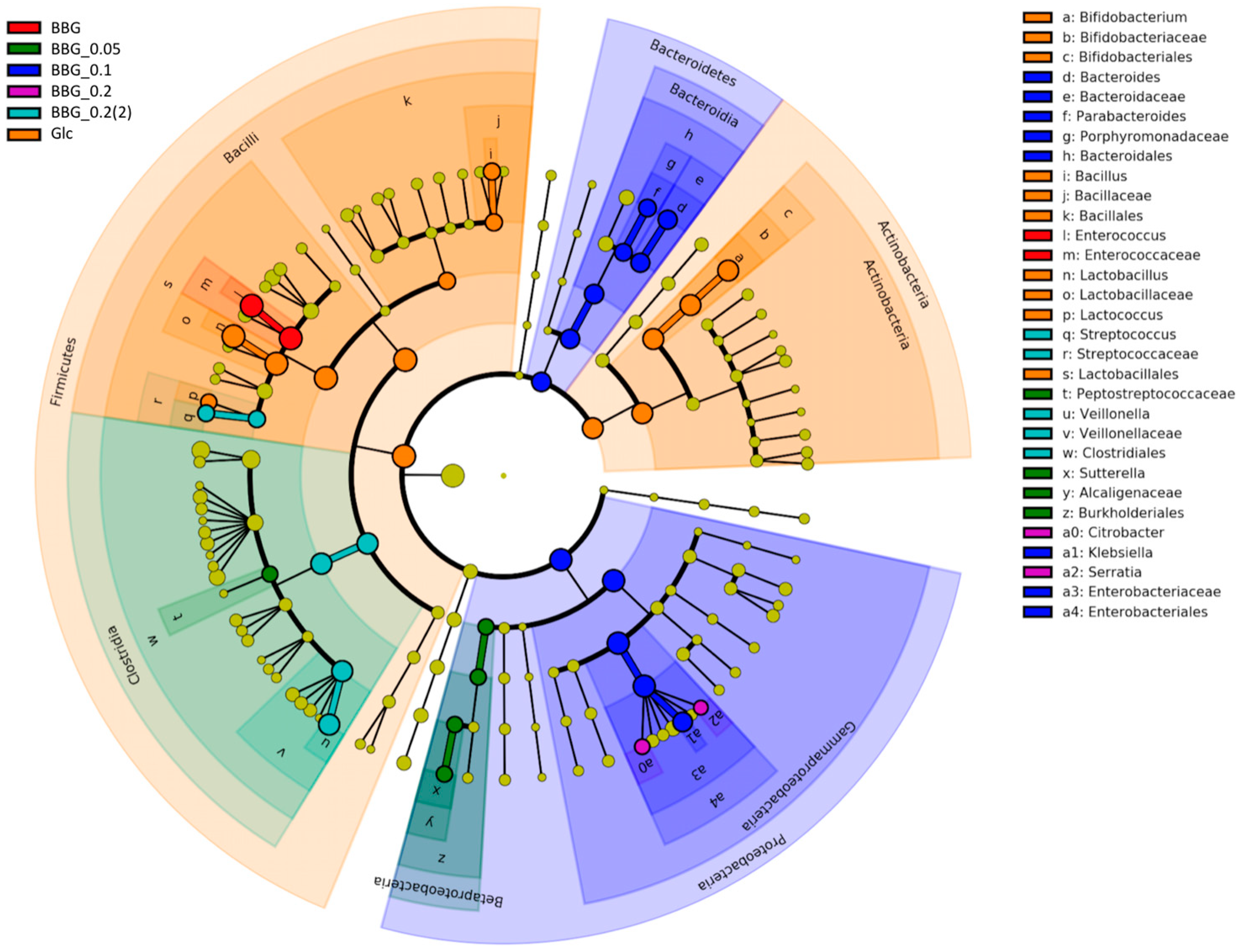
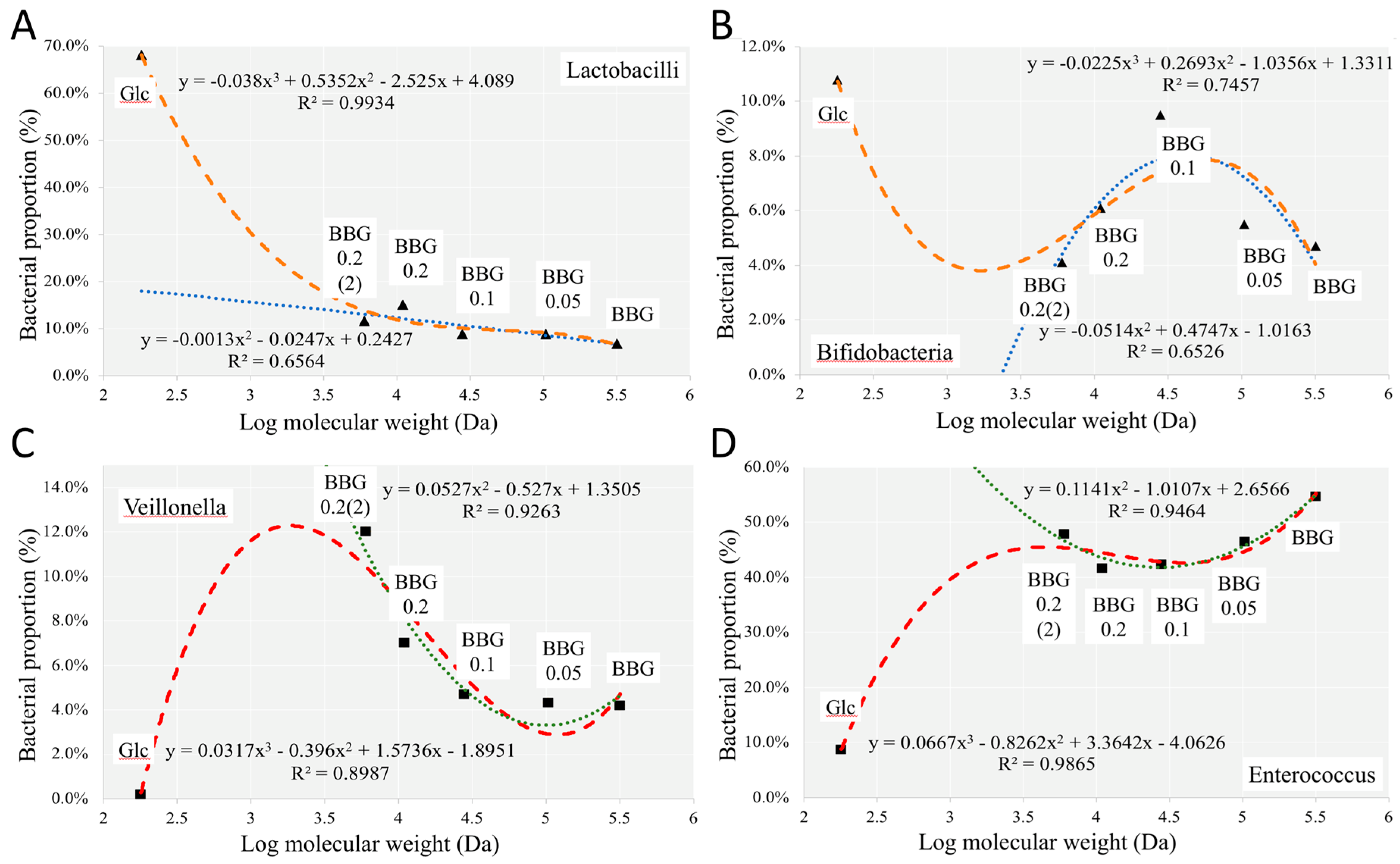
2.3.1. Change of Infant Faecal Microbiome Profile by Fermentation of BBG Samples
2.3.2. Metagenome Prediction of KEGG Functional Annotation
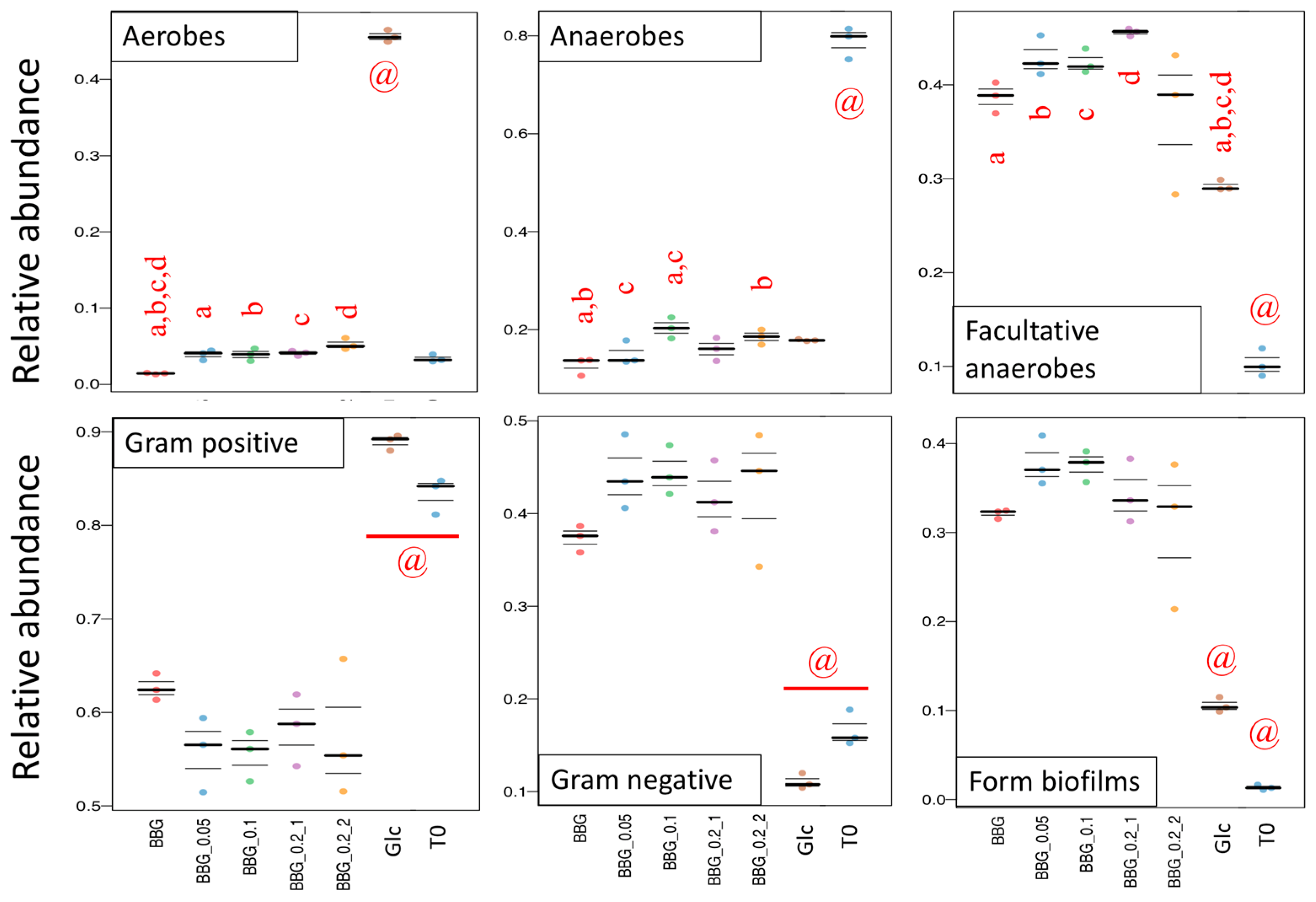
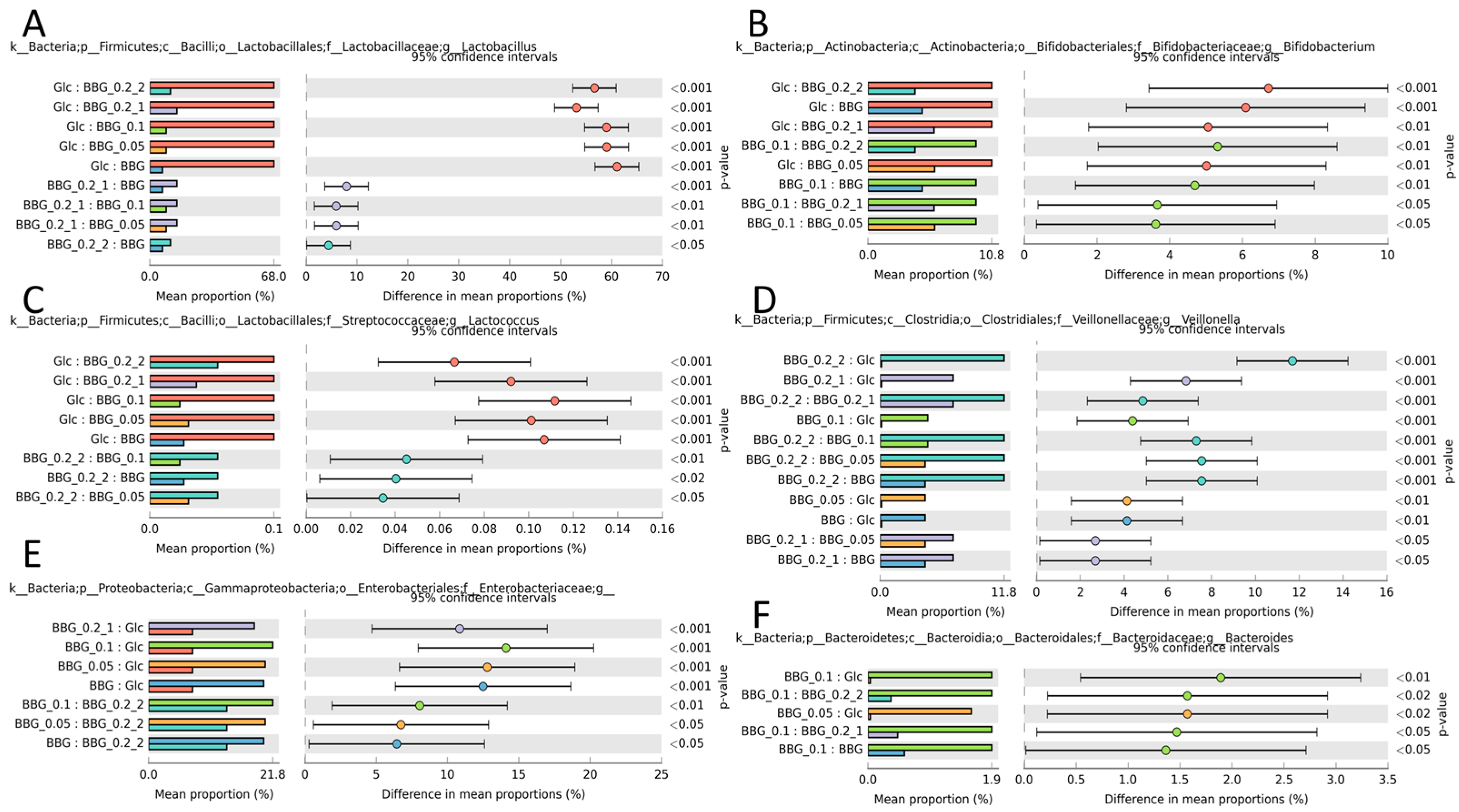
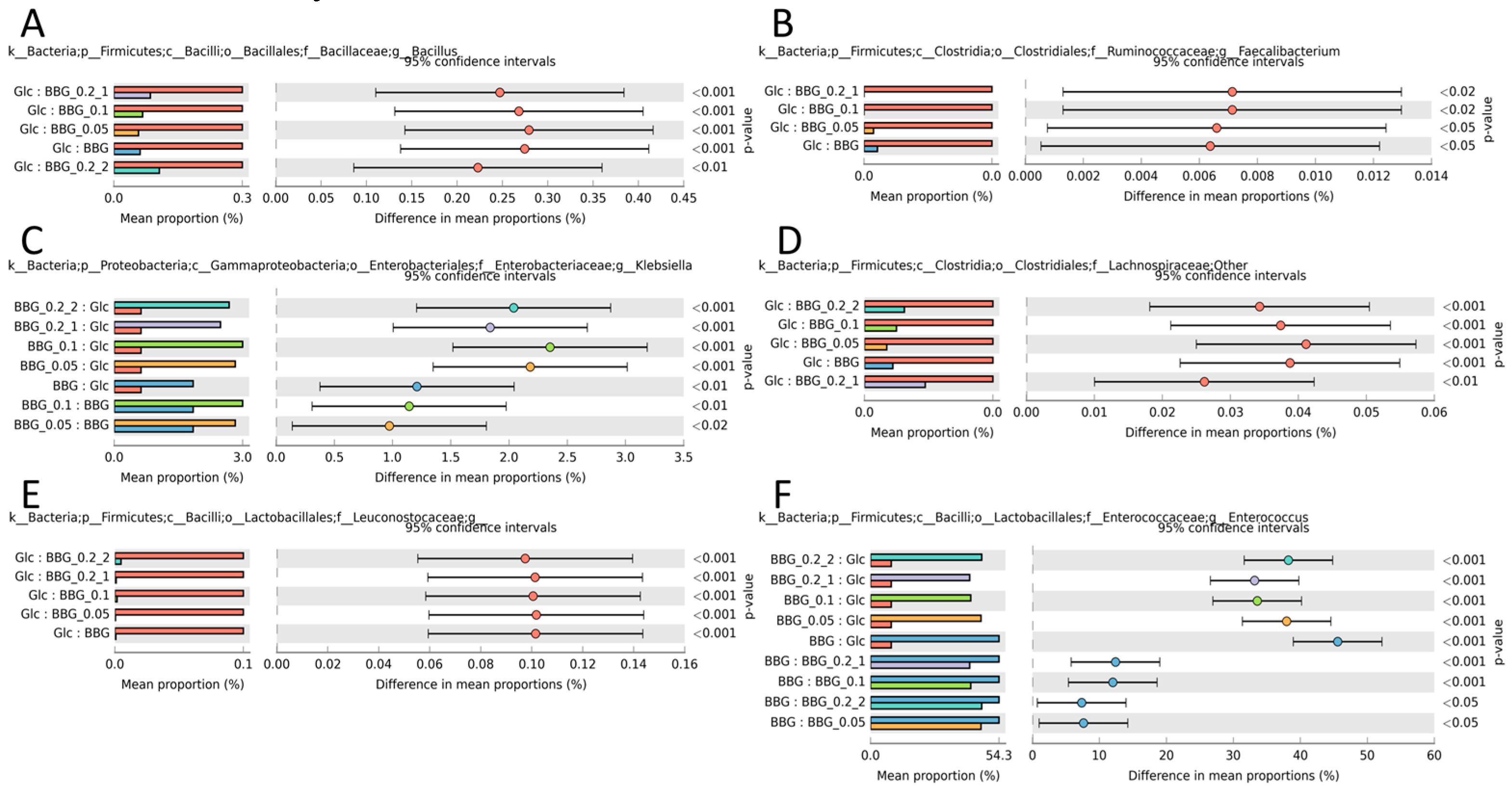
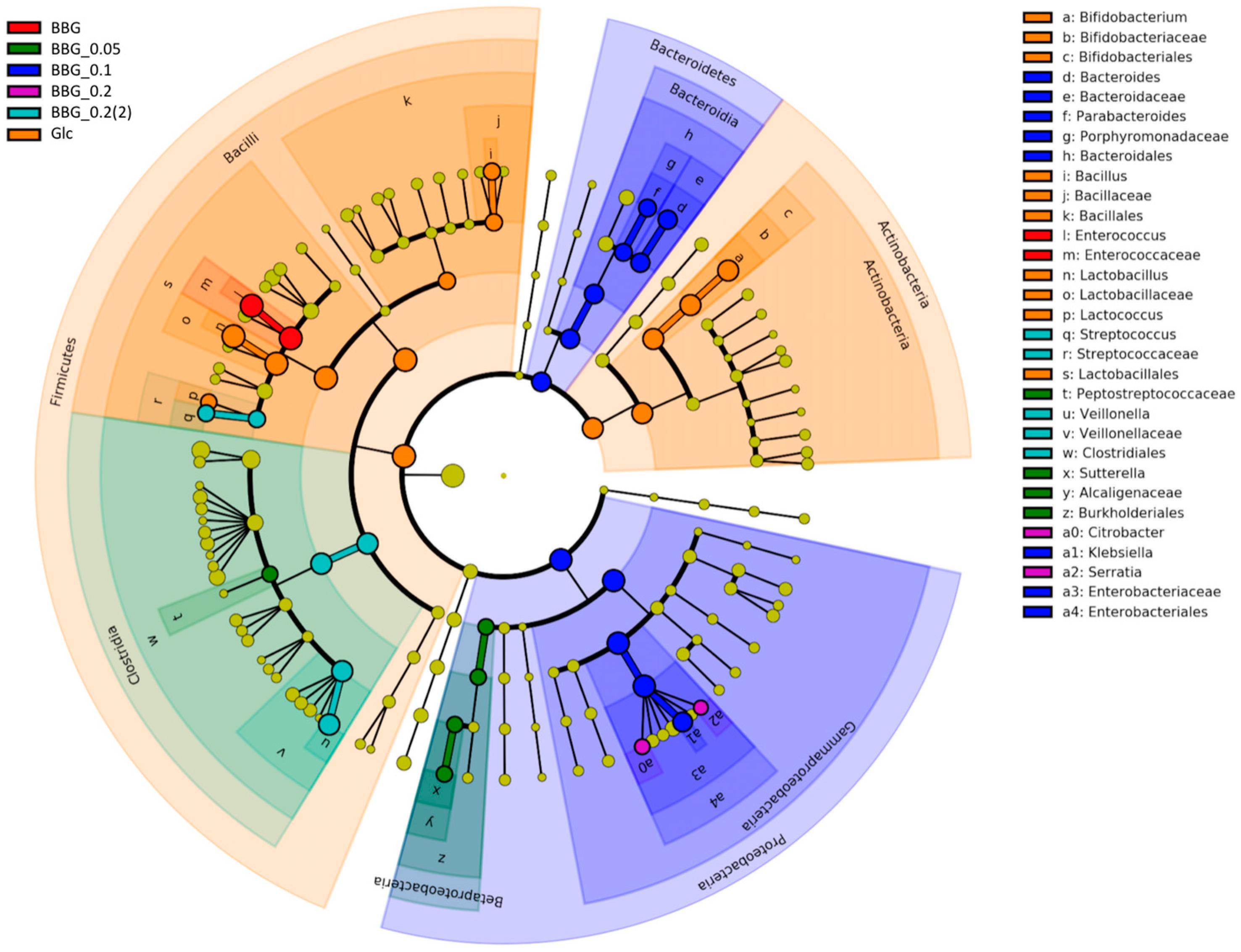
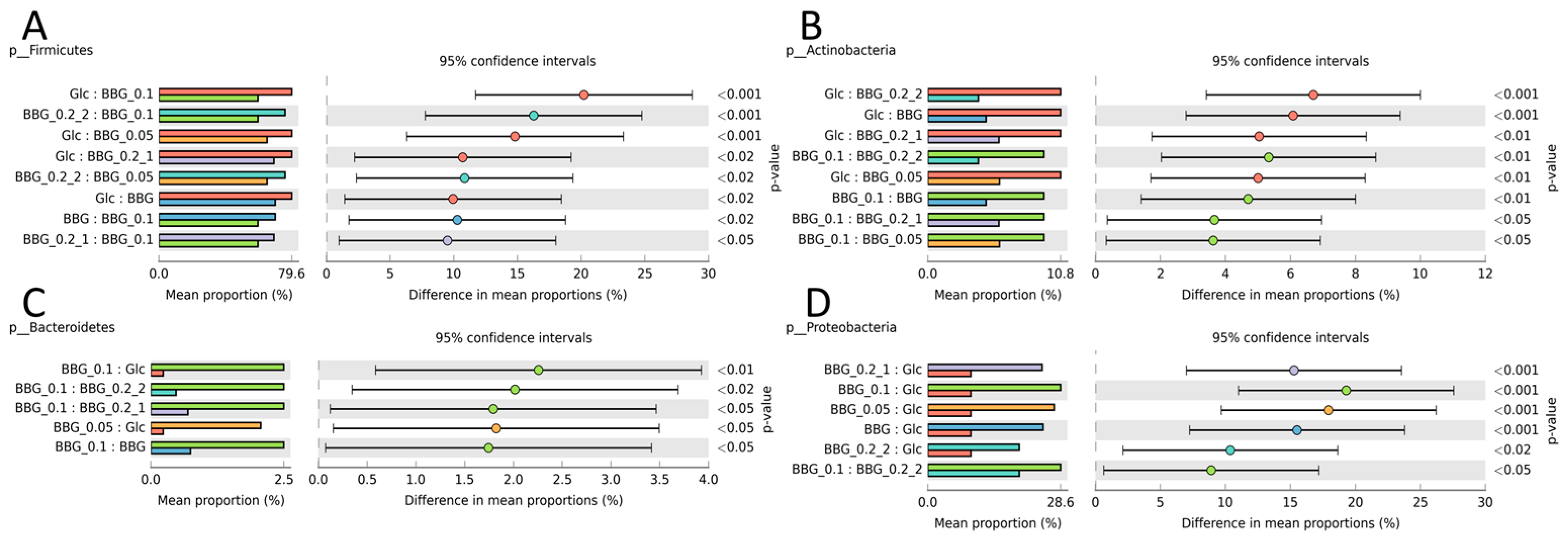
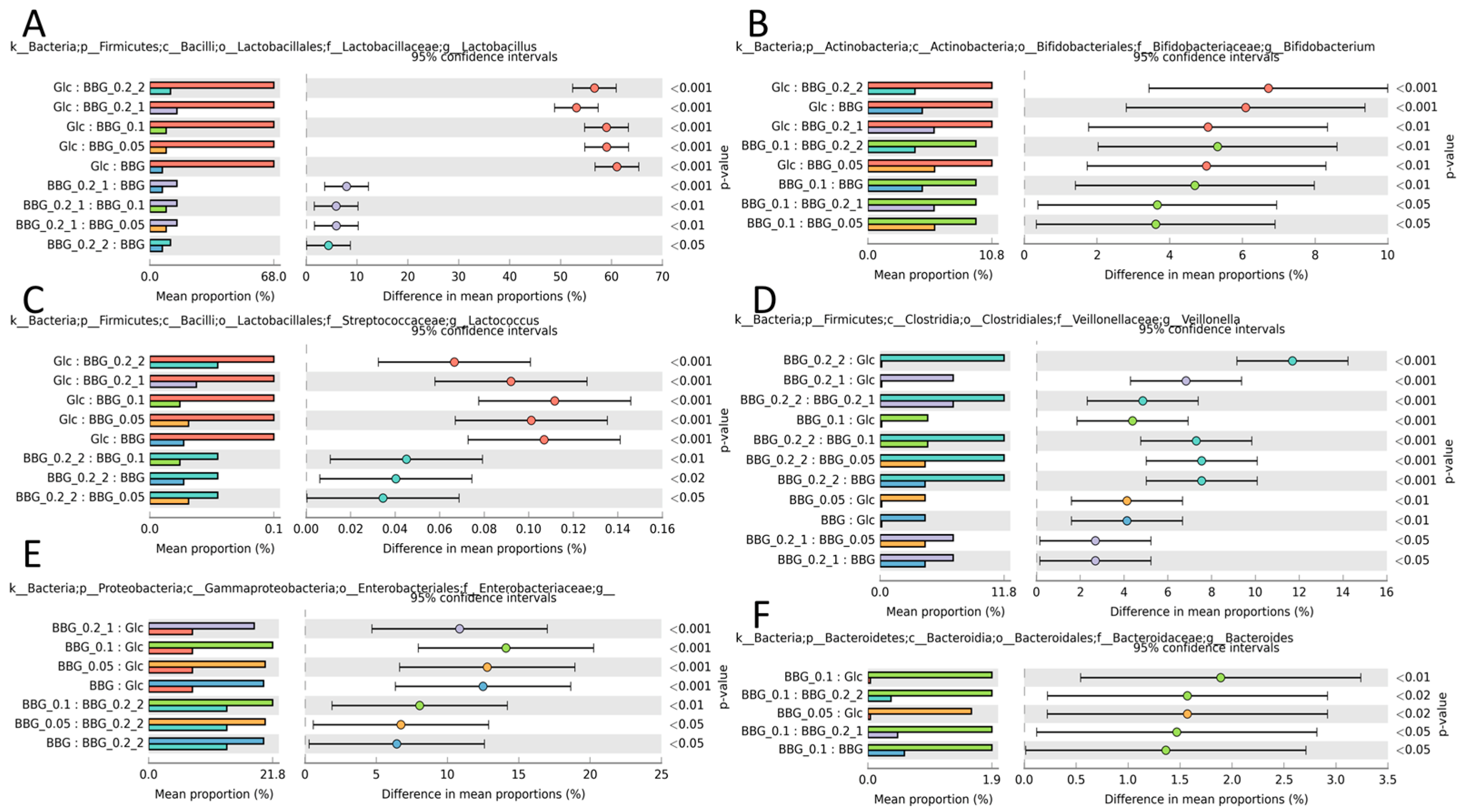
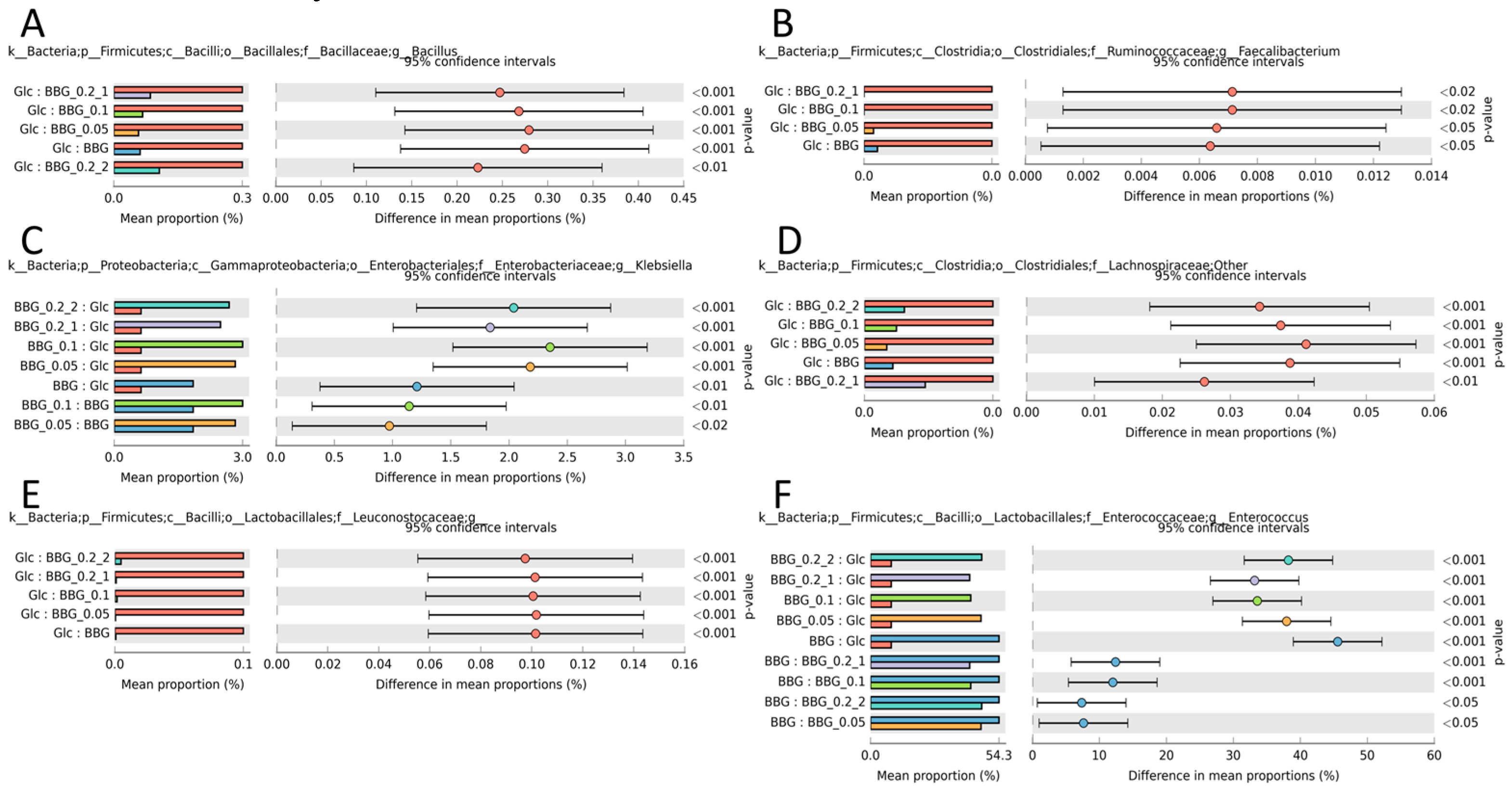
2.3.3. Microbial Biomarkers Identification
3. Discussion
3.1. Efficacy of Controlled Acid Hydrolysis in MW Reduction
3.2. Polymeric BBGs and Monomeric Glucose on Microbial Taxa
3.3. Effect of Polymeric BBGs and Monomeric Glucose on Saccharolytic and Proteolytic Profiles
3.4. Insights from the Phenotypic Data, Microbial Profiles against MW Reduction of BBGs
4. Materials and Methods
4.1. Materials
4.2. Infant Faecal Sample Collection
4.3. Depolymerization of Barley β-Glucans
4.4. Chemical Composition and Linkage Analysis of BBGs
4.5. Molecular Weight Distribution of BBGs
4.6. Spectroscopic Methods of BBGs
4.7. In Vitro Deep Well-Plate Fermentation
4.8. Standard Bacterial Plate Count
4.9. Short Chain Fatty Acids and Dissolved Ammonia Determination
4.10. DNA Extraction, 16S Amplicon Sequencing and Bioinformatics
4.11. Integration and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grain: World Markets and Trade (December report by United States Department of Agriculture Foreign Agricultural Service). Available online: https://downloads.usda.library.cornell.edu/usda-esmis/files/zs25x844t/8g84mr14m/0k225g11x/grain.pdf (accessed on 1 February 2019).
- Lam, K.L.; Cheung, P.C.K. Non-digestible long chain β-glucans as novel prebiotics. Bioact. Carbohydr. Dietary Fibre. 2013, 2, 45–64. [Google Scholar] [CrossRef]
- Jayachandran, M.; Chen, J.; Chung, S.S.M.; Xu, B. A critical review on the impacts of β-glucans on gut microbiota and human health. J. Nutr. Biochem. 2018, 61, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Du, B.; Xu, B. A critical review on production and industrial applications of β-glucans. Food Hydrocoll. 2016, 52, 275–288. [Google Scholar] [CrossRef]
- Hughes, S.A.; Shewry, P.R.; Gibson, G.R.; McCleary, B.V.; Rastall, R.A. In vitro fermentation of oat and barley derived β-glucans by human faecal microbiota. FEMS Microbiol. Ecol. 2008, 64, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Mikkelsen, D.; le Paih, L.; Gidley, M.J. In vitro fermentation kinetics and end-products of cereal arabinoxylans and (1,3;1,4)-β-glucans by porcine faeces. J. Cereal Sci. 2011, 53, 53–58. [Google Scholar] [CrossRef]
- Mikkelsen, M.S.; Jensen, M.G.; Nielsen, T.S. Barley β-glucans varying in molecular mass and oligomer structure affect cecal fermentation and microbial composition but not blood lipid profiles in hypercholesterolemic rats. Food Funct. 2017, 8, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K. Diet, microbiota, and metabolic health: Trade-off between saccharolytic and proteolytic fermentation. Annu. Rev. Food Sci. Technol. 2018, 9, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Olson, B.H. Rapid method for direct extraction of DNA from soil and sediments. Appl. Environ. Microbiol. 1991, 57, 1070–1074. [Google Scholar] [PubMed]
- Vogt, S.L.; Pena-Díaz, J.; Finlay, B.B. Chemical communication in the gut: Effects of microbiota generated metabolites on gastrointestinal bacterial pathogens. Anaerobe 2015, 34, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Hemsworth, G.R.; Dejean, G.; Rogers, T.E.; Pudlo, N.A.; Urs, K.; Jain, N.; Davies, G.J.; Martens, E.C.; Brumer, H. Molecular mechanism by which prominent human gut bacteroidetes utilize mixed-linkage β-glucans, major health-promoting cereal polysaccharides. Cell Rep. 2017, 21, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Riíos-Coviaán, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de los Reyes-Gavilaán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.; Haller, S.; Eckmanns, T.; Harder, T. Routine screening for colonization by Gram-negative bacteria in neonates at intensive care units for the prediction of sepsis: Systematic review and meta-analysis. J. Hosp. Infect. 2018, 99, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; a Reyes, J.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Ishibashi, K.; Yamanaka, D.; Adachi, Y.; Kanzaki, K.; Iwakura, Y.; Ohno, N. Production of low-molecular weight soluble yeast β-glucan by an acid degradation method. Int. J. Biol. Macromol. 2018, 107, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.; Virkki, L.; Anttila, H.; Esselstrom, H.; Tuomainen, P.; Sontag-Strohm, T. Hydrolysis of β-glucan. Food Chem. 2006, 97, 71–79. [Google Scholar] [CrossRef]
- Cho, H.J.; Jang, W.J.; Moon, S.Y.; Lee, J.M.; Kim, J.H.; Han, H.S.; Kim, K.W.; Lee, B.J.; Kong, I.S. Immobilization of β-1,3-1,4-glucanase from Bacillus sp. on porous silica for production of β-glucooligosaccharides. Enzyme Microb. Technol. 2018, 110, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, J.; Yan, Q.; You, X.; Yang, S.; Jiang, Z. In vitro digestibility and prebiotic potential of curdlan (1-3)-β-d-glucan oligosaccharides in Lactobacillus species. Carbohydr. Polym. 2018, 188, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Lee, E.; Oh, M.J.; Kim, Y.; Park, H.Y. High-glucose or-fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.K.; Caruso, T.; Buscot, F.; Fischer, M.; Hancock, C.; Maier, T.S.; Meiners, T.; Muller, C.; Obermaier, E.; Prati, D.; et al. Choosing and using diversity indices: Insights for ecological applications from the german biodiversity exploratories. Ecol. Evol. 2014, 4, 3514–3524. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Deehan, E.C.; Walter, J.; Backhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; White, P.J. Optimizing the molecular weight of oat β-glucan for In vitro bile acid binding and fermentation. J. Agric. Food Chem. 2011, 59, 10322–10328. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yu, X.; Dong, L.; Shen, R. In vitro fermentation of oat β-glucan and hydrolysates by fecal microbiota and selected probiotic strains. J. Sci. Food Agric. 2017, 97, 4198–4203. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A. What is microbial community ecology? ISME J. 2009, 3, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the teddy study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, J.; Liu, C.; Xiong, Q.; Duan, H.; Cheung, P.C.K. Stable and biocompatible mushroom β-glucan modified gold nanorods for cancer photothermal therapy. J. Agric. Food Chem. 2017, 65, 9529–9536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Cheung, P.C. Fermentation of β-glucans derived from different sources by bifidobacteria: Evaluation of their bifidogenic effect. J. Agric. Food Chem. 2011, 59, 5986–5992. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Choi, M.W.; Li, X.; Cheung, P.C.K. Immunomodulatory effect of structurally-characterized mushroom sclerotial polysaccharides isolated from Polyporus rhinocerus on human monoctyes THP-1. J. Funct. Foods 2018, 41, 90–99. [Google Scholar] [CrossRef]
- Liu, C.R.; Chen, J.L.; Chen, L.; Huang, X.S.; Cheung, P.C.K. Immunomodulatory activity of polysaccharide-protein complex from the mushroom sclerotia of Polyporus rhinocerus in murine macrophages. J. Agric. Food Chem. 2016, 64, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, B.; Chen, J.; Cheung, P.C.K. Cell wall structure of mushroom sclerotium (Pleurotus tuber-regium): Part 2. Fine structure of a novel alkali-soluble hyper-branched cell wall polysaccharide. Food Hydrocoll. 2014, 38, 48–55. [Google Scholar] [CrossRef]
- Lam, K.L.; Keung, H.Y.; Ko, K.C.; Kwan, H.S.; Cheung, P.C.K. In vitro fermentation of β-glucans and other selected carbohydrates by infant fecal inoculum: An evaluation of their potential as prebiotics in infant formula. Bioact. Carbohydr. Dietary Fibre 2018, 14, 20–24. [Google Scholar] [CrossRef]
- Milani, C.; Hevia, A.; Foroni, E.; Duranti, S.; Turroni, F.; Lugli, G.A.; Sanchez, B.; Martin, R.; Gueimonde, M.; van Sinderen, D.; et al. Assessing the fecal microbiota: An optimized ion torrent 16S rRNA gene-based analysis protocol. PLoS ONE 2013, 8, 68739. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Walron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism level microbiome phenotypes. bioRxiv 2017, 133462. [Google Scholar] [CrossRef]
- R Core Team. A language and environment for statistical computing. R Foundation for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 1 February 2019).
Sample Availability: Samples of the compounds BBGs are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples * | Carbohydrate (% by wt) | Protein (% by wt) | Molecular Weight (kDa) |
|---|---|---|---|
| BBG | 98.0 ± 0.7 | 0.2 ± 0.5 | 319 |
| BBG_0.05 | 98.2 ± 0.3 | 0.2 ± 0.7 | 104 |
| BBG_0.1 | 97.6 ± 0.3 | 0.2 ± 0.1 | 28 |
| BBG_0.2 | 98.1 ± 0.5 | 0.2 ± 0.3 | 11 |
| BBG_0.2(2) | 97.9 ± 0.4 | 0.2 ± 0.1 | 6 |
| Samples | Peak Area (%) a | Relative Molar Ratio c T-Glcp:1,3-Glcp:1,4-Glcp | Relative Molar Ratio c T-Glcp:1,3-Glcp:1,4-Glcp | ||
|---|---|---|---|---|---|
| T-Glcp b | 1,3-Glcp | 1,4-Glcp | |||
| BBG | 3.99 ± 1.00 | 14.25 ± 0.15 | 81.76 ± 0.85 | 1:3.57:20.49 | 0.28:1:5.74 |
| BBG_0.05 | 3.76 ± 0.94 | 14.15 ± 0.14 | 82.09 ± 0.80 | 1:3.77:21.84 | 0.27:1:5.80 |
| BBG_0.1 | 13.22 ± 2.99 | 13.11 ± 0.45 | 73.68 ± 2.54 | 1:0.99:5.58 | 1.01:1:5.62 |
| BBG_0.2 | 22.46 ± 4.55 | 9.94 ± 0.58 | 67.60 ± 3.97 | 1:0.44:3.01 | 2.26:1:6.80 |
| BBG_0.2(2) | 32.28 ± 5.73 | 9.24 ± 0.78 | 58.48 ± 4.94 | 1:0.29:1.81 | 3.49:1:6.33 |
| Samples | Total Plate Count (CFU) | Ratio of CFUS (Relative to Glucose) | [DNA] (µg/mL) * | Ratio of [DNA] (Relative to Glucose) |
|---|---|---|---|---|
| BBG | 8.7 × 104 | 3.7 | 67.3 ± 7.4 a,b | 3.4 |
| BBG_0.05 | 2.3 × 104 | 1.0 | 76.4 ± 4.0 c,d | 3.9 |
| BBG_0.1 | 5.4 × 104 | 2.3 | 83.5 ± 9.1 a,e,f | 4.2 |
| BBG_0.2 | 6.8 × 104 | 2.9 | 71.9 ± 4.1 g,h | 3.6 |
| BBG_0.2(2) | 5.2 × 104 | 2.2 | 54.8 ± 0.6 c,g,i | 2.8 |
| GLC | 2.4 × 104 | 1 | 19.7 ± 2.4 b,d,e,f,h,i | 1 |
| T0 | 6.4 × 103 | 0.3 | 4.3 ± 0.2 @ | 0.2 |
| Samples | Phylogeny-Based | Non-Phylogeny-Based | ||||
|---|---|---|---|---|---|---|
| PD Whole Tree | Fisher Alpha | Berger Parker D | Shannon | Simpson_E | Simpson Reciprocal | |
| BBG | 17.68 ± 0.77 a,b | 90.81 ± 4.66 a,bc | 0.18 ± 0.01 a,b,c,d,e | 4.87 ± 0.08 a,b,c,d | 0.026 ± 0.001 a | 13.17 ± 0.84 a,b,c,d |
| BBG_0.05 | 18.69 ± 0.50 | 100.40 ± 1.89 | 0.14 ± 0.01 a,f | 5.22 ± 0.07 a,e | 0.031 ± 0.002 b | 16.96 ± 0.92 a,e |
| BBG_0.1 | 19.95 ± 0.59 a | 103.76 ± 5.26 a | 0.15 ± 0.01 b,g | 5.31 ± 0.11 b,f | 0.033 ± 0.001 a,c | 18.27 ± 1.15 b,f |
| BBG_0.2 | 19.41 ± 0.95 | 105.66 ± 2.98 b | 0.13 ± 0.01 c,h | 5.21 ± 0.04 c,g | 0.031 ± 0.000 d | 17.69 ± 0.51 c,g |
| BBG_0.2(2) | 19.85 ± 0.53 b | 107.50 ± 5.94 c | 0.15 ± 0.01 d,i | 5.12 ± 0.08 d | 0.028 ± 0.004 | 16.14 ± 1.50 d,h |
| GLC | 19.34 ± 0.81 | 101.11 ± 5.89 | 0.22 ± 0.01 e,f,g,h,i | 4.93 ± 0.02 e,f,g | 0.023 ± 0.002 b,c,d | 12.63 ± 0.25 e,f,g,h |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, K.-L.; Ko, K.-C.; Li, X.; Ke, X.; Cheng, W.-Y.; Chen, T.; You, L.; Kwan, H.-S.; Cheung, P.C.-K. In Vitro Infant Faecal Fermentation of Low Viscosity Barley β-Glucan and Its Acid Hydrolyzed Derivatives: Evaluation of Their Potential as Novel Prebiotics. Molecules 2019, 24, 828. https://doi.org/10.3390/molecules24050828
Lam K-L, Ko K-C, Li X, Ke X, Cheng W-Y, Chen T, You L, Kwan H-S, Cheung PC-K. In Vitro Infant Faecal Fermentation of Low Viscosity Barley β-Glucan and Its Acid Hydrolyzed Derivatives: Evaluation of Their Potential as Novel Prebiotics. Molecules. 2019; 24(5):828. https://doi.org/10.3390/molecules24050828
Chicago/Turabian StyleLam, Ka-Lung, Kin-Chun Ko, Xiaojie Li, Xinxin Ke, Wai-Yin Cheng, Tianfeng Chen, Lijun You, Hoi-Shan Kwan, and Peter Chi-Keung Cheung. 2019. "In Vitro Infant Faecal Fermentation of Low Viscosity Barley β-Glucan and Its Acid Hydrolyzed Derivatives: Evaluation of Their Potential as Novel Prebiotics" Molecules 24, no. 5: 828. https://doi.org/10.3390/molecules24050828
APA StyleLam, K.-L., Ko, K.-C., Li, X., Ke, X., Cheng, W.-Y., Chen, T., You, L., Kwan, H.-S., & Cheung, P. C.-K. (2019). In Vitro Infant Faecal Fermentation of Low Viscosity Barley β-Glucan and Its Acid Hydrolyzed Derivatives: Evaluation of Their Potential as Novel Prebiotics. Molecules, 24(5), 828. https://doi.org/10.3390/molecules24050828